
A mechanistic study of carbonyl activation under solvent-free conditions: evidence drawn from the synthesis of imidazoles†
Kiran Pradhana,
Bipransh Kumar Tiwaryb,
Mossaraf Hossaina,
Ranadhir Chakrabortyb and
Ashis Kumar Nanda*a
aDepartment of Chemistry, University of North Bengal, Darjeeling, India 734013. E-mail: ashis_nanda@hotmail.com; Tel: +91-9474329191
bDepartment of Biotechnologyy, University of North Bengal, Darjeeling, India 734013
First published on 21st January 2016
Abstract
Syntheses of various imidazoles and their derivatives, imidazole N-oxides and 1-hydroxyimidazole 3-oxides, from sterically different dicarbonyl moieties provided insights into the self-catalytic effect of the condensed phase reactions of carbonyl compounds. The self-catalytic activity in solvent-free multi-component syntheses was investigated using a combination of methods viz., reactivity, spectroscopy and theory. While IR spectroscopic studies revealed that reacting molecules were polarised in bulk, quantum mechanical calculations of associated HCHO monomers suggest an increase in the average dipole moment of each monomer and provide evidence for the presence of cooperative effects. A comparative study of the kinetics of un-catalysed and catalysed reactions with the help of HPLC provided insights into the mechanism.
1 Introduction
The application of a solvent-free synthetic methodology for organic compounds that were conventionally synthesized in a solvent medium has become more common in recent years.1 Thus, well-known organic reactions such as the aldol,2 Claisen,3 Stobbe4 and Knoevenagel condensations,5 the Thorpe,6 Tischenko,7 Reformatsky and Luche reactions,8 the Baeyer–Villiger oxidation,9 the pinacol rearrangement10 and the oxidative coupling of phenols,11 just to name a few, have been found not only to occur under solvent-free conditions but are more efficient than reactions in solution. In addition to the simplicity and cleanness of the procedure, the absence of any media has been found to lead to uncommon reactivities. Apart from these efforts, over the past two decades organic synthesis has been devoted to extending the scope of solvent free reactions, notably for newer multi-component reaction (MCR) strategies.12 Solvent-free protocols have already been developed for almost all the classical multi-component reactions namely the Strecker,13 Hantzsch,14 Biginelli,15 Mannich,16 Passerini,17 Ugi,18 Gewald,19 Petasis,20 Radziwenski synthesis,21 and so on. And to impart greater reactivity to the substrates and the reagents, these synthetic protocols have constructively exploited the use of transition metal catalysts and the very recent organocatalysts.Lewis acids and organocatalysts have been shown to be excellent catalysts for C–C bond forming reactions, and through carbonyl activation provide effective avenues for upgrading classical MCRs. In particular, close attention has to be paid to condensation reaction mechanisms that involve the activation of carbonyl-containing electrophiles. The vast number of investigations related to chemical transformation mediated by organocatalysts22 or by Lewis acids in anhydrous organic media,23 aqueous media24 and in the absence of any media25 has provided a basic understanding of the intricate dynamics of adduct formation between the acid and the carbonyl moieties and has shed light on the role that these adducts have in accelerating catalytic transformations. A noteworthy example is the dramatic increase in reaction rates for the Radziwenski synthesis attained by the activation of a carbonyl group by a Lewis acid center25 or by a Lewis base center26 through the formation of adducts.
In the present study we have attempted to explore in some details the mechanistic insight for better reaction under solvent-free condition compared to reactions in solution. Here we have chosen a well-studied method for imidazole synthesis.
2 Experimental
The compounds 1a–1i, 2a–2l, 3a–3g, 4a–4g and 5a–5e were all prepared under solvent-free conditions. All other reagents are commercially available and were used as purchased from the supplier. Grinding experiments were performed with an agate mortar and pestle, which was acetone rinsed and dried prior to use. The yield of the reaction was determined by the product peak area count in HPLC analysis with respect to the purified product. NMR spectra were recorded on Bruker Avance 300 spectrometer chemical shifts (δ in ppm) were referenced to external SiMe4. IR spectra were recorded on a FTIR-8300 SHIMADZU spectrophotometer. Analytical thin-layer chromatography was performed using silica gel aluminium sheets (Merck, TLC silica gel 60 F254). HPLC was performed on a Waters-2487 Dual Lambda absorber with a RP-18 (Symmetry Shield) column. The solvent used was methanol with a flow rate of 0.5 ml min−1. In all experiments the same column and same flow rate was maintained. The electrospray mass spectra were recorded on a MICROMASS QUATTRO II triple quadruple mass spectrometer. The ESI capillary was set at 3.5 kV and cone voltage was 40 V. X-Ray diffraction data was collected on a Bruker APEX CCD-II diffractometer at 293 K. The structure was solved by a direct method using the program SHELXS-97.272.1 Computational details
The optimized geometry and the energy of each monomer were calculated using the CBS-QB3 model chemistry. The CBS-QB3 model chemistry28 was employed for the calculation of the molecular structures since this method is known to predict thermochemical parameters with high accuracy.29 Molecular geometry was fully optimized and harmonic vibrational frequencies were calculated using the analytic second derivatives to confirm the convergence to minima on the potential surface. All the calculations were carried out using the Gaussian 98 suite of programs.302.2 General procedure for the synthesis of 2,4,5-trisubstituted imidazoles (1a–1i)
210 mg (1 mmol) of benzil, 1 mmol of the corresponding aldehyde and 770 mg (10 mmol) of ammonium acetate was taken in an agate mortar and thoroughly ground. The contents were transferred to a test tube and heated to 150–160 °C for 4 minutes. The contents were cooled and water was added to the test tube and filtered. The product was recrystallised from ethanol. Completion of the reaction was checked by TLC.2.3 General procedure for the synthesis of 1,2,4,5-tetrasubstituted imidazoles (2a–2l)
210 mg (1 mmol) of benzil, 1 mmol of aldehyde, 1 mmol of primary amine and 385 mg (5 mmol) of ammonium acetate was taken in an agate mortar and thoroughly ground. The contents were transferred to a test tube and heated to 150–160 °C for 4 minutes. The contents were cooled and water was added to the test tube and filtered. The product was recrystallised from ethanol. Completion of the reaction was checked by TLC.2.4 General procedure for the synthesis of imidazole N-oxide (3a–3g)
1 mmol of the monoxime, 1 mmol of the aldehyde and 385 mg (5 mmol) of ammonium acetate was ground into an intimate mixture in an agate mortar and pestle. The mixture was then heated to 115–120 °C in an oil bath with constant shaking. A black solution resulted which was cooled when a black sticky precipitate formed. To the black precipitate was then added a small volume of diethyl ether when a brown precipitate separated. The precipitate was then thoroughly washed with ethyl acetate, dissolved in ethanol and crystallized by addition of water to yield pure products.2.5 General procedure for the synthesis of N-substituted imidazole-1-oxide (4a–4g)
1 mmol of the monoxime, 1 mmol of the aldehyde and 1.5 mmol of the amine was ground for 2 minutes and subsequently heated in an oil bath at 115–120 °C, when a melt was formed. After a further 8 minutes of heating, the completion of the reaction was indicated by TLC. On cooling the melt slowly solidified and to the product so formed was added a little amount of ether whereby a precipitate was obtained. The precipitate was further washed with hot ethyl acetate. Recrystallization from ethanol gave products with the same melting points.2.6 General procedure for the synthesis of 1-hydroxy imidazole-3-oxide (5a–5e)
2 mmol each of the monoxime and the aldehyde was thoroughly ground with 695 mg (10 mmol) of hydroxylamine hydrochloride in an agate mortar and pestle for a period of ca. 3 minutes during which it melts and then gets hardened slowly. The mixture was then transferred to a test tube and heated in an oil bath maintained at 110–120 °C when it started to melt. Constant shaking for another 7 minutes gave the product which remained in the melt form even at room temperature. On completion of the reaction, checked by TLC, addition of 5 ml of diethyl ether or 5 ml of ethyl acetate precipitated the product. The water insoluble products were then washed with water and ethyl acetate to get the pure products.3 Results and discussion
The Radziwenski Imidazole (R-I) synthesis is a very useful preparative method for imidazole and its derivatives. The manifold utilizes a diketone, an aldehyde and ammonium acetate as the ammonia source (Scheme 1). The pure products have been synthesized in quantitative yields in 4 minutes at the given temperature by heating in an oil bath under solvent-free conditions. The temperature conditions and product yield were optimised by HPLC studies (ESI†).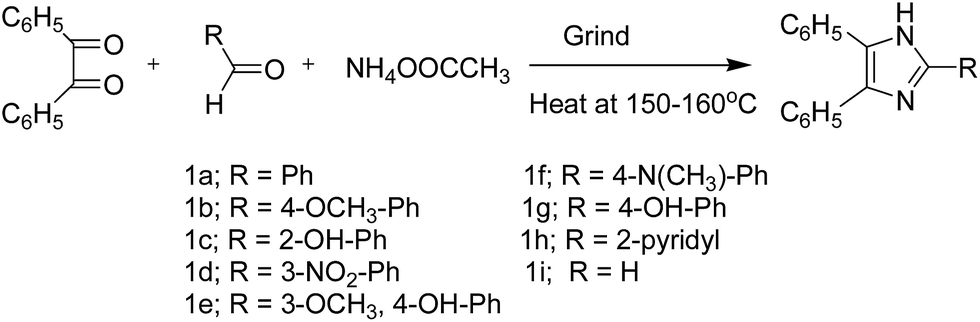 | ||
| Scheme 1 R-I synthesis of tri-substituted imidazoles (1a–1i). | ||
Besides usual spectroscopic methods for determining the structure, we have used single crystal X-ray diffraction (Sc-XRD) data of one of the representative compounds, 4,5-diphenyl-1H-imidazole, 1i, to confirm the structure. The single crystals of the compound suitable for Sc-XRD were obtained by the slow evaporation from methanol/hexane mixture. The compound crystallizes in a monoclinic crystal system with the space group P21/c (Hall group −P2ybc); a = 11.0471(4) Å, b = 9.2483(3) Å, c = 11.5780(4) Å, α = 90°, β = 93.921(3)°, γ = 90°, Z = 4, μ = 0.577 mm−1, F000 = 468.0 and Kα = 1.54184 Å. The ORTEP diagram is presented in Fig. 1. The heterocyclic ring is planar. One interesting point in the diagram is the attachment of one proton in each nitrogen atom which reflects overall an excess of one proton. The N7–C11 and N9–C10 bond distances are identical (1.380 Å and 1.3799 Å respectively), similarly N7–C8 and N9–C8 bond distances are also close (1.3157 Å and 1.3462 Å respectively); corresponding N7 and N9 centered bond angles are also close (107.83° and 105.91°).
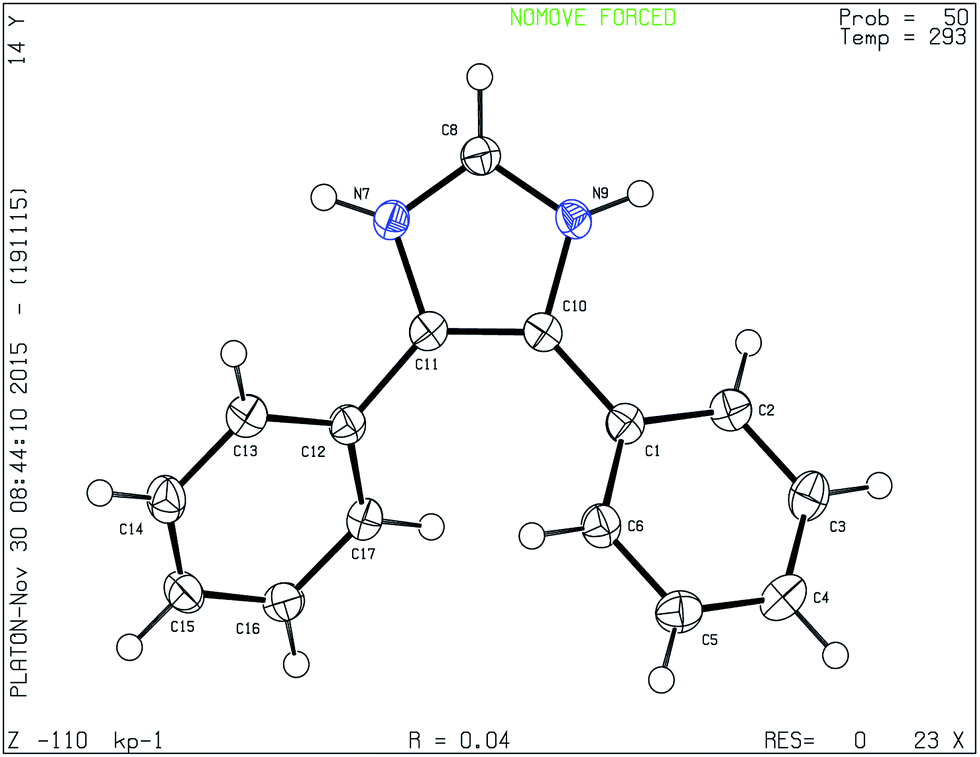 | ||
| Fig. 1 ORTEP diagram of 1i derived from single crystal data. | ||
Intermolecular extra proton transfer rate in imidazole is reported in the order of 0.3 × 10−12 second31 but there is no such data for intramolecular N to N proton transfer due to the shift of N–C double bond in imidazole; intuitively it is difficult to say that the process could be faster than X-ray diffraction time (10−18 second). However, the bond lengths are in between C–N single bond (1.47 Å) and double bond (1.25 Å). Since both the nitrogen atoms are identical, and on probability basis H atoms might have added to both the atoms during computation. The extra proton does not originate from the imidazolinium salt because in such case the counter ion should be in the unit cell; and non equivalence of the nitrogen atoms would have been observed.
However, these reactions are not very efficient in solution, and the yield of product is not very high. Existing literature reveals that the Debus–Radziszewski imidazole synthesis in solution state takes around 24 h to achieve moderate to good yield.25 By contrast, solvent-free reactions proceed efficiently under much simpler conditions to give products in quantitative yield (Scheme 2).
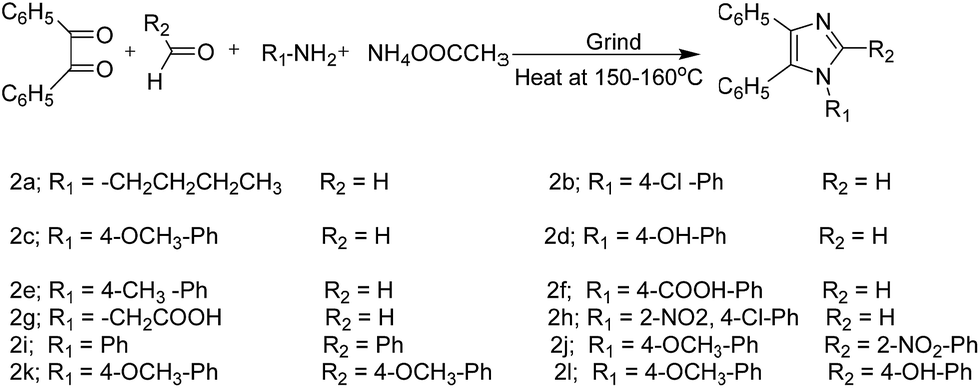 | ||
| Scheme 2 R-I synthesis of tetra-substituted imidazoles (2a–2l). | ||
Interestingly, very similar solvent-free reactions of various ammonia derivatives and alkyl/aryl monoximes at 115–120 °C for 10 min also gave the corresponding imidazole N-oxides (3a–3g), N-substituted imidazole N-oxides (4a–4g) and 1-hydroxy imidazole 3-oxide derivatives (5a–5e) in quantitative yields (Scheme 3). This is the first report of a solvent-free condensed phase protocol for the oxide and hydroxyl oxide derivatives of imidazoles. Apart from the usual characterizations of the compounds, further spectroscopic investigations, vibrational assignments, HOMO–LUMO, NBO and MEP analysis of two of the synthesized compounds 3a32 and 3c33 have already been done and reported. The syntheses also proves that acetic acid, generated in situ in the reaction, is not the only catalysing factor for enhancing the reaction rates under solvent-free conditions.
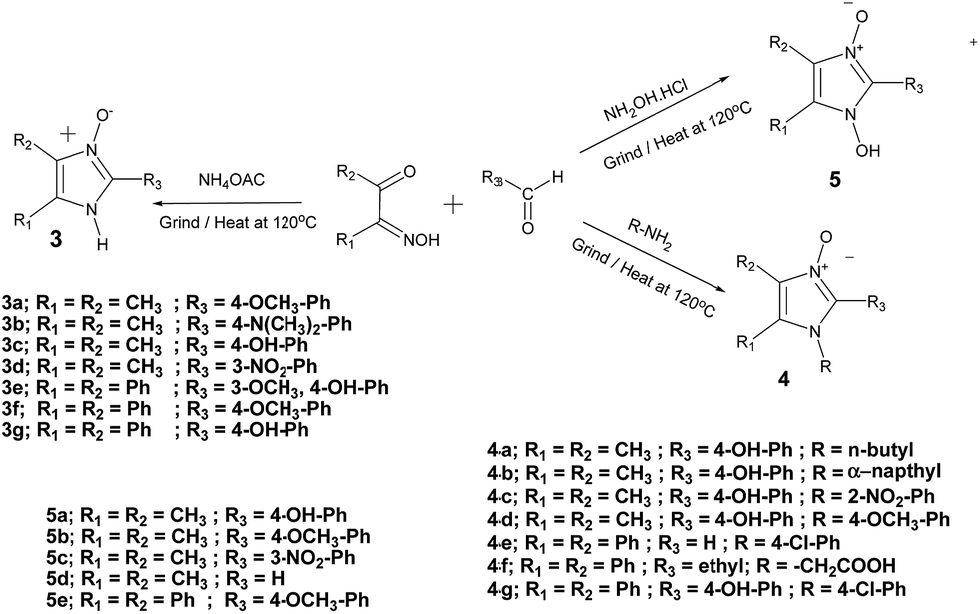 | ||
| Scheme 3 Analogous R-I synthesis of imidazole N-oxides (3a–3g), N-substituted imidazole N-oxides (4a–4g) and 1-hydroxy imidazole 3-oxides (5a–5e). | ||
In order to understand, at least partially, why these reactions proceed so efficiently under solvent-free conditions, a combination of methods viz., reactivity, spectroscopy and theory have been used.
3.1 Kinetic studies
Firstly, the reaction kinetics of imidazoles 1b and 1c were studied through HPLC monitoring of benzil and aldehyde (reactants) consumption along with product (imidazoles) formation. A good linearity was observed in each case with the plots of natural logarithm values (−ve for the reactants and +ve for the products) of peak area against time. From the slope of these curves the first order rate constants and half lives (t1/2) were determined. The observed dependence of reactants' concentrations (logarithm) and product formation with the variation of time (at reaction temperature 125 °C, temperature optimised for maximum conversion) is shown in Fig. 2. The product peak in HPLC was not much prominent within the first 10 minutes. This information helps to understand more about the sequences of reactions which manifest together as a multi-component reaction.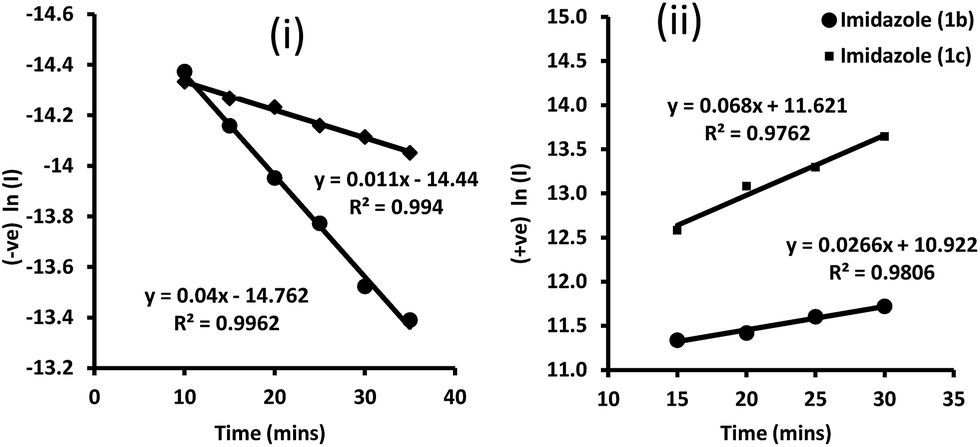 | ||
| Fig. 2 (−ve)/(+ve) ln(I) vs. time (mins) of [i] benzil during formation of imidazoles (1b) and (1c); [ii] rate curve of the product imidazoles (1b) and (1c) formation; I = peak area. | ||
The catalytic effect of some metal salts (5 mol%) at the same reaction temperature (125 °C) was also compared. The corresponding rate constant and half life values are shown in Table 1.
| Catalyst | Rate of benzil consumption (t1/2)a | Rate of aldehyde consumption (t1/2) | Rate of product formation (t1/2) |
|---|---|---|---|
| a Note: half-life (t1/2) in minutes. | |||
| Solvent-free (no catalyst) | 0.011 (63.01) | 0.008 (86.64) | 0.026 (26.66) |
| Sm(NO3)3·6H2O | 0.012 (57.76) | 0.008 (86.64) | 0.023 (30.13) |
| Yb(SO3CF3)3 | 0.033 (21.00) | 0.009 (77.02) | 0.115 (6.03) |
| ZrO(NO3)2 | 0.058 (11.95) | 0.017 (40.77) | 0.034 (20.38) |
| (NH4)2Ce(NO3)6 | 0.035 (19.80) | 0.024 (28.88) | 0.112 (6.19) |
| NiCl2·6H2O | 0.032 (21.66) | 0.021 (33.00) | 0.041 (16.90) |
The general catalytic process at the molecular level maybe represented as in Scheme 4.
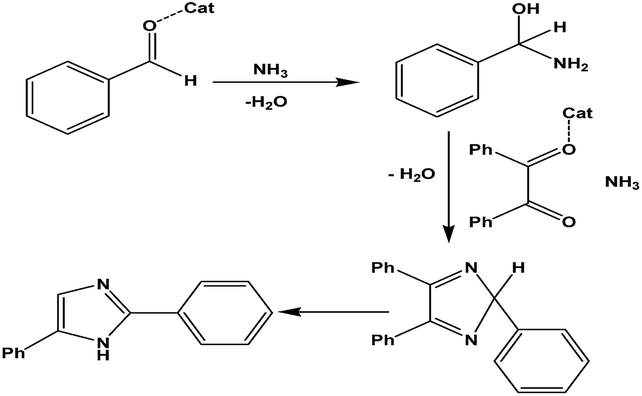 | ||
| Scheme 4 Mechanism of the R-I synthesis. | ||
The compiled results indicate that the catalysts acted at different stages of the reaction sequences of the MCR reaction. To cite a representative case of ytterbium triflate, a threefold increase in the rate of benzil consumption and fivefold increase in imidazole formation as against the catalyst free reaction was observed. Considering that in a reaction, catalysts in general, are used in very small proportions. Therefore only a small mole-fraction of the substrate would have the chance to be associated with the catalyst and thus the probability of the substrate taking part in adduct formation would be further reduced. Hence with only traces of a catalyst (5 mol% in the present case) added to a reaction, its rate should have hardly been affected as only a few molecules would engage in the activated complex in comparison to the vast number of un-associated molecules in the reaction media. We also studied the reactions at different elevated temperatures and found that at the temperature range 150–160 °C, almost quantitative products formed in a very short time of 4 minutes; and that too without using any catalyst. This is a landmark record in imidazole synthesis (ESI†).
3.2 Infrared studies
In order to contemplate the cause behind the efficacy in solvent-free procedure; we studied the IR-spectra of pure benzil in thin film as well as that with catalyst. The carbonyl region of a benzil thin film and that with 5 mmol% of ytterbium triflate has been presented in Fig. 3. It was observed that the free carbonyl band (with catalyst) at 1676 cm−1 has apparently increased in intensity with concomitant shift to lower frequency compared to the corresponding band for benzil. It was earlier reported34 that C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching band of benzil appears at 1676 cm−1 in the crystalline state and at 1685 cm−1 in solution.
O stretching band of benzil appears at 1676 cm−1 in the crystalline state and at 1685 cm−1 in solution.
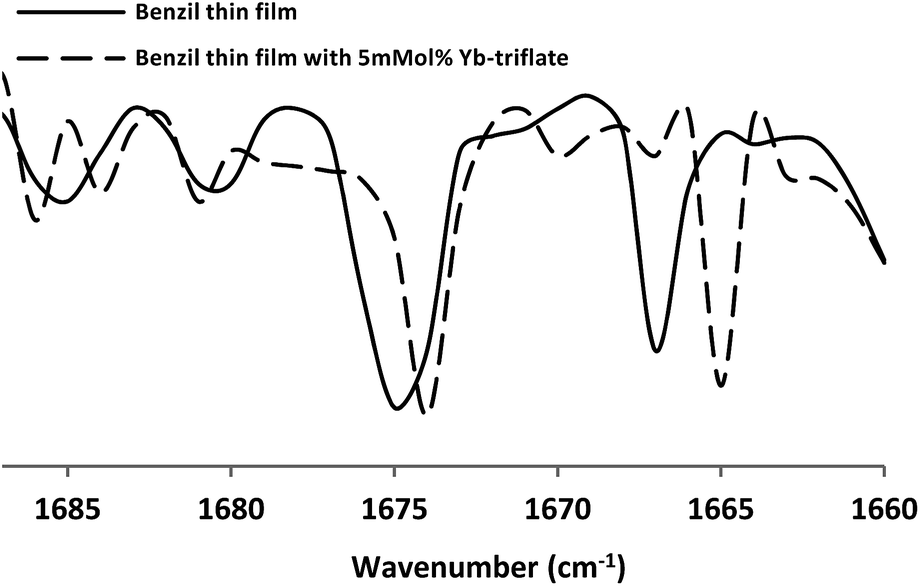 | ||
| Fig. 3 IR spectra of benzil thin film, with and without a catalyst. | ||
As the catalyst caused a red-shift and brought about enhancement in intensity, we have considered this phenomenon as a marker of catalytic effect. Similar shift of carbonyl stretching frequency for benzophenone in TiO2 surface has also been reported.35 The difference in stretching frequency (9 cm−1 red-shift) indicates a greater degree of single bond character in the CO bond (polarisation enhancement) in the solid state compared to that in the solution state for benzil. The red-shift of carbonyl stretching provides an indication of bulk polarization of benzil in the solid state and similar effect is also observed with catalysts in the solid state.
The extent of the shift caused by the presence of trace amounts of zirconyl nitrate and ytterbium triflate on benzil carbonyl stretching frequency in hexane solution was also studied. The IR spectra in the carbonyl range have been presented in Fig. 4.
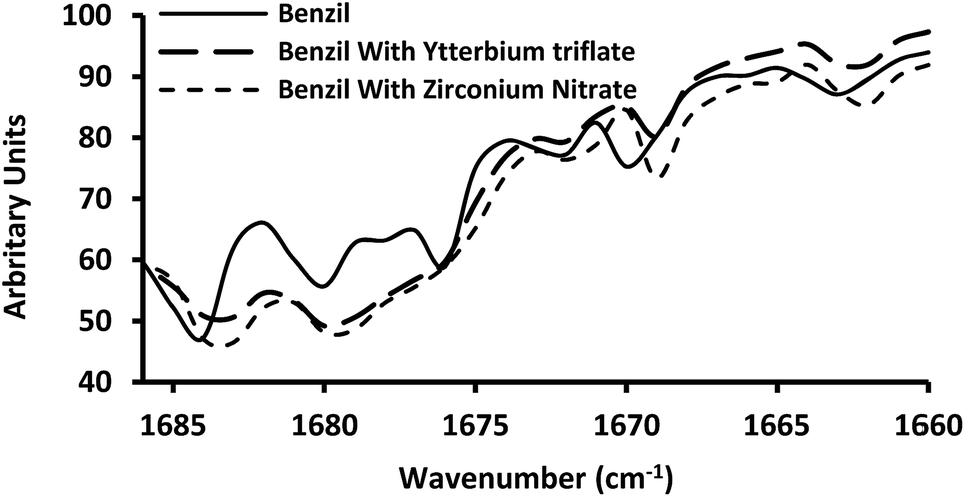 | ||
| Fig. 4 Solution IR spectra in the carbonyl range of benzil in hexane. | ||
It was found that in the solution spectra, the free carbonyl peak at 1685 cm−1 and all other associated peaks showed a little amount of red-shift in the presence of a catalyst. In addition to this, there was concomitant diminishing of the intensity of the free carbonyl peak with increasing intensities of some other peaks at lower frequencies (particularly the peak at 1680 cm−1) in the presence of traces of catalyst. It is also apparent that the catalyst, zirconyl nitrate, caused a little bit more polarization than that with ytterbium triflate. Notably we have found that the catalytic effect of zirconyl nitrate is a bit greater for benzil consumption rate as well (Table 1). The conspicuous enhancement of the band at 1680 cm−1 in the presence of catalyst in both solution state and in solid state is observed. The peak at 1680 cm−1 has been described as another CO stretching band associated with the different symmetry of the molecule.36 Enhancement of the band near 1680 cm−1 in presence of catalyst suggests that the catalysts not only polarize the carbonyl but also influence the conformation of benzil. The IR spectroscopic investigation thus strongly provides evidences in favour of bulk polarization in the condensed phase which consequently leads to the enhancement of the electrophilicity of the carbonyl carbon. The above observation suggests that catalysts bind to the carbonyl oxygen and the weak interaction activates the carbonyl group with the enhancement of polarization.
3.3 Computational studies
As further proof of concept, the CBS-QB3 model chemistry calculations of a HCHO monomer and trimer were performed. Geometry optimization for a linear arrangement of the trimer was done as shown in Fig. 5. In molecular association a number of minimum energy conformations with different geometries are feasible, we searched for minimum energy in linear arrangement and our calculation terminated on convergence to such minima.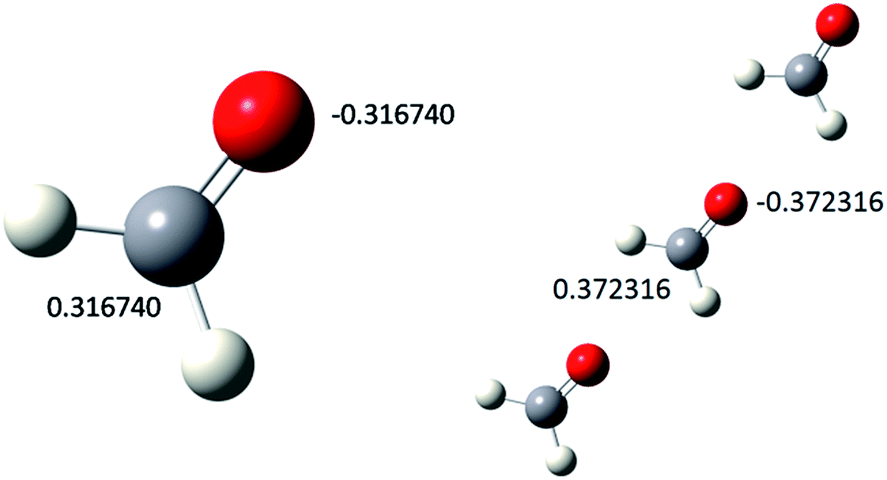 | ||
| Fig. 5 Minimum energy models with partial charges. | ||
The partial charges on the carbon and the oxygen atom and the dipole moment of the associated monomer are found to be enhanced as we move from the monomer to the trimer (Table 2).
| Parameter | Monomer | Trimer |
|---|---|---|
| a Note: [a]atomic charges with hydrogens summed into heavy atoms; [b]average value. | ||
| Mulliken charge at C (au)[a] | 0.316740 | 0.372316 |
| Mulliken charge at O (au)[a] | −0.316740 | −0.372316 |
| Dipole moment (Debye) | 2.8542 | 3.2133[b] |
| C–O bond distance (Å) | 1.20001 | 1.20259 |
| Symmetry | C2v | C1 |
It is evident that the atomic charges in the associated monomers are different to those in the isolated free monomer, indicating a sizeable charge reorganisation due to the association. These may be interpreted as atomic expressions of sizeable cooperative effects. This charge reorganization in the carbonyls, as a consequence, brings about enhanced polarization in the carbonyl groups in their environment as the partial charges increase in each atom. A seminal report has already established the possibility of a dipole–dipole type of intermolecular interaction and the occurrence of little or no H-bond in formaldehyde itself.37
The theoretical ab initio calculation of conformationally similar formaldehyde dimer was done by Smith et al.,38 whereby they have shown that an excess electron can attach to the system forming a dipole bound anion. This work indirectly supports our proposition as well, to the cause of enhancement in electrophilic behavior of the carbonyl group under solvent-free condition. The theoretical results also suggest that, as a result of the cooperative effect of very weak forces a bulk amount of carbonyl groups get activated. Therefore, the activation can be attributed to a unique spatial organization of the carbonyl moieties. This would be best understood if we consider an arbitrary molecular assembly where polarization transfer through a non-covalent bond (weak bond interactions) could well be conceived of as shown in Scheme 5.
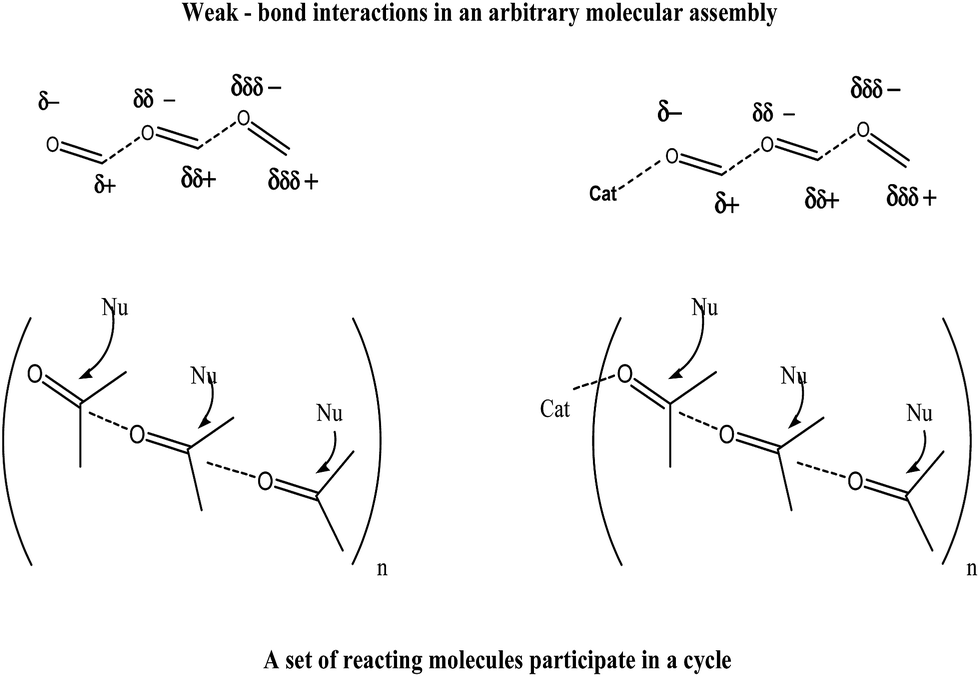 | ||
| Scheme 5 Polarization transfer through a non-covalent bond without and with a catalyst. | ||
This pseudo-conjugated pi-system in the proposed supramolecular assembly makes the system more chemically soft (more polarizable) compared to a free carbonyl which is also responsible for better reactivity in solvent free reaction media. Hence, considerable catalytic action became viable in condensed phase. Since, in such an assembly a set of reacting molecules participates in a cycle, the process undoubtedly becomes faster. In addition to this self-activating effect, if some oxyphilic substance were present in catalytic amount it bonded to the terminal carbonyl oxygen affecting further activation of the trail of carbonyls and thereby resulting in further enhancement of the catalytic effect (Scheme 5). The present work emphasizes on meeting this challenge of using weak forces as a design tool for imparting new properties and performance in molecules and materials.
The present work incorporates the concept of constitutional dynamic chemistry to resolve the existing paradox in the process of catalysis (involving carbonyl activation). According to Prof. Lehn, supramolecular entities are assembled entities of discrete number of molecular sub-units held together reversibly through weak interactions (non-covalent interactions).39 When extended to the carbonyl system, one could conceive the carbonyl molecular sub-units to be held together through similar type of weak interactions, by virtue of it being polar. In our opinion this hypothesis unequivocally embraces all the catalytic effects on carbonyls in solution state as well as in solvent-free molten state. As a proof of concept, we have reported herein the un-catalyzed Radziwenski synthesis of imidazole and its derivatives under solvent-free conditions employing a multicomponent protocol.
4 Conclusions
The polarizability of organized carbonyl functionalities in condensed phase contributes for the observed self-catalysis. High yields of many different imidazoles were obtained from the simply mechanical grinding and heating of MCR starting materials, even in the absence of Lewis acid catalysts. The very weak dipole of carbonyls can induce polarization in bulk because the carbonyl bonds are very much polarisable and the net result is the enhancement of electrophilicity of carbonyls. In polar solvents, the weak but favorable conformation of the carbonyl cluster expectedly breaks due to stronger solute–solvent interactions. Thus solvents act adversely to the self-catalytic effect. This phenomenon can be well utilized to generate a self-catalytic effect without using any catalytic substance.Acknowledgements
The author gratefully acknowledges IISER, Kolkata for the Single crystal X-ray studies and CDRI, Lucknow for elemental analysis and mass spectra. The authors are also thankful to Dr Anirban Misra, Professor and Mr Tamal Goswami, Reseach Associate, Department of Chemistry, N.B.U for the computational calculations.References
- K. Tanaka and F. Toda, Chem. Rev., 2000, 100, 1025 CrossRef CAS PubMed; F. Toda, CrystEngComm, 2002, 4, 215 RSC; G. W. V. Cave, C. L. Raston and J. L. Scott, Chem. Commun., 2001, 2159 RSC; G. Rothenberg, A. P. Downie, C. L. Raston and J. L. Scott, J. Am. Chem. Soc., 2001, 123, 8701 CrossRef PubMed; G. W. V. Cave and C. L. Raston, Chem. Commun., 2000, 2199 RSC; V. P. Balema, J. W. Wiench, M. Pruski and V. K. Pecharsky, Chem. Commun., 2002, 724 RSC.
- F. Toda, K. Tanaka and K. Hamai, J. Chem. Soc., Perkin Trans. 1, 1990, 3207 RSC.
- K. Yoshizawa, S. Toyota and F. Toda, Tetrahedron Lett., 2001, 42, 7983 CrossRef CAS.
- K. Tanaka, T. Sugino and F. Toda, Green Chem., 2000, 2, 303 RSC.
- G. Kaupp, M. R. Naimi-Jamal and J. Schmeyers, Tetrahedron, 2003, 59, 375 CrossRef.
- K. Yoshizawa, S. Toyota and F. Toda, Green Chem., 2002, 4, 68 RSC.
- D. C. Waddell and J. Mack, Green Chem., 2009, 11, 79 RSC.
- K. Tanaka, S. Kishigami and F. Toda, J. Org. Chem., 1991, 56, 4333 CrossRef CAS.
- F. Toda, M. Yagi and K. Kiyoshige, J. Chem. Soc., Chem. Commun., 1988, 958 RSC.
- F. Toda and T. Shigemasa, J. Chem. Soc., Perkin Trans. 1, 1989, 209 RSC.
- F. Toda, K. Tanaka and S. Iwata, J. Org. Chem., 1989, 54, 13 CrossRef.
- N. Elders, D. van der Born, L. J. D. Hendrickx, B. J. J. Timmer, A. Krause, E. Janssen, F. J. J. de Kanter, E. Ruijter and R. V. A. Orru, Angew. Chem., Int. Ed., 2009, 48, 5856 CrossRef CAS PubMed.
- B. Karimi and D. Zareyee, J. Mater. Chem., 2009, 19, 8665 RSC; H. Wang, X. Zhao, Y. Li and L. Lu, Org. Lett., 2006, 8(7), 1379 CrossRef CAS PubMed; P. Galletti, M. Pori and D. Giacomini, Eur. J. Org. Chem., 2011, 3896 CrossRef.
- M. A. Zolfigol, E. Kolvari, A. Abdoli and M. Shiri, Mol. Diversity, 2010, 14(4), 809 CrossRef CAS PubMed; V. Sivamurugan, R. S. Kumar, M. Palanichamy and V. Murugesan, J. Heterocycl. Chem., 2005, 42(5), 969 CrossRef; G. V. M. Sharma, K. L. Reddy, P. S. Lakshmi and P. R. Krishna, Synthesis, 2006, 55 CrossRef.
- F. Bigi, S. Carloni, B. Frullanti, R. Maggi and G. Sartori, Tetrahedron Lett., 1999, 40(17), 3465 CrossRef CAS; J. Peng and Y. Deng, Tetrahedron Lett., 2001, 42(34), 5917 CrossRef; R. Wang and Z. Liu, J. Org. Chem., 2012, 77(8), 3952 CrossRef PubMed.
- L. el Kaim, L. Gautier, L. Grimaud, L. M. Harwood and V. Michaut, Green Chem., 2003, 5, 477 RSC; Y. Hayashi, T. Urushima, S. Aratake, T. Okano and K. Obi, Org. Lett., 2008, 10(1), 21 CrossRef CAS PubMed.
- D. Koszelewski, W. Szymanski, J. Krysiak and R. Ostaszewski, Synth. Commun., 2008, 38(7), 1120 CrossRef CAS; T. Bousquet, M. Jida, M. Soueidan, R. Deprez-Poulain, F. Agbossou-Niedercorn and L. Pelinski, Tetrahedron Lett., 2012, 53(3), 306 CrossRef.
- N. Liu, S. Cao, J. Wu, J. Yu, L. Shen, X. Feng and X. Qian, Tetrahedron, 2008, 64, 3966 CrossRef CAS; L. El Kaim, L. Grimaud and S. Hadrot, Tetrahedron Lett., 2006, 47(23), 3945 CrossRef; M. Jida, S. Malaquin, R. Deprez-Poulain, G. Laconde and B. Deprez, Tetrahedron Lett., 2010, 51(39), 5109 CrossRef.
- J. S. B. Forero, E. M. de Carvalho, J. J. Junior and F. M. da Silva, Heterocycl. Lett., 2011, 1(1), 61 Search PubMed; K. Wang, D. Kim and A. Domling, J. Comb. Chem., 2010, 12, 111 CrossRef CAS PubMed.
- P. Nun, J. Martinez and F. Lamaty, Synthesis, 2010, 12, 2063 Search PubMed.
- B. Jiang, X. Wang, F. Shi, S.-J. Tu, T. Ai, A. Ballew and G. Li, J. Org. Chem., 2009, 74, 9486 CrossRef CAS PubMed; G. Bratulescu, Synthesis, 2009, 2319 CrossRef; X. Diao, Y. Wang, Y. Jiang and D. Ma, J. Org. Chem., 2009, 74, 7974 CrossRef PubMed; T. Horneff, S. Chuprakov, N. Chernyak, V. Gevorgyan and V. V. Fokin, J. Am. Chem. Soc., 2008, 130, 14972 CrossRef PubMed; P. Saha, T. Ramana, N. Purkait, M. A. Ali, R. Paul and T. Punniyamurthy, J. Org. Chem., 2009, 74, 8719 CrossRef PubMed; M. M. Heravia, M. Zakeria, N. Karimia, M. Saeedia, H. A. Oskooiea and N. T. Hosienib, Synth. Commun., 2010, 40, 1998 CrossRef; K. Hirano, S. Urban, C. Wang and F. Glorius, Org. Lett., 2009, 11, 1019 CrossRef PubMed.
- P. I. Dalko and L. Moisan, Angew. Chem., Int. Ed., 2004, 43, 5138 CrossRef CAS PubMed; D. Basavaiah, A. J. Rao and T. Satyanarayana, Chem. Rev., 2003, 103, 811 CrossRef PubMed; P. Langer, Angew. Chem., Int. Ed., 2000, 39, 3049–3052 CrossRef; H. Groger and J. Wilken, Angew. Chem., Int. Ed., 2001, 40, 529 CrossRef; J. W. J. Kennedy and D. G. Hall, Angew. Chem., Int. Ed., 2003, 42, 4732 CrossRef PubMed; B. List, Tetrahedron, 2002, 58, 5573 CrossRef.
- M.-O. Simon and C.-J. Li, Chem. Soc. Rev., 2012, 41, 1415 RSC.
- C.-J. Li, Chem. Rev., 2005, 105, 3095 CrossRef CAS PubMed; Y. R. Leshkov and M. E. Davis, ACS Catal., 2011, 1, 1566 CrossRef; F. Cruz-Acosta, A. Santos-Exposito, P. de Armas and F. Garcia-Tellado, Chem. Commun., 2009, 6839 RSC; R. N. Butler and A. G. Coyne, Chem. Rev., 2010, 110, 6302 CrossRef PubMed.
- S. D. Sharma, P. Hazarika and D. Konwar, Tetrahedron Lett., 2008, 49, 2216 CrossRef.
- S. Samai, G. C. Nandi, P. Singh and M. S. Singh, Tetrahedron, 2009, 65, 10155 CrossRef CAS.
- G. M. Sheldrick, SHELXS-97, University of Gottingen, 1997 Search PubMed.
- J. A. Montgomery, M. J. Frisch, J. W. Ochterski and G. A. Petersson Jr, J. Chem. Phys., 2000, 112, 6532 CrossRef CAS; J. A. Montgomery, M. J. Frisch, J. W. Ochterski and G. A. Petersson Jr, J. Chem. Phys., 1999, 110, 2822 CrossRef.
- L. A. Curtiss, K. Raghavachari, G. W. Trucks and J. A. Pople, J. Chem. Phys., 1991, 94, 7221 CrossRef CAS; L. A. Curtiss, K. Raghavachari, P. C. Redfern, V. Rassolov and J. A. Pople, J. Chem. Phys., 1998, 109, 7764 CrossRef; J. W. Ochterski, G. A. Petersson and J. A. Montgomery, J. Chem. Phys., 1996, 104, 2598 CrossRef; G. A. Petersson, D. K. Malick, W. G. Wilson, J. W. Ochterski, J. A. Montgomery and M. J. Frisch, J. Chem. Phys., 1998, 109, 10570 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria and M. A. Robb, et al., Gaussian 09, Revision A.11.4, Gaussian, Inc., Pittsburg PA, 2002 Search PubMed.
- W. Munch, et al., Solid State Ionics, 2001, 145, 437 CrossRef CAS.
- K. B. Benzon, H. T. Varghese, C. Y. Panicker, K. Pradhan, B. K. Tiwary, A. K. Nanda and C. V. Alsenoy, Spectrochim. Acta, Part A, 2015, 151, 965 CrossRef CAS PubMed.
- K. B. Benzon, H. T. Varghese, C. Y. Panicker, K. Pradhan, B. K. Tiwary, A. K. Nanda and C. V. Alsenoy, Spectrochim. Acta, Part A, 2015, 146, 307 CrossRef CAS PubMed.
- E. C. Lee, D. Kim, P. Jurecka, P. Tarakeshwar, P. Hobza and K. S. Kim, J. Phys. Chem. A, 2007, 111, 3446 CrossRef CAS PubMed.
- L. Colombo, D. Kirin, V. Volovsek, N. E. Lindsay, J. F. Sullivan and J. R. Durig, J. Phys. Chem., 1989, 93, 6290 CrossRef CAS.
- L. M. Babkov, T. V. Bezrodnaya, G. A. Puchkovskaya, K. E. Uspenskii and V. V. Shimanovaskaya, J. Struct. Chem., 2008, 49, 517 CrossRef CAS.
- T. Bercovici, J. King and R. S. Becker, J. Chem. Phys., 1972, 56, 3956 CrossRef CAS; J. E. D. Bene, J. Chem. Phys., 1971, 60, 3812 CrossRef.
- M. A. Dayle, J. S. Smith, Y. Elkadi and L. Adanokicz, Chem. Phys. Lett., 1999, 305, 169 CrossRef.
- J. M. Lehn, Angew. Chem., Int. Ed., 1990, 29, 1304 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra16386b |
| This journal is © The Royal Society of Chemistry 2016 |