
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGrowth of N-doped graphene from nitrogen containing aromatic compounds: the effect of precursors on the doped site†
Tokio
Katoh
a,
Gaku
Imamura
bc,
Seiji
Obata
b and
Koichiro
Saiki
*ab
aDepartment of Chemistry, School of Science, The University of Tokyo, Kashiwanoha 5-1-5, Kashiwa, Chiba 277-8561, Japan. E-mail: saiki@k.u-tokyo.ac.jp
bDepartment of Complexity Science and Engineering, Graduate School of Frontier Sciences, The University of Tokyo, Kashiwanoha 5-1-5, Kashiwa, Chiba 277-8561, Japan. Fax: +81-4-7136-3903; Tel: +81-4-7136-5526
cWorld Premier International (WPI) Research Center, International Center for Materials Nanoarchitectonics (MANA), National Institute for Materials Science (NIMS), 1-1 Namiki, Tsukuba, Ibaraki 305-0044, Japan
First published on 27th January 2016
Abstract
Doping of nitrogen into the graphene lattice tunes and tailors its electronic properties. To elucidate the doping mechanism and develop a method of controlling the doped structure, nitrogen doped graphene was synthesized from four kinds of nitrogen-containing aromatic compounds: quinoline, pyridine, pyrrole, and pyrimidine on Pt(111) at a variety of temperatures. The doped site and amount of nitrogen in the synthesized graphene depended considerably on the source molecule. Especially, nitrogen doped graphene with pyridinic-N and pyrrolic-N were predominantly obtained from quinoline and pyrrole sources. Comparison between the graphenes from different molecules indicated that structure and thermal stability of the source molecules together with their structural affinity to the honeycomb lattice determined the doping amount and the doped site.
1. Introduction
Graphene is a single layer of graphite which consists of sp2 carbon atoms packed in a hexagonal lattice. Since its first isolation in 2004,1 graphene has attracted the attention of many scientists due to its fascinating properties.2–5 Doping of graphene with hetero atoms has been expected to bring further intriguing properties to graphene.6,7 Among a variety of such hetero atoms nitrogen is a fertile candidate for modifying the properties of graphene due to its comparable atomic size and additional valence electron. First principles calculations predicted the presence of a band-gap, which would realize a large on/off ratio in field effect transistors.8 Moreover, catalytic activity in the oxygen reduction reaction has been reported for nitrogen doped graphene.9,10 A most interesting feature of N-doping is that the doped site has a significant effect on the electronic properties of the graphene.11,12 Three types of N-doping sites in graphene are often considered: graphitic-, pyridinic-, and pyrrolic-N. Both theoretical and experimental studies have proved that graphitic-N causes n-type doping whereas pyridinic- and pyrrolic-N give rise to p-type doping.12–14 Considering applications, pyridinic-N improved the performance of Li-ion batteries by enhancing Li-ion intercalation.14,15 Although many studies have reported the synthesis of N-doped graphene, efficient methods for controlling the amount and site of nitrogen atoms in graphene are yet to be developed.The synthesis methods of N-doped graphene are generally divided into two types: top-down and bottom-up approaches.16,17 A top-down approach produces N-doped graphene via post treatment to graphene or graphene oxide.18,19 Nitrogen incorporation via UV-irradiation in ammonia,20,21 and nitrogen plasma treatment,12,22–24 for example, have been reported. In a bottom-up approach, on the other hand, nitrogen atoms are incorporated into graphene during the growth process. Chemical vapor deposition (CVD) is the most typical and the most promising bottom-up fabrication method. In this process, graphene formation and N-doping occur simultaneously. CVD synthesis of N-doped graphene have been performed mostly on transition metal substrates such as Ni,25,26 Cu,27–34 and Pt.35–37 Especially, platinum is an outstanding substrate due to its chemical stability; platinum is not easily oxidized compared to other transition metals. Owing to such stability, graphene grows on Pt without help of other gases, which enables us to synthesize graphene from a single source molecule.35–39 Single-source CVD uses a molecule that could have suitable chemical composition and structure. The single-source CVD has an advantage in observing the effect of source molecules directly. Furthermore, it might be expected that N atoms are doped into graphene reflecting the structure of source molecules, leading to the control of doped sites. Therefore, the single-source CVD is considered to be effective not only for controlling the structure, but also for understanding the growth mechanism.
In the present study, N-doped graphene is synthesized on Pt(111) by the single-source CVD in ultra-high vacuum (UHV) and analyzed in situ. In such well-controlled conditions, the product is free from contamination such as oxygen, which assured the precise evaluation of doped site and content. Our previous study reported the effect of source molecule on the nitrogen doping.35,37 Structural control of N-doped graphene is, however, yet to be accomplished, or the nature of nitrogen doping via single-molecule CVD is still unclear. To achieve the structure control of N-doped graphene, we have conducted the single-source CVD using four kinds of aromatic-N compounds. With this approach, we have accomplished site-selective N-doping to graphene. Considering the relationship between the structure of N-doped graphene and the source molecule, we discuss the growth mechanism of N-doped graphene.
2. Experimental
All the experiments were performed under UHV conditions (<10−7 Pa). N-Doped graphene was synthesized on Pt(111) by low-pressure CVD (LPCVD). A Pt substrate was mechanically polished, and then cleaned by sonication in acetone for 30 min. A clean Pt(111) surface was prepared through repeated cycles of Ar ion bombardment and annealing at 1000 °C in UHV. Cleanness of the Pt(111) surface was confirmed by low energy electron diffraction (LEED) and X-ray photoelectron spectroscopy (XPS). In the LPCVD process, the Pt(111) substrate kept at a desired temperature was exposed to the ambience of source molecules, which were introduced into the UHV chamber through a variable leak valve. Fig. 1 shows the structures of source molecules: (a) quinoline, (b) pyridine, (c) pyrrole, and (d) pyrimidine. The pressure was kept at 0.1 Pa for 60 min which supplied sufficient amount of source molecules for obtaining a monolayer graphene with the full coverage. The temperature of the substrate (Ts) was determined with an infrared pyrometer. The as-grown films were characterized with in situ XPS and LEED measurements. XPS measurements were performed with an X-ray source (Thermo VG Scientific XR3E2) and a hemispherical analyzer (SPECS, PHOIBOS-100). LEED measurements were performed by BDL800IR-MCP2 spectrometer (OCI Vacuum Microengineering Inc.) with a beam energy of 100 eV. After the XPS and LEED measurements, the film was transferred to a SiO2 substrate by a bubbling method40 and characterized with Raman spectroscopy in air. The Raman spectrum was measured by NRS-3100 (JASCO Corporation) with an excitation wavelength of 532 nm. | ||
| Fig. 1 Molecular structures of the source molecules: (a) quinoline, (b) pyridine, (c) pyrrole, and (d) pyrimidine. The structures were optimized with density-functional-theory calculations. (a) Quinoline (b) pyridine (c) pyrrole (d) pyrimidine. | ||
3. Results
First, N-doped graphene was synthesized on Pt(111) kept at 500 °C. Fig. 2 shows the LEED patterns of the films deposited at 500 °C from the source molecules: (a) quinoline, (b) pyridine, (c) pyrrole, and (d) pyrimidine. Two noticeable features are observed in the diffraction patterns; sharp spots with a six-fold rotational symmetry, and arc-shaped spots with twelve-fold rotational symmetry. The sharp spots with six-fold symmetry originated from the Pt(111) surface. The arc-shape spots outside the Pt spots were previously observed for the graphene on Pt.37,41 The lattice constant of 2.46 Å calculated from the radius of arc-shape spots in Fig. 2 is equal to that of graphene (2.46 Å). The diffraction spots from graphene were observed for all the films synthesized at 500 °C. The intensity of the arc-shape spots depends on the source molecule. The arc-shape spots are sharp for the quinoline-derived graphene (Fig. 2(a)), whereas that are blurred for the pyrrole-derived graphene (Fig. 2(c)). The difference in sharpness reflects the domain size of graphene. Judging from the intensity of the arc-shape spots and the moiré structures around Pt spots, the domain size increases in sequence, pyrrole, pyridine, pyrimidine, and quinoline. | ||
| Fig. 2 LEED patterns of the films synthesized at 500 °C from (a) quinoline, (b) pyridine, (c) pyrrole, and (d) pyrimidine. | ||
Raman spectroscopy is a powerful method for evaluating the structure of graphene. Fig. 3 shows the Raman spectra of the graphenes synthesized from four kinds of molecules at 500 °C. The G band at ∼1580 cm−1 originates from the sp2 carbon network. The D band located at ∼1350 cm−1 indicates the presence of a certain amount of defects in the sp2 carbon network.42 In addition, a component for amorphous carbon is identified between the D band and the G band.43,44 The D′ band at ∼1620 cm−1 is also activated by defects in graphene and enhanced especially by boundaries and vacancies.45 The spectra shown in Fig. 3 are normalized with the peak height of the G band. Absence of amorphous components (between D and G bands) in the quinoline-derived graphene indicates its highest crystallinity of the four. For the graphenes synthesized from the other source molecules, the intensity of the 2D band (∼2700 cm−1) might be a good index to evaluate the domain size of the graphene; the more I2D/IG is, the larger the domain size is.46,47 Judging from the Raman spectra, the domain size of the N-doped graphene increases in sequence, pyrrole, pyridine, pyrimidine, and quinoline, which is consistent with the results of LEED measurements (Fig. 2).
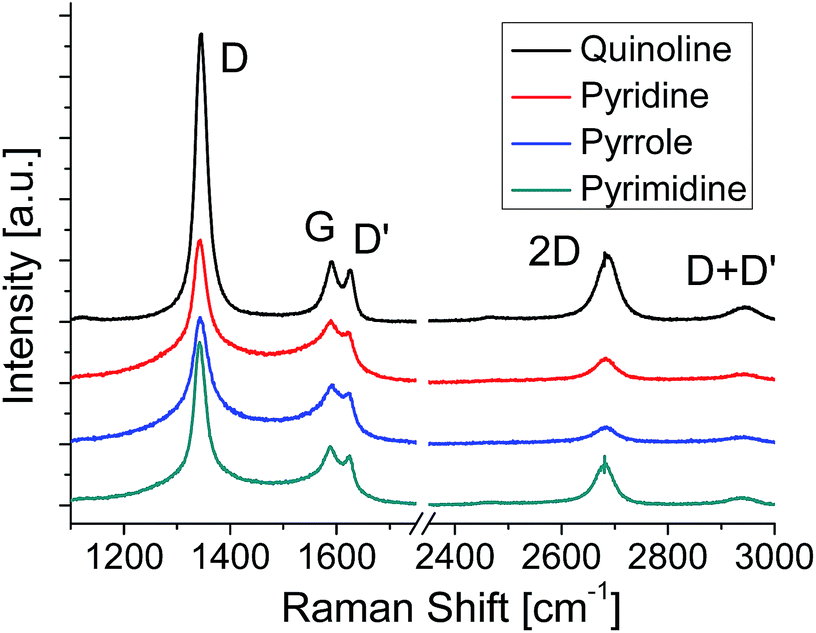 | ||
| Fig. 3 Raman spectra of N-doped graphenes synthesized at 500 °C. | ||
The amount and chemical state of nitrogen in graphene were evaluated by XPS measurement. The absence of oxygen atom was confirmed for all the specimens by the XPS in the O 1s region. Fig. 4 shows the XPS spectra in the region of N 1s for the graphenes synthesized from (a) quinoline, (b) pyridine, (c) pyrrole, and (d) pyrimidine at 500 °C. The spectra were fitted with three Gaussian functions, which corresponded to pyridinic- (398.6 eV), pyrrolic- (400.1 eV), and graphitic- (401.0 eV) N states. The component at around 402 eV can be assigned to positively charged nitrogen since there is no oxygen detected in XPS measurement (Fig. S1†).48 The nitrogen content, defined as [N]/([C] + [N]), was evaluated from the areas of the N 1s and the C 1s peaks. The quinoline-derived graphene (Fig. 4(a)) is dominated by pyridinic-N (0.35%). The pyridine- and pyrimidine-derived graphenes, on the other hand, consist of both pyridinic- and graphitic-N states (Fig. 4(d)). It is noteworthy that the pyrimidine-derived graphene contains less nitrogen (1.78%) than the pyridine-derived one (2.41%), even though the N content of pyrimidine is higher than that of pyridine. The pyrrole-derived graphene contains a largest amount of N atoms (4.28%) doped predominantly at pyrrolic-N sites (Fig. 4(c)). There are several reports which synthesized nitrogen-doped graphene on Cu foil by CVD with using pyridine as source molecule.29,34 According to these literatures, nitrogen-doped graphene with around 2.4% of N atoms has obtained at 1000 °C. By comparing them with our method, they need twice the temperature to synthesize graphene with same N concentration. The difference is considered as due to strong catalytic activity of Pt substrate.
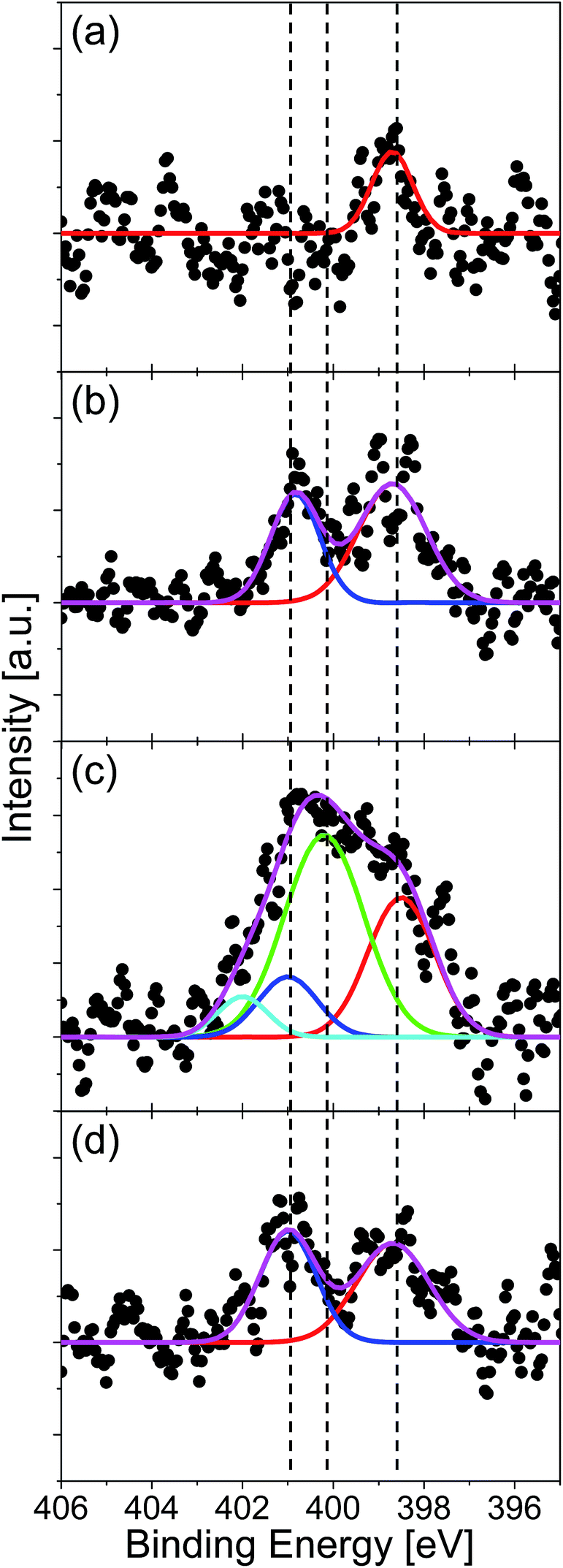 | ||
| Fig. 4 XPS N 1s spectra of N-doped graphenes synthesized at 500 °C from (a) quinoline, (b) pyridine, (c) pyrrole, and (d) pyrimidine. Each spectrum is fitted with three Gaussian functions: pyridinic- (red line), pyrrolic- (green line), and graphitic-N (blue line). | ||
XPS measurement revealed that the N content increased in sequence, quinoline, pyrimidine, pyridine, and pyrrole. This order correlates inversely with that of the domain size evaluated from the sharpness of LEED and the 2D intensity of Raman spectra. The nitrogen atoms doped into the graphene lattice seemed to decrease the domain size. The nitrogen atom bonded at grain edge would hamper the lateral growth of graphene, leading to the smaller domain size.31 Therefore, the larger the N content was, the smaller the domain size became.
The incorporation of nitrogen atoms affected also the C 1s spectra. Fig. S2† shows the XPS C 1s spectra of graphene synthesized at 500 °C. The peak intensity ratio of C 1s to Pt 4f indicates that the as-grown graphenes were monolayer.36,37 A satellite peak observe around 286 eV originated from C–N bonds. The intensity of this peak increases in sequence, quinoline, pyrimidine, pyridine, and pyrrole, which is consistent with the N content determined from N 1s spectra.
The N content (2.4%) in the pyridine-derived graphene at 500 °C was different from that observed in the previous study (4.0%), although the growth procedure was the same except the annealing temperature (Tanneal) of Pt before graphene growth.35 The increase of Tanneal from 827 °C to 1000 °C might have improved the surface cleanness to result in enhancement of catalytic activity of Pt surface, leading to the decrease in N content at 500 °C.
To investigate the growth mechanism of N-doped graphene, we changed the growth temperature. Fig. 5(a) shows the XPS N 1s spectra of the graphenes synthesized from the source molecules: (a) quinoline, (b) pyridine, (c) pyrrole, and (d) pyrimidine at 600 °C. Nitrogen doping was observed only for the pyridine-derived graphene, in which graphitic-N and pyridinic-N peaks are observed with the total N content of 1.64%. Fig. S3† shows the C 1s region spectra for the graphenes grown at 600 °C. Only the pyridine-derived graphene shows a broad peak with a shoulder at the higher binding energy side. The graphenes synthesized from other molecules have only a narrow peak at 284.1 eV, indicating absence of nitrogen in the graphene lattice. Exclusion of nitrogen atoms during the CVD growth suggests that source molecules were decomposed and nitrogen-containing fragment was detached from the surface. Fig. 5(b) shows the temperature dependence of the nitrogen content for each source molecule. At 600 °C, N-doped graphene can be obtained only from pyridine. At higher temperature (over 700 °C), LEED and XPS revealed that non-doped graphene was formed regardless of the source molecules.
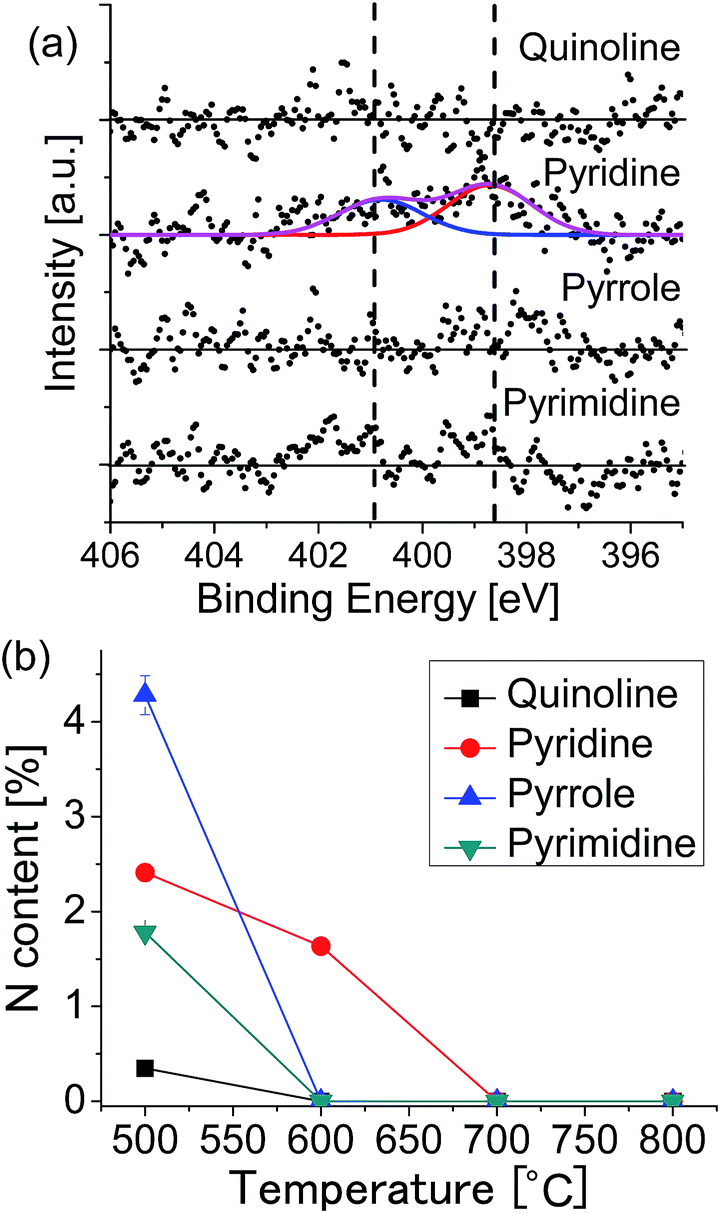 | ||
| Fig. 5 (a) XPS N 1s spectra of the graphenes synthesized at 600 °C. (b) Nitrogen content as a function of growth temperature for each of the source molecules. | ||
Fig. 6 shows the Raman spectra of the graphenes formed at 600 °C. Compared to the graphenes formed at 500 °C, the relative intensity of 2D band increases for all the graphenes, which means the increase of the domain size. In the LEED images the blurred arc-shape spots observed for the graphenes formed at 500 °C (Fig. 2(c)) become clear six- or twelve-fold symmetrical spots for the graphenes formed at 600 °C (Fig. S4(c)†), which also means the increase of the domain size. At 600 °C the pyridine-derived graphene shows the smallest domain size of the four. This incorporation of nitrogen atoms impedes the evolution of domain size in the pyridine-derived graphene. The other three samples are free from N-dopants, though 600 °C is not sufficient to produce defect free graphene on Pt by CVD.
 | ||
| Fig. 6 Raman spectra of the graphenes synthesized at 600 °C. | ||
4. Discussion
Nitrogen doped graphene was synthesized from four kinds of aromatic molecules. Table 1 summarizes the experimental results: the nitrogen content for the films deposited at 500 °C and 600 °C together with the major doped sites. Nmole means the nitrogen content in each molecule. The nitrogen content N500 seems increasing with Nmole but it decreases for pyrimidine. N600 is finite only for the pyridine-derived graphene. With respect to doped sites, pyrrole-derived and quinoline-derived graphenes have a single-kind of site, while two kinds of site are observed for pyridine-derived and pyrimidine-derived graphenes. In this section we discuss the growth mechanism of nitrogen doped graphene focusing on the two points: doped amount and doped site.| Source molecule | [N]mole | [N]500 | ([N]500/[N]mole) | [N]600 | E C–N (kJ mol−1) | N-Doped sites |
|---|---|---|---|---|---|---|
| Quinoline | 0.100 | 0.004 | 0.035 | 0 | 188 (ref. 49) | Pyridinic |
| Pyridine | 0.167 | 0.024 | 0.145 | 0.016 | 320 (ref. 50) | Pyridinic, graphitic |
| Pyrrole | 0.200 | 0.043 | 0.214 | 0 | 318 (ref. 51 and 52) | Pyrrolic |
| Pyrimidine | 0.333 | 0.018 | 0.053 | 0 | 275 (ref. 53) | Pyridinic, graphitic |
The nitrogen content is much smaller than that of source molecule in any case. This means decomposition occurred for the graphene growth even at temperatures as low as the present case. It has been reported that a major component decomposed thermally from these aromatic molecules is HCN.52,54–56 The CVD process on metal substrates would desorb such a volatile species as HCN from the surface before graphene formation, which resulted in exclusion of nitrogen atoms from graphene.35 In previous works, thus we considered that a bond reforming process played a role for nitrogen atoms to be incorporated to graphene during CVD growth.35–37 In the bond reforming process partially decomposed or deformed aromatic molecules coalesce with each other, leading to the formation of graphene.57,58 Less decomposition might be favorable for the remaining of nitrogen atoms in the synthesized graphene.
The activation energy of ring opening reaction (EC–N) in the source aromatic compounds is shown in Table 1. The order of remaining nitrogen amount qualitatively agrees with that of decomposition energy. Pyrrole and pyridine have a large activation energy and a large nitrogen content among the four molecules. Thus the amount of molecules participating the graphene formation via bond-reforming process is relatively large for these molecules. This could explain why the pyridine-derived graphene has a larger amount of nitrogen than the pyrimidine-derived one although the amount of nitrogen is half that of pyrimidine in molecule.
For the growth at 600 °C, only the pyridine-derived graphene contains the nitrogen in the lattice. The pyrrole-derived graphene grew excluding the nitrogen, although the activation energy of decomposition was close to that of pyridine. This difference could be ascribed to the difference in the intermediate species produced by the ring-opening reaction. A major product of the ring-opening reaction is N-terminated radicals such as ˙CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCHCHC
CHCHCHC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N. C-Terminated radical species such as ˙CHCHCHN–CCH are, however, also produced by cleavage of C–C bond in pyridine.50,56 Such C-terminated species were less likely to be converted into volatile species and could be attached to the growing graphene edges. At higher temperatures at which most source molecules are decomposed, N atoms could be thus incorporated into graphene synthesized from pyridine.
N. C-Terminated radical species such as ˙CHCHCHN–CCH are, however, also produced by cleavage of C–C bond in pyridine.50,56 Such C-terminated species were less likely to be converted into volatile species and could be attached to the growing graphene edges. At higher temperatures at which most source molecules are decomposed, N atoms could be thus incorporated into graphene synthesized from pyridine.
In contrast, C-terminated species are hardly produced from pyrrole. The activation energy of C–C bond cleavage was theoretically evaluated to be greater than that of C–N bond by 164 kJ mol−1 for pyrrole,59 while only 31.5 kJ mol−1 for pyridine.50 Thus C-terminated radical species are rarely produced from pyrrole at 600 °C, while partly produced from pyridine at the same temperature. Thus it can be concluded that the thermal stability of source molecules affect strongly the doped amount in the graphenes synthesized from N-containing aromatic molecules.
Nitrogen atoms were incorporated mostly via bond reforming process and thus the doped site tended to reflect the site the nitrogen atom occupied in the source molecule. Nitrogen atoms resided at a pyridinic site and a pyrrolic site in the quinoline-derived graphene and pyrrole-derived graphene, respectively. Pyridine- and pyrimidine-derived graphenes, however, had graphitic nitrogen atoms in addition to pyridinic ones. We consider that the presence of graphitic-N is due to the structural affinity of pyridine and pyrimidine for the honeycomb lattice of graphene. These two molecules can be easily incorporated into graphene lattice due to their hexagonal molecular shape. Such structural affinity increases the probability of the formation of graphitic-N during the coalescent process.26,30,33 In contrast, it is difficult for pyrrole to be incorporated into the inside of the honeycomb lattice of graphene retaining their original molecular shape. Because of its low structural affinity, a pyrrole molecule can only be incorporated at the edges of graphene, terminating the growth of graphene by pentagonal edges. Focusing on the skeletal structure, quinoline has only two-fold symmetry, whereas pyridine and pyrimidine have six-fold symmetry. Due to such lower structural symmetry and larger molecular size of quinoline, the probability of coalescence in the bond-reforming process might be smaller than those of pyridine and pyrimidine. Therefore, quinoline was not incorporated into the inside of graphene, but at the edge of graphene domains, resulting in the dominant pyridinic-N doping with lower N concentration.
5. Conclusions
Four kinds of nitrogen-containing aromatic molecules were deposited on a heated Pt(111) substrate. Nitrogen doped graphenes were formed from all molecules at 500 °C, while at 600 °C nitrogen doping was observed only in the pyridine-derived graphene. The nitrogen content and doped site in the synthesized graphene depended on the source molecule, the origin of which could be interpreted by a bond-reforming model taking account of structural stability against heating. The pyridinic-N and pyrrolic-N were exclusively obtained from quinoline and pyrrole sources, while the pyridine-derived and pyrimidine-derived graphenes contained both pyridinic-N and graphitic-N in the lattices. The present result would open a way of controlling the structure of N-doped graphene, leading to the improvement in graphene-based electronic devices and catalysts.Acknowledgements
This work was partially supported by a Grant-in-Aid for Scientific Research from MEXT of Japan (No. 25107002). The DFT calculations were performed using Research Center for Computational Science, Okazaki, Japan.Notes and references
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- K. I. Bolotin, K. J. Sikes, Z. Jiang, M. Klima, G. Fudenberg, J. Hone, P. Kim and H. L. Stormer, Solid State Commun., 2008, 146, 351–355 CrossRef CAS.
- A. A. Balandin, S. Ghosh, W. Bao, I. Calizo, D. Teweldebrhan, F. Miao and C. N. Lau, Nano Lett., 2008, 8, 902–907 CrossRef CAS PubMed.
- M. D. Stoller, S. Park, Z. Yanwu, J. An and R. S. Ruoff, Nano Lett., 2008, 8, 3498–3502 CrossRef CAS PubMed.
- C. Lee, X. Wei, J. W. Kysar and J. Hone, Science, 2008, 321, 385–388 CrossRef CAS PubMed.
- T. B. Martins, R. H. Miwa, A. J. R. Da Silva and A. Fazzio, Phys. Rev. Lett., 2007, 98, 3–6 Search PubMed.
- Z.-H. Sheng, H.-L. Gao, W.-J. Bao, F.-B. Wang and X.-H. Xia, J. Mater. Chem., 2012, 22, 390 RSC.
- A. Lherbier, A. R. Botello-Méndez and J. C. Charlier, Nano Lett., 2013, 13, 1446–1450 CrossRef CAS PubMed.
- L. Qu, Y. Liu, J. B. Baek and L. Dai, ACS Nano, 2010, 4, 1321–1326 CrossRef CAS PubMed.
- L. Zhang and Z. Xia, J. Phys. Chem. C, 2011, 115, 11170–11176 CAS.
- Y. F. Lu, S. T. Lo, J. C. Lin, W. Zhang, J. Y. Lu, F. H. Liu, C. M. Tseng, Y. H. Lee, C. Te Liang and L. J. Li, ACS Nano, 2013, 7, 6522–6532 CrossRef CAS PubMed.
- K. Akada, T. Terasawa, G. Imamura, S. Obata and K. Saiki, Appl. Phys. Lett., 2014, 104, 131602 CrossRef.
- T. Schiros, D. Nordlund, L. Pálová, D. Prezzi, L. Zhao, K. S. Kim, U. Wurstbauer, C. Gutiérrez, D. Delongchamp, C. Jaye, D. Fischer, H. Ogasawara, L. G. M. Pettersson, D. R. Reichman, P. Kim, M. S. Hybertsen and A. N. Pasupathy, Nano Lett., 2012, 12, 4025–4031 CrossRef CAS PubMed.
- X. Kong and Q. Chen, Phys. Chem. Chem. Phys., 2013, 15, 12982 RSC.
- A. L. M. Reddy, A. Srivastava, S. R. Gowda, H. Gullapalli, M. Dubey and P. M. Ajayan, ACS Nano, 2010, 4, 6337–6342 CrossRef CAS PubMed.
- H. Wang, T. Maiyalagan and X. Wang, ACS Catal., 2012, 2, 781–794 CrossRef CAS.
- C. N. R. Rao, K. Gopalakrishnan and A. Govindaraj, Nano Today, 2014, 9, 324–343 CrossRef CAS.
- S. Sandoval, N. Kumar, A. Sundaresan, C. N. R. Rao, A. Fuertes and G. Tobias, Chem.–Eur. J., 2014, 20, 11999–12003 CrossRef CAS PubMed.
- S. Sandoval, N. Kumar, J. Oro-Solé, A. Sundaresan, C. N. R. Rao, A. Fuertes and G. Tobias, Carbon, 2015, 96, 594–602 CrossRef.
- G. Imamura and K. Saiki, Chem. Phys. Lett., 2013, 587, 56–60 CrossRef CAS.
- G. Imamura and K. Saiki, RSC Adv., 2015, 5, 70522–70526 RSC.
- Y. Wang, Y. Shao, D. W. Matson, J. Li and Y. Lin, ACS Nano, 2010, 4, 1790–1798 CrossRef CAS PubMed.
- Y. Shao, S. Zhang, M. H. Engelhard, G. Li, G. Shao, Y. Wang, J. Liu, I. A. Aksay and Y. Lin, J. Mater. Chem., 2010, 20, 7491 RSC.
- S. H. Park, J. Chae, M.-H. Cho, J. H. Kim, K.-H. Yoo, S. W. Cho, T. G. Kim and J. W. Kim, J. Mater. Chem. C, 2014, 2, 933 RSC.
- C. Zhang, L. Fu, N. Liu, M. Liu, Y. Wang and Z. Liu, Adv. Mater., 2011, 23, 1020–1024 CrossRef CAS PubMed.
- D. Usachov, O. Vilkov, A. Grüneis, D. Haberer, A. Fedorov, V. K. Adamchuk, A. B. Preobrajenski, P. Dudin, A. Barinov, M. Oehzelt, C. Laubschat and D. V. Vyalikh, Nano Lett., 2011, 11, 5401–5407 CrossRef CAS PubMed.
- D. Wei, Y. Liu, Y. Wang, H. Zhang, L. Huang and G. Yu, Nano Lett., 2009, 9, 1752–1758 CrossRef CAS PubMed.
- Z. Luo, S. Lim, Z. Tian, J. Shang, L. Lai, B. MacDonald, C. Fu, Z. Shen, T. Yu and J. Lin, J. Mater. Chem., 2011, 21, 8038 RSC.
- Z. Jin, J. Yao, C. Kittrell and J. M. Tour, ACS Nano, 2011, 5, 4112–4117 CrossRef CAS PubMed.
- Y. Xue, B. Wu, L. Jiang, Y. Guo, L. Huang, J. Chen, J. Tan, D. Geng, B. Luo, W. Hu, G. Yu and Y. Liu, J. Am. Chem. Soc., 2012, 134, 11060–11063 CrossRef CAS PubMed.
- T. Terasawa and K. Saiki, Jpn. J. Appl. Phys., 2012, 51, 055101 CrossRef.
- J. Li, Z. Ren, Y. Zhou, X. Wu, X. Xu, M. Qi, W. Li, J. Bai and L. Wang, Carbon, 2013, 62, 330–336 CrossRef CAS.
- J. Zhang, J. Li, Z. Wang, X. Wang, W. Feng, W. Zheng, W. Cao and P. Hu, Chem. Mater., 2014, 26, 2460–2466 CrossRef CAS.
- Y. Ito, C. Christodoulou, M. V. Nardi, N. Koch, H. Sachdev and K. Müllen, ACS Nano, 2014, 8, 3337–3346 CrossRef CAS PubMed.
- G. Imamura and K. Saiki, J. Phys. Chem. C, 2011, 115, 10000–10005 CAS.
- G. Imamura, C. W. Chang, Y. Nabae, M. A. Kakimoto, S. Miyata and K. Saiki, J. Phys. Chem. C, 2012, 116, 16305–16310 CAS.
- T. Katoh, G. Imamura, S. Obata, M. Bhanuchandra, G. Copley, H. Yorimitsu and K. Saiki, Phys. Chem. Chem. Phys., 2015, 17, 14115–14121 RSC.
- S. Entani, S. Ikeda, M. Kiguchi, K. Saiki, G. Yoshikawa, I. Nakai, H. Kondoh and T. Ohta, Appl. Phys. Lett., 2006, 88, 153126 CrossRef.
- M. Yamamoto, S. Obata and K. Saiki, Surf. Interface Anal., 2010, 42, 1637–1641 CrossRef CAS.
- L. Gao, W. Ren, H. Xu, L. Jin, Z. Wang, T. Ma, L.-P. Ma, Z. Zhang, Q. Fu, L.-M. Peng, X. Bao and H.-M. Cheng, Nat. Commun., 2012, 3, 699 CrossRef PubMed.
- M. Gao, Y. Pan, L. Huang, H. Hu, L. Z. Zhang, H. M. Guo, S. X. Du and H. J. Gao, Appl. Phys. Lett., 2011, 98, 033101 CrossRef.
- L. G. Cançado, A. Jorio, E. H. M. Ferreira, F. Stavale, C. A. Achete, R. B. Capaz, M. V. O. Moutinho, A. Lombardo, T. S. Kulmala and A. C. Ferrari, Nano Lett., 2011, 11, 3190–3196 CrossRef PubMed.
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14095–14107 CrossRef CAS.
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 64, 075414 CrossRef.
- A. Eckmann, A. Felten, A. Mishchenko, L. Britnell, R. Krupke, K. S. Novoselov and C. Casiraghi, Nano Lett., 2012, 12, 3925–3930 CrossRef CAS PubMed.
- C. Y. Su, Y. Xu, W. Zhang, J. Zhao, A. Liu, X. Tang, C. H. Tsai, Y. Huang and L. J. Li, ACS Nano, 2010, 4, 5285–5292 CrossRef CAS PubMed.
- M. Jin, T. H. Kim, S. C. Lim, D. L. Duong, H. J. Shin, Y. W. Jo, H. K. Jeong, J. Chang, S. Xie and Y. H. Lee, Adv. Funct. Mater., 2011, 21, 3496–3501 CrossRef CAS.
- S. Wang, X. Wang and S. P. Jiang, Phys. Chem. Chem. Phys., 2011, 13, 6883–6891 RSC.
- A. E. Axworthy, V. H. Dayan and G. B. Martin, Fuel, 1978, 57, 29–35 CrossRef CAS.
- Y. Ninomiya, Z. Dong, Y. Suzuki and J. Koketsu, Fuel, 2000, 79, 449–457 CrossRef CAS.
- J. C. Mackie, M. B. Colket III, P. F. Nelson and M. Esler, Int. J. Chem. Kinet., 1991, 23, 733–760 CrossRef CAS.
- G. B. Bacskay, M. Martoprawiro and J. C. Mackie, Chem. Phys. Lett., 1999, 300, 321–330 CrossRef CAS.
- A. Doughty and J. C. Mackie, J. Chem. Soc., Faraday Trans., 1994, 90, 541 RSC.
- A. Lifshitz, C. Tamburu and A. Suslensky, J. Phys. Chem., 1989, 93, 5802–5808 CrossRef CAS.
- J. C. Mackie, M. B. Colket III and P. F. Nelson, J. Phys. Chem., 1990, 94, 4099–4106 CrossRef CAS.
- N. R. Hore and D. K. Russell, J. Chem. Soc., Perkin Trans. 2, 1998, 269–276 RSC.
- A. L. Pinardi, G. Otero-Irurueta, I. Palacio, J. I. Martinez, C. Sanchez-Sanchez, M. Tello, C. Rogero, A. Cossaro, A. Preobrajenski, B. Gómez-Lor, A. Jancarik, I. G. Stará, I. Starý, M. F. Lopez, J. Méndez and J. A. Martin-Gago, ACS Nano, 2013, 7, 3676–3684 CrossRef CAS PubMed.
- Y. Lu and X. Yang, Carbon, 2015, 81, 564–573 CrossRef CAS.
- M. Martoprawiro, G. B. Bacskay and J. C. Mackie, J. Phys. Chem., 1999, 103, 3923–3934 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra22664c |
| This journal is © The Royal Society of Chemistry 2016 |