
Improved coulombic efficiency and cycleability of SnO2–Cu–graphite composite anode with dual scale embedding structure†
Abstract
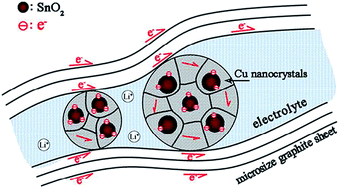
To improve the coulombic efficiency (CE) and cycle life of SnO2 anode in lithium ion batteries, SnO2–Cu–graphite composites with dual scale embedding structure are synthesized by ball milling. The SnO2–Cu composite, in which SnO2 nanoparticles with grain size less than 10 nm are uniformly dispersed in inactive nanocrystalline Cu matrix, is firstly obtained by milling the mixture of SnO2 and Cu nanopowders (molar ratio 1 : 2), and then further milling with graphite (C) to obtain SnO2–Cu–C composite with microsized graphite sheets as matrix of SnO2–Cu composite. The 50 h-milled SnO2–Cu composite exhibits higher initial CE (76.0 ± 1.5%) and subsequent CE than 50 h-milled SnO2. Furthermore, the SnO2–Cu–C composite anode is capable of retaining a maximum charge capacity of 450.8 mA h g−1 at 100 mA g−1 after 80 cycles with a capacity retention ratio of 74.4%, displaying superior cyclic stability to as-milled SnO2, SnO2–Cu and SnO2–C composites. The improved CE and cycleability are attributed to the unique dual scale embedding structure that offers good conductivity of electron and lithium ion as well as the nanostructure stability of active materials. This unique composite structure might be extended to other high-capacity anode materials, to achieve high performance lithium ion batteries.
 Please wait while we load your content...
Please wait while we load your content...

