
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCopper(II) quinolinonato-7-carboxamido complexes as potent antitumor agents with broad spectra and selective effects†
Radka Křikavová,
Ján Vančo,
Zdeněk Trávníček*,
Roman Buchtík and
Zdeněk Dvořák
Regional Centre of Advanced Technologies and Materials, Division of Biological Active Complexes and Molecular Magnets, Faculty of Science, Palacký University, 17. listopadu 12, CZ-771 46 Olomouc, Czech Republic. E-mail: zdenek.travnicek@upol.cz
First published on 14th December 2015
Abstract
A series of five copper(II) mixed-ligand complexes with the composition [Cu(quix)(phen)]NO3·yH2O (1 to 5), where Hquix stands for 2-(4-amino-3,5-dichlorophenyl)-3-hydroxy-4(1H)-quinolinone-7-carboxamides with different N-substitutions: Hqui1 = N-propyl (1), Hqui2 = N-isobutyl (2), Hqui3 = N-cyclohexyl (3), Hqui4 = N-benzyl (4), and Hqui5 = N-p-xylyl (5); phen = 1,10-phenanthroline and y = 0 or 1, were synthesized, characterized and screened for in vitro antitumor activity on a panel of six human cancer cell lines, including osteosarcoma (HOS), breast adenocarcinoma (MCF7), malignant melanoma (G361), cervix carcinoma (HeLa), ovarian carcinoma (A2780) and cisplatin-resistant ovarian carcinoma (A2780R). All the complexes, except for limitedly soluble complex 4, showed very potent cytotoxicity (IC50 ≈ 1–7 μM); the best IC50 value was found for complex 5 against A2780, with IC50 = 0.6(1) μM. Moreover, complex 5 was found to be non-toxic up to 50 μM against non-malignant lung fibroblast cells (MRC-5); therefore, showing a promising selectivity index [IC50(MRC-5)/IC50(A2780)] that was higher than 80. The complexes were also shown to bind to calf thymus DNA, interact with physiological levels of L-cysteine and act as chemical nucleases. It was additionally suggested that the species responsible for the biological activities of the prepared complexes were the [Cu(qui)(phen)]+ cations, or similar cationic species containing the {Cu(phen)} residue. The results clearly demonstrated that targeted structural optimization of the quinolinonato ligand in this class of complexes leads to compounds with high-level and broad-spectrum anticancer activity along with significantly increased selectivity.
1. Introduction
Metal-based cancer therapy has hitherto exclusively exploited the antitumour properties of platinum(II) complexes. Concretely, cisplatin and its derivatives (oxaliplatin and carboplatin) are currently well-established anticancer drugs used worldwide, whose therapeutic effects are based on the inhibition of cancer cell proliferation through binding to nuclear DNA.1–3 However, these platinum(II) complexes possess several limitations that complicate chemotherapy such as negative side effects (e.g. neurotoxicity, nephrotoxicity and emetogenesis) and intrinsic and/or acquired resistance phenomena.4,5 However, it is unambiguously the high therapeutic efficiency of these platinum(II)-based therapeutics, which has inspired many scientists to focus on the development of the next generation of transition metal complexes with both improved anticancer profiles and reduced negative side effects.6 One of the investigated approaches to decreasing the toxicity with respect to the platinum(II) complexes involves using essential transition metals as central atoms. Copper has been identified as a promising candidate, as it is an essential trace element found in all living organisms. Copper is involved in multiple functions of crucial enzymes7 and works as a fundamental component of the redox system (transition between the CuI and CuII oxidation states).8 Furthermore, an additional benefit of copper being an endogenous essential metal lies in the already established metabolic pathways for copper-containing compounds, which could also play a role in modulating the general toxicity of prospective copper-based therapeutics.The advantages of using copper as the central atom for the preparation of potential metallotherapeutics have already been exemplified in a number of copper complexes, particularly those involving planar polyheterocyclic ligands; these have shown notable cytotoxicity, with mechanisms of action related to varied modes of DNA interaction and redox metabolism alterations.9–12 The first reported CuII complex of this group that was able to inhibit tumour growth in vivo (studied with the Landschutz ascites tumour model in mice) was shown to have the composition [Cu(tmphen)2]Cl2, where tmphen stands for 3,4,7,8-tetramethyl-1,10-phenanthroline.13 Since then, many CuII complexes containing 1,10-phenanthroline (phen) and other bidentate N-donor heterocyclic ligands (N–N) and bidentate O-donor ligands (O–O) of the type [Cu(N–N) (O–O)]n=0/1+ have been synthesized and studied for their anticancer properties. Among these compounds, the Casiopeínas® series of CuII mixed-ligand bis-chelated antineoplastic agents, containing phen, 2,2′-bipyridine (bpy) or their substituted derivatives, as well as essential amino acids or acetylacetone,14–18 stand out. Their remarkable antitumour activity in vitro and in vivo, as well as promising results from preclinical studies, have classified these complexes as auspicious candidates for clinical trials.19
In our recent studies, we have focused on Casiopeínas®-like mixed-ligand CuII complexes containing bidentate N-donor heterocyclic ligands (phen, bpy, or their N–N derivatives), the 2-phenyl-3-hydroxy-4(1H)-quinolinonato ligand (qui) and nitrate or tetrafluoroborate counter anions (Y) of the general composition [Cu(qui)(N–N)]Y·yH2O].20–22 These complexes were identified as promising cytotoxic agents against a broad spectrum of human cancer cell lines, with IC50 reaching micromolar to sub-micromolar values, e.g. the IC50 value against A2780 ovarian cancer cells equalled 0.36 ± 0.05 μM for [Cu(qui)(mphen)]BF4·H2O (mphen = 5-methyl-1,10-phenanthroline). Moreover, markedly lower cellular toxicities of the complexes to healthy cells were observed; the ratios of the effective concentration against cancer cells to the toxic concentration to primary human hepatocytes were up to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40. In general, the best selectivity index was observed for the complexes containing non-substituted phen as the N-donor ligand.
:40. In general, the best selectivity index was observed for the complexes containing non-substituted phen as the N-donor ligand.
Quite recently, an extensive structure-cytotoxic activity relationship study was performed for 2-phenylsubstituted-3-hydroxyquinolin-4(1H)-one-carboxamides, which showed that the carboxamide substitution plays a positive role in the tuning of cytotoxicity (the best derivatives showed submicromolar IC50 values against the CEM leukaemia cell line) and results in very favourable therapeutic indices (up to 40 for the derivatives used in this study).23 These encouraging results inspired us to extend our previous studies and elucidate whether and how the 2-(3,5-dichloro-4-amino)phenyl- and 7-alkylcarboxamido-substitutions on the quinolinone skeleton may influence the biological activity of mixed-ligand complexes of the [Cu(quix)(phen)]NO3·yH2O (1–5) type. Herein, we report the synthesis, thorough characterization, and results of biological activity evaluations, i.e. cytotoxicity screening against a panel of seven human cancer and non-malignant cell lines, as well as investigations of their interactions with DNA and sulphur-containing biomolecules, of a series of copper(II) complexes of the general composition [Cu(quix)(phen)]NO3·yH2O (1–5, where x = 1–5, y = 0 or 1).
2. Experimental
2.1. Materials and methods
Chemicals and solvents were purchased from Sigma-Aldrich Co. and Acros Organics Co. and were used as received. The 2-phenyl-3-hydroxy-4(1H)-quinolinone-7-carboxamides (Hqui1–5) with N-substitution: Hqui1 = N-propyl, Hqui2 = N-isobutyl, Hqui3 = N-cyclohexyl, Hqui4 = N-benzyl, Hqui5 = N-p-xylyl, used as the starting materials for the preparation of complexes 1 to 5, were synthesized following a previously reported procedure;24 their purity was verified by elemental analysis, FTIR and NMR spectroscopy (Fig. S1 and S2, ESI†).Elemental analysis (C, H, N) was carried out on a Flash EA-2000 Elemental Analyser (Thermo Finnigan). FTIR spectra were obtained on a Nexus 670 FTIR (ThermoNicolet) using the ATR technique in the range of 200–4000 cm−1. Electronic absorption spectra of 10−3 and 10−5 M N,N-dimethylformamide (DMF) solutions and diffuse-reflectance UV-Vis spectra were obtained with a Lambda 40 spectrometer (Perkin Elmer Instruments) in the range of 300–900 nm. Mass spectra (MS) were obtained on an LCQ Fleet (ThermoFisher Scientific) spectrometer using the positive electrospray ionisation (ESI+) and full scan modes for 10−5 M methanol solutions of 1–5. NMR spectra of Hqui1–5 were measured in DMF-d7 on a Varian 400 MHz NMR spectrometer at 298 K. Tetramethylsilane (TMS) was used as the internal reference standard for the 1H and 13C NMR experiments. Simultaneous thermogravimetric (TG) and differential thermal (DTA) analyses were performed on an Exstar TG/DTA 6200 thermal analyser (Seiko Instruments Inc.). TG/DTA studies were carried out in platinum pans from ambient temperature to 800 °C with a 2.5 °C min−1 temperature gradient in dynamic air atmosphere (100 mL min−1). Conductivity experiments were performed on a Cond 340i/SET (WTW) in DMF (10−3 M, at 25 °C). The quantum chemical calculations were performed at the DFT level using the hybrid B3LYP functional with the LACVP+** basis set using Spartan’10 (version 1.1.0v4) software.25–27
2.2. Synthesis of [Cu(qui1)(phen)]NO3 (1), [Cu(qui2)(phen)]NO3 (2), [Cu(qui3)(phen)]NO3 (3), [Cu(qui4)(phen)]NO3·H2O (4) and [Cu(qui5)(phen)]NO3 (5)
1,10-Phenanthroline (phen) monohydrate (198 mg, 1 mmol) was dissolved in ethanol (10 mL), and the corresponding quinolinone (Hqui1–5) (1 mmol) dispersed in ethanol (30 mL) was then added with stirring. A solution of Cu(NO3)2·3H2O (242 mg, 1 mmol) in distilled water (5 mL) was slowly added to the resulting mixture with stirring. The reaction mixture was stirred at room temperature for a few hours until a solid product formed. The obtained solid was filtered, washed with a small amount of cold water and ethanol, and dried at 40 °C under an infrared lamp.:1). λmax (solid state)/nm 445, 739. λmax (10−5 M DMF solution)/nm 425 (ε/M−1 cm−1 9288). FTIR (ν, cm−1): 3307, 3200, 3070, 2961, 2934, 2874, 1664, 1623, 1572, 1541, 1492, 1452, 1429, 1376, 1314, 1254, 1184, 1148, 1040, 943, 895, 878, 847, 742, 720, 697, 653, 601, 584, 561, 513, 432, 337, 311, 244. ESI + MS: m/z 647 ([M − (NO3)]+, 100%), 243 ([Cu(phen)]+, 10%), 1296 ([{M − (NO3)}2]+, 2%).:1). λmax (solid state)/nm 445, 751. λmax (10−5 M DMF solution)/nm 437 (ε/M−1 cm−1 8470). FTIR (ν, cm−1): 3311, 3238, 3200, 3068, 2959, 2930, 2872, 1624, 1575, 1543, 1490, 1453, 1431, 1372, 1312, 1257, 1184, 1146, 1087, 1042, 942, 894, 878, 847, 744, 720, 693, 655, 600, 582, 560, 501, 434, 335, 311. ESI + MS: m/z 661 ([M − (NO3)]+, 100%), 1322 ([{M − (NO3)}2]+, 30%), 243 ([Cu(phen)]+, 18%).:1). λmax (solid state)/nm 425, 789. λmax (10−5 M DMF solution)/nm 424 (ε/M−1 cm−1 12660). FTIR (ν, cm−1): 3319, 3109, 3068, 2930, 2855, 1620, 1573, 1544, 1520, 1494, 1453, 1431, 1376, 1314, 1255, 1182, 1150, 1111, 1082, 1040, 943, 892, 876, 848, 830, 789, 742, 722, 698, 653, 619, 588, 563, 495, 471, 436, 332, 310, 259. ESI + MS: m/z 687 ([M − (NO3)]+, 100%), 1376 ([{M − (NO3)}2+, 15%), 243 ([Cu(phen)]+, 9%).:1). λmax (solid state)/nm 451, 778. λmax (10−5 M DMF solution)/nm 427 (ε/M−1 cm−1 10129). FTIR (ν, cm−1): 3456, 3319, 3198, 3084, 3063, 3034, 3007, 2967, 2930, 1619, 1572, 1542, 1378, 1308, 1253, 1226, 1180, 1145, 1109, 1082, 1039, 946, 889, 876, 845, 790, 740, 720, 698, 653, 603, 565, 557, 514, 501, 460, 435, 413, 303, 247. ESI + MS: m/z 695 ([M − (NO3)]+, 100%), 1390 ([{M − (NO3)}2+, 20%), 243 ([Cu(phen)]+, 10%).:1). λmax (solid state)/nm 448, 773. λmax (10−5 M DMF solution)/nm 424 (ε/M−1 cm−1 10941). FTIR (ν, cm−1): 3331, 3066, 2946, 2924, 2863, 1621, 1575, 1545, 1492, 1455, 1429, 1381, 1312, 1279, 1252, 1227, 1181, 1147, 1111, 1040, 947, 894, 878, 848, 785, 759, 742, 722, 694, 655, 615, 584, 565, 492, 477, 436, 344, 318, 246. ESI + MS: m/z 709 ([M − (NO3)]+, 100%), 1418 ([{M − (NO3)}2+, 23%), 243 ([Cu(phen)]+, 11%).2.3. Biological activity
:1, v/v) were used as reference samples. The interacting system was analysed immediately after preparation (0 h), and was then analysed 1, 12, and 24 h after the solutions of complex 5 and the sulphur-containing biomolecules were mixed.3. Results and discussion
3.1. General properties
Yellow-green copper(II) complexes of the general composition [Cu(qui1–5)(phen)]NO3·yH2O (1–5; x = 0–1) were synthesized via the reaction of 1,10-phenanthroline (phen) monohydrate and the corresponding 2-(4-amino-3,5-dichlorophenyl)-3-hydroxy-4(1H)-quinolinone-7-carboxamides with N-substitution, i.e. Hqui1 = N-propyl (1), Hqui2 = N-isobutyl (2), Hqui3 = N-cyclohexyl (3), Hqui4 = N-benzyl (4), and Hqui5 = N-p-xylyl (5), with Cu(NO3)2·3H2O in an ethanol/water mixture (11:1, v/v) in a 1:1:1 molar ratio, with yields of 68–85% (Scheme 1). According to the conductivity measurements, complexes 1–5 behaved as electrolytes of the 1:1 type in 10−3 M DMF solution with conductivity values of ca. 70 S cm2 mol−1. Furthermore, all the ESI+ mass spectra of the studied compounds contained the peaks of the corresponding complex [Cu(qui1–5)(phen)]+ cations with a relative intensity of 100%, thus providing evidence regarding the composition of 1 to 5. In addition, the peaks of the fragments of the dimeric species [{Cu(qui1–5)(phen)}2]+ as well as a peak corresponding to [Cu(phen)]+ at 243.0 m/z (calc. 243.0) were detected in the spectra of all the complexes, thus suggesting the same fragmentation patterns for the studied compounds.
 | ||
| Scheme 1 The general structural formula of complexes 1–5. | ||
The presence of the water molecule of crystallization in the monohydrated complex 4 was confirmed by simultaneous TG/DTA analysis. The calculated and experimental data regarding the crystal water elimination were found to be in good accordance (calcd/found 2.4/2.3%). All the other complexes were found to be thermally stable up to ca. 180–200 °C. The course of the thermal degradation of all the complexes above 200 °C was analogous, proceeding without the formation of any thermally stable intermediates, and characterized by four exothermic effects on the DTA curve. The final products of degradation (probably associated with the formation of CuO) formed at ca. 540–580 °C. The thermal decomposition data are listed in Section 2.3.
3.2. UV-Vis and FTIR spectral characterizations
The electronic spectra of the CuII complexes 1 to 5 were measured in the 300 to 900 nm region, both in the solid state and in DMF solutions (10−3 and 10−5 M). The diffuse-reflectance spectra of the complexes exhibited one intensive well-defined absorption maximum centred at 425–451 nm, assignable to the charge-transfer transitions (CT), and one very broad maximum in the range of 739–789 nm, which can be attributed to d–d transitions of the 2B1g → 2Eg, 2B1g → 2B2g, and 2B1g → 2A1g types, characteristic of copper(II) complexes with distorted square-planar arrangements.31 The measured UV-Vis spectra of the 10−5 M DMF solutions of 1–5 were dominated by CT transitions observed in a similar region, at 425 nm (1), 437 nm (2), 424 nm (3), 427 nm (4) and 424 nm (5). The maxima corresponding to the d–d transitions with low absorbance values could be detected as unintensive shoulder bands only in the DMF solutions with c ≥ 10−3 M. In comparison with the solid state spectra, these maxima were shifted to lower wavelengths ranging from 599 to 611 nm (molar absorption coefficients equalling 81–156 M−1 cm−1) (Fig. 1). This shift is most likely connected with a change in the coordination geometry around the metal as a result of the coordination of solvent molecules to the copper(II) centre in the solution, which was confirmed by EPR spectroscopy for structurally analogous complexes in our previous study.20 | ||
| Fig. 1 UV-Vis spectra of complex 1 in the range of 300–800 nm: diffuse reflectance spectrum (solid black line); 10−5 M DMF solution spectrum (violet dashed line); 10−3 M DMF solution spectrum (blue dash-dotted line). λmax (solid state, nm): 445, 739. λmax (10−5 M DMF solution, nm)/ε (M−1 cm−1): 425/9288. λmax (10−3 M DMF solution, nm)/ε (M−1 cm−1): 605/156. Maxima are indicated with black arrows. | ||
FTIR spectra of the complexes were obtained in the region of 150–4000 cm−1. The interpretation of the spectra, made also in comparison with the spectra of Hqui1–5 and phen, indirectly confirmed the coordination of both types of organic ligands to the copper(II) atom. In the region of 2800–3500 cm−1, bands for the characteristic stretching vibrations of ν(N–H) were identified at 3307–3331 cm−1, ν(C–H)ar at 3063–3109 cm−1, and ν(C–H)aliph at 2855–2959 cm−1. The spectrum of monohydrated complex 4 additionally showed a broad band at 3456 cm−1, assignable to ν(O–H) originating from the water molecule of crystallization. Furthermore, two characteristic carbonyl group bands were clearly observed in the spectra of 1–5. A weak to medium intensity band in the region of 1642–1664 cm−1 could be assigned to the stretching amide carbonyl vibration, whereas the strong bands at 1619–1624 cm−1 are assignable to the qui carbonyl group in the position 4 of the heterocycle. The aromatic ν(C⋯C)ring vibrations of the phen ring can be found between 1572 and 1575 cm−1 and in the region of 1429–1431 cm−1. Very strong characteristic bands of the qui aromatic ν(C⋯C)ring vibrations appeared at 1490–1494 cm−1. The medium intensity band in the region of 1372–1381 cm−1 should be assigned to the qui ν(CNH) vibration. The uncoordinated nitrate group ν3(NO3) bands were observed between 1308 and 1314 cm−1. Two bands were also detected at 845–848 cm−1 and 720–722 cm−1, which are characteristic of the phen ligand (Fig. S2, ESI†). In the far-IR region, the medium bands at 303–318 cm−1 could be attributed to ν(Cu–N), whereas the peaks at 560–565 and 492–513 cm−1 could be connected with the stretching vibrations of ν(Cu–O).32–34
3.3. Quantum chemical calculations
The X-ray structures of two complexes, [Cu(qui)(phen)]NO3·H2O and [Cu(qui)(ambpy)]NO3 (ambpy = bis(2-pyridyl)amine), with analogous compositions of 1–5 have been already reported.20 Several attempts (including slow evaporation of the reaction solution or mother liquor at varied temperatures and diffusion of diethyl ether to saturated ethanol/DMF solution) were made to obtain suitable crystals of 1–5 for single crystal X-ray analysis; however, all employed methods were unsuccessful. In an effort to confirm the composition of 1–5 suggested by the results of the various analytical methods described above and to predict the molecular structure of the prepared complexes, a model for DFT calculation was built on the basis of the structural similarities of the complexes with [Cu(qui)(phen)]NO3·H2O and [Cu(qui)(ambpy)]NO3.20 The geometry of the complex cation of 3 was optimized at the DFT level using the hybrid B3LYP functional with the LACVP+** basis set. The optimized geometry of [Cu(qui3)(phen)]+ is shown in Fig. 2. The copper(II) atom is tetra-coordinated by one bidentate O,O-coordinated qui3 (Hqui3 = 2-(4-amino-3,5-dichlorophenyl)-N-cyclohexyl-3-hydroxy-4(1H)-quinolinone-7-carboxamide) and one bidentate N,N-coordinated phen. The Cu–N bonds (Cu–N1 = 2.043 Å; Cu–N2 = 2.043 Å) were found to be rather longer than the Cu–O bonds (Cu–O1 = 1.916 Å; Cu–O2 = 1.941 Å), which is in agreement with the bond length comparison in the vicinity of the metal centre in the structurally related complex [Cu(qui)(phen)]NO3·H2O (further abbreviated as CuQ).20 However, the calculated bond lengths in the vicinity of the central atom were found to be slightly greater than those found in the X-ray structure of CuQ (Cu–N1 = 1.988(2) Å; Cu–N2 = 1.978(2) Å; Cu–O1 = 1.892(2) Å; Cu–O2 = 1.916(2) Å). The geometry around the copper(II) atom in 4 could be described as distorted square-planar; the O1–Cu–N1 and O2–Cu–N2 angles were equal to 177.28° and 175.34°, respectively. | ||
| Fig. 2 The geometry of the complex [Cu(qui3)(phen)]+ cation of 3 optimized at the B3LYP/LACVP+** level of theory. H-atoms were omitted for clarity. | ||
3.4. In vitro cytotoxicity
In our previous study, dealing with mixed-ligand CuII complexes20–22 involving the CuN2O2 donor set, it was demonstrated that the 2-phenyl-3-hydroxy-4(1H)-quinolinone skeleton (Hqui) represents a promising O,O-donor ligand for Casiopeínas®-like anticancer complexes, which represented the motivation for the study of complexes 1–5 presented herein.Complexes 1–5 and cisplatin, used as a standard, were screened for in vitro cytotoxicity against osteosarcoma (HOS), breast adenocarcinoma (MCF7), cervix epithelioid carcinoma (HeLa), malignant melanoma (G361), ovarian carcinoma (A2780), cisplatin-resistant ovarian carcinoma (A2780R) and non-malignant human fibroblast (MRC5) cells. In the case of 4, the testing was limited by the low solubility of the complex in the medium used; in this case, the IC50 was expressed as >5.0 μM. Otherwise, all the tested complexes exhibited dose-dependent cytotoxicity against all the cancer cell lines, reaching low-micromolar or sub-micromolar IC50 values. Complexes 1–3 and 5 were significantly more cytotoxic (ANOVA, p < 0.05) against HOS than cisplatin (IC50 = 18.9 ± 1.7 μM). Against MCF7, only 1 and 2 showed IC50 values significantly lower than cisplatin (IC50 = 17.9 ± 3.5 μM). These values are comparable with those obtained for the structurally related complex [Cu(qui)(phen)]NO3·H2O (CuQ), involving non-substituted qui, with IC50 = 4.3 ± 0.1 μM (HOS) and 7.3 ± 2.3 μM (MCF7).20 The viability of HeLa cells, which were not sensitive to cisplatin up to the highest tested concentration (IC50 > 50 μM), was reduced comparably by all the complexes, including CuQ (IC50 = 2.9 ± 0.3 μM). In the case of G361, the IC50 values were practically identical for all the tested compounds, including cisplatin (5.8 ± 2.4 μM). On the other hand, the previously reported CuQ was significantly more active on this cell line, with IC50 = 0.8 ± 0.2 μM. Furthermore, all the tested complexes were found to possess mutually comparable and significantly higher cytotoxicities (ANOVA, p < 0.05) than cisplatin against A2780. The results showed that the complexes were up to ca. 35 times more effective than cisplatin; the best IC50 value was determined for 5, i.e. 0.6 ± 0.1 μM (cisplatin: 21.8 ± 3.9 μM). Cytotoxicity against A2780R was tested with the aim of uncovering the possible ability of the complexes to overcome the resistance of these cells towards cisplatin (IC50 > 50 μM). The complexes showed activities of low micromolar IC50 (≈3 μM) on this cell line. The difference between the IC50 values determined for A2780 and A2780R was non-significant (ANOVA, p < 0.05) for 1 and 3, which means that the cytotoxic effect was practically the same against both cell lines (Table 1).
| Compound | HOS | MCF7 | HeLa | G361 | A2780 | A2780R | MRC-5 |
|---|---|---|---|---|---|---|---|
| a n.t. = not tested (due to inactivity in the primary cytotoxicity testing against HOS and MCF7 up to the concentration limited by solubility in the used medium). | |||||||
| 1 | 3.3 ± 1.3 | 5.5 ± 2.5 | 3.2 ± 0.3 | 3.5 ± 0.1 | 1.0 ± 0.6 | 2.9 ± 1.2 | 4.9 ± 2.7 |
| 2 | 4.6 ± 0.2 | 7.1 ± 2.6 | 3.0 ± 1.1 | 3.7 ± 0.5 | 1.3 ± 0.4 | 4.4 ± 0.6 | 15.9 ± 1.2 |
| 3 | 7.1 ± 1.6 | >10 | 4.1 ± 0.8 | 3.1 ± 0.4 | 1.9 ± 0.2 | 2.5 ± 0.7 | >25 |
| 4 | >5 | >5 | n.t. | n.t. | n.t. | n.t. | n.t. |
| 5 | 6.7 ± 2.5 | 31.9 ± 6.4 | 2.7 ± 0.5 | 3.2 ± 0.6 | 0.6 ± 0.1 | 3.4 ± 0.2 | >50 |
| Cisplatin | 18.9 ± 1.7 | 17.9 ± 3.5 | >50 | 5.8 ± 2.4 | 21.8 ± 3.9 | >50 | >50 |
To be considered as an anticancer therapeutic candidate, a compound must be selective for transformed cells over healthy non-transformed cells. To assess the cytotoxicity profiles of the presented complexes, their toxicity towards non-malignant MRC-5 cells was evaluated. It was found that MRC-5 cells showed rather different sensitivities towards the complexes. Complex 1 was comparably cytotoxic against both transformed and healthy cells, with IC50 = 4.9 ± 2.7 μM for MRC-5. Complex 2 showed ca. 3× lower toxicity for MRC-5 than 1 (IC50 = 15.9 ± 1.2 μM). The IC50 value for complex 3 could not be determined due to its limited solubility in the medium used; therefore, its toxicity can be expressed as IC50 > 25 μM. However, the result for 3 suggests that its cytotoxicity against cancer cells is ca. an order of magnitude higher than its cytotoxicity against healthy cells. The best cytotoxicity profile was found for 5, which was non-toxic to MRC-5 up to the highest tested concentration (IC50 > 50 μM). The selectivity index (SI) for 5, calculated as the ratio of IC50(non-malignant cells)/IC50(A2780), is therefore higher than ca. 80, as compared to ca. 2 for cisplatin and SI ≈ 40 for [Cu(qui)(phen)]BF4 (with IC50 = 20.4 ± 1.2 μM against primary human hepatocytes).22 Based on the presented results, it can be suggested that the 2-(3,5-dichloro-4-amino)phenyl and 7-alkylcarboxamido substitutions introduced on the qui skeleton resulted in a beneficial cytotoxic profile in the cases of compounds 3 and 5, but not in the cases of 1 and 2, therefore highlighting the importance of suitable structural modifications of the O,O-ligand in this type of copper(II) complex. The results of this study clearly showed that optimization of the composition of Cu-quinolinonato complexes can lead to compounds with increased anticancer effects and simultaneously increased selectivity, which is connected with the reduction of negative side effects.
3.5. Interaction with CT-DNA assessed by fluorescence ethidium bromide (EB) displacement assay
Ethidium bromide (EB) is a typical intercalator of DNA, because its planar phenanthridine ring is inserted between two adjacent base pairs. The formation of the intercalation manifests itself by the increase of the fluorescence intensity of the formed EB–DNA supramolecular complex up to 24-fold.35,36 This interaction is, however, competitive; when a suitable intercalator is added to the EB–DNA supramolecular complex, some of the EB molecules are substituted and fluorescence quenching is observed.37 Therefore, CuII complexes 1–5 were gradually added to the EB–DNA system with increasing concentration (1–50 μM). The emission of the EB–DNA system upon the addition of complex 1 is shown in Fig. 3. The results of mathematical analysis of the quenching curves (Table 2) indicated that all the complexes were able to bind to CT-DNA, with apparent binding constants (Kapp) between 1.6 × 106 M−1 (5) and 2.7 × 107 M−1 (1).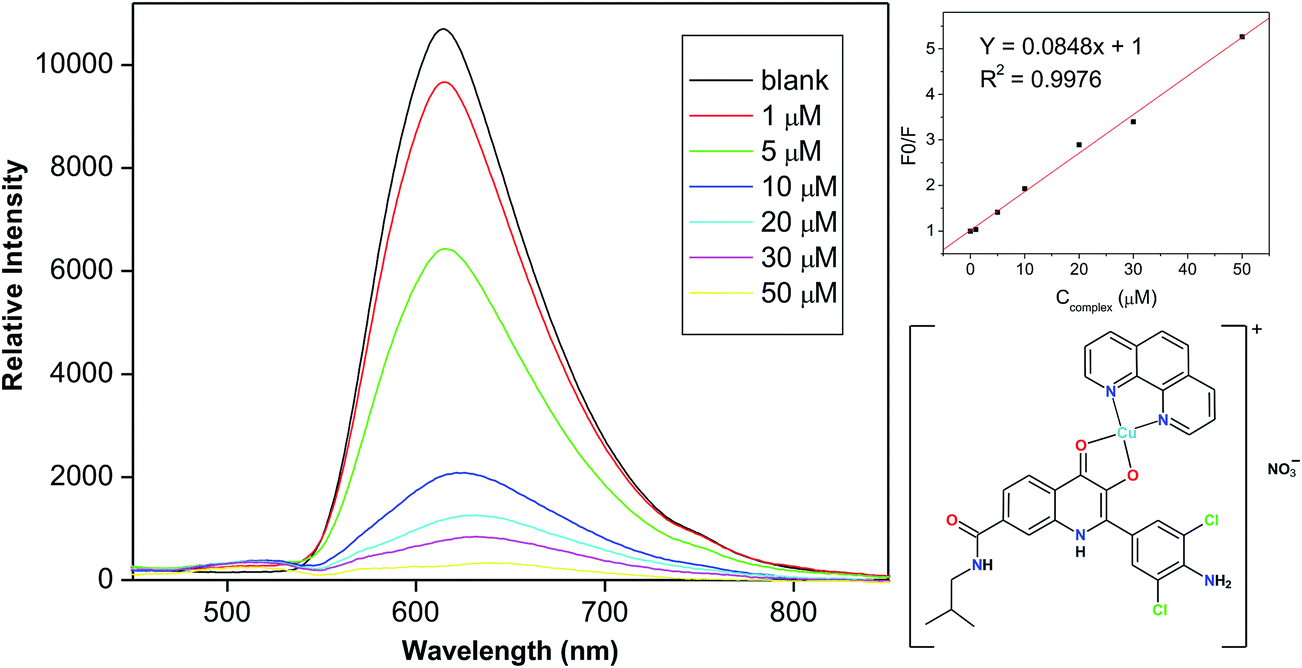 | ||
| Fig. 3 The results of the fluorescence ethidium bromide (EB) displacement titration assay showing the fluorescence quenching of the EB–DNA system upon the addition of complex 2. | ||
3.6. pUC-19 plasmid DNA cleavage
To obtain deeper insight into the mechanisms responsible for the cytotoxicity of the tested copper(II) complexes, the representative complex 5 was tested to determine its ability to behave as a chemical nuclease under conditions of oxidative stress, in this case represented by a mixture of hydrogen peroxide and a reducing agent (L-ascorbic acid). This model was selected based on the well-known fact that the overdriven metabolism in tumour cells generates considerable amounts of reactive oxygen and nitrogen species,38 such as superoxide anionic radicals or hydrogen peroxide, which can generate highly-toxic hydroxyl species and/or other highly reactive metal-containing species by the Fenton reaction.39 These highly-reactive species such as hydroxyl are able to cause irreversible damage to the structure of double-stranded DNA (dsDNA); for example, they can induce the decoupling of nitrogen bases, nick one polynucleotide chain, or cleave both strands of DNA.40 A relatively small circular dsDNA sequence from the pUC19 plasmid was used as a model object, and the ability of the selected representative complex 5 to oxidatively cleave the polynucleotide chains in the presence of a large excess of hydrogen peroxide was monitored by gel electrophoresis, which allowed qualitative and quantitative evaluation of the different forms of modified DNA (see Fig. 4). The quantitative parameter of total DNA cleavage was calculated as the percentage of the integrated densities of the cleaved forms (OC-form + 2× L-form) from the sum of integrated densities of all the identified forms of DNA (CCC-form + OC-form + 2× L-form). The results of the quantification are presented in Table 3. | ||
| Fig. 4 The electrophoretogram depicting the cleavage of pUC19 plasmid DNA by complex 5 in the presence of hydrogen peroxide. The three basic forms of plasmid DNA are identified as supercoiled DNA (CCC-form), open circular form (OC-form), and linear form (L-form). Lanes 1–3, 7–9 and 13–15 represent the products of the cleavage of pUC19 plasmid induced by complex 5 applied in concentrations of 20 μM, 100 μM, and 200 μM, respectively, without the addition of ascorbic acid as a reducing agent. Lanes 4–6, 10–12 and 16–18 represent the products of the cleavage of pUC19 plasmid induced by complex 5 applied in concentrations of 20 μM, 100 μM, and 200 μM, respectively, with the addition of ascorbic acid as a reducing agent (complete cleavage in all cases). Lane 19 represents a blank sample (the mixture of pUC19 plasmid and hydrogen peroxide) and lane 20 represents the native pUC19 plasmid. | ||
| Lane | Concentration of 5 | Conditions | CCC [%] | L [%] | OC [%] | Average DNA cleavage [%] |
|---|---|---|---|---|---|---|
| 1 | 20 μM | W/o L-ascorbic acid | 57.1% | 6.9% | 36.0% | 46.3 |
| 2 | 58.1% | 5.7% | 36.2% | |||
| 3 | 56.8% | 7.7% | 35.5% | |||
| 4 | 20 μM | With L-ascorbic acid | 0% | 0% | 0% | 100 |
| 5 | 0% | 0% | 0% | |||
| 6 | 0% | 0% | 0% | |||
| 7 | 100 μM | W/o L-ascorbic acid | 28.7% | 13.8% | 57.5% | 75.6 |
| 8 | 24.7% | 18.0% | 57.3% | |||
| 9 | 29.7% | 10.3% | 60.0% | |||
| 10 | 100 μM | With L-ascorbic acid | 0% | 0% | 0% | 100 |
| 11 | 0% | 0% | 0% | |||
| 12 | 0% | 0% | 0% | |||
| 13 | 200 μM | W/o L-ascorbic acid | 0% | 0% | 0% | 100 |
| 14 | 0% | 0% | 0% | |||
| 15 | 0% | 0% | 0% | |||
| 16 | 200 μM | With L-ascorbic acid | 0% | 0% | 0% | 100 |
| 17 | 0% | 0% | 0% | |||
| 18 | 0% | 0% | 0% | |||
| 19 | Blank | 68.1% | 0% | 31.9% | 31.9 | |
| 20 | Native plasmid | 94.7% | 1.2% | 4.1% | 6.4 | |
The results of the cleavage experiments showed the remarkable ability of complex 5 to cleave the DNA by an oxidative mechanism in the presence of a reducing agent (L-ascorbic acid); even at the smallest concentration used (20 μM), the complete cleavage of DNA molecules to small fragments occurred, leaving no trace of uncleaved DNA in the electrophoretograms (see lanes 4–6 and 10–12 in Fig. 4). At higher concentrations of the complex, the DNA fragments were cross-linked into structures of higher order (and lower electrophoretic mobility); therefore, a smear can be detected in the electrophoretogram (see lanes 13–18 in Fig. 4). Complex 5, however, showed the ability to cleave the polynucleotide chains of dsDNA also without the addition of the reducing agent, reaching DNA cleavage levels of up to ca. 80% at 100 μM concentration. At a concentration of 200 μM, even in the absence of reducing agent, complete cleavage of the DNA occurred (see lanes 16 to 18 in Fig. 4).
In conjunction with the abovementioned presented results of the EB-displacement method, in which complex 5 showed the ability to effectively substitute ethidium bromide at the intercalating sites in the minor groove of the DNA double helix, it can be speculated that the mechanism of DNA-cleavage mediated by this complex is based on sequence non-specific interactions between the cations [Cu(qui)(phen)]+, or similar cationic species containing the {Cu(phen)} residue (see Section 3.7, interactions with sulphur-containing biomolecules assessed by ESI-MS) and the phosphate moieties; the mechanism may also be partly due to intercalation in the minor groove of the double helical DNA,41,42 the reduction of Cu(II) to Cu(I), and the Fenton reaction-mediated production of reactive oxygen species that are able to cleave the polynucleotide chains, probably at the C1′ or C4′ sites of the adjacent deoxyribose moieties.40
3.7. Interactions with sulphur-containing biomolecules assessed by ESI-MS
With the aim of describing the interactions of the complexes (using the example of the representative complex 5) in solutions that mimic the conditions of the real physiological environment, we performed mass spectrometric evaluations of the interacting systems, containing 10 μM of complex 5 and a mixture of L-cysteine and L-glutathione at physiological concentrations (290 μM and 6 μM, respectively).30 The mass spectra of this system were obtained immediately after mixing the components (0 h) and then after 1, 12, and 24 h. The most pronounced changes were found after 24 h of interaction (see Fig. 5). The mass spectrum of the interacting system was compared to that of complex 5, dissolved in methanol:water (1:1, v/v) (see Fig. 5B), which contained a rich variety of peaks corresponding to the ionic species derived from the phen ligand (at 181.17 m/z) and solvated {Cu(phen)} residues, such as [Cu(phen)2 + H]2+ at 211.67 m/z, [Cu(phen)]+ at 243.08 m/z, [Cu(phen) + H2O + OH]+ at 278.08 m/z, [Cu(phen) + H2O + NO3 + CH3OH + H]+ at 356.00 m/z, [Cu(phen)2 + H]+ at 423.17 m/z, [Cu(phen)2 + H2O + NO3 + CH3OH + H]+ at 536.00 m/z, and [M − NO3]+ at 711.08 m/z. In addition to the abovementioned ionic species, the mass spectrum of the interacting system (see Fig. 5C) containing L-cysteine and L-glutathione showed one additional set of peaks corresponding to the interacting species containing the L-cysteine residue, with the composition [Cu(qui5) + Cys + H]+, at 667.92 m/z.
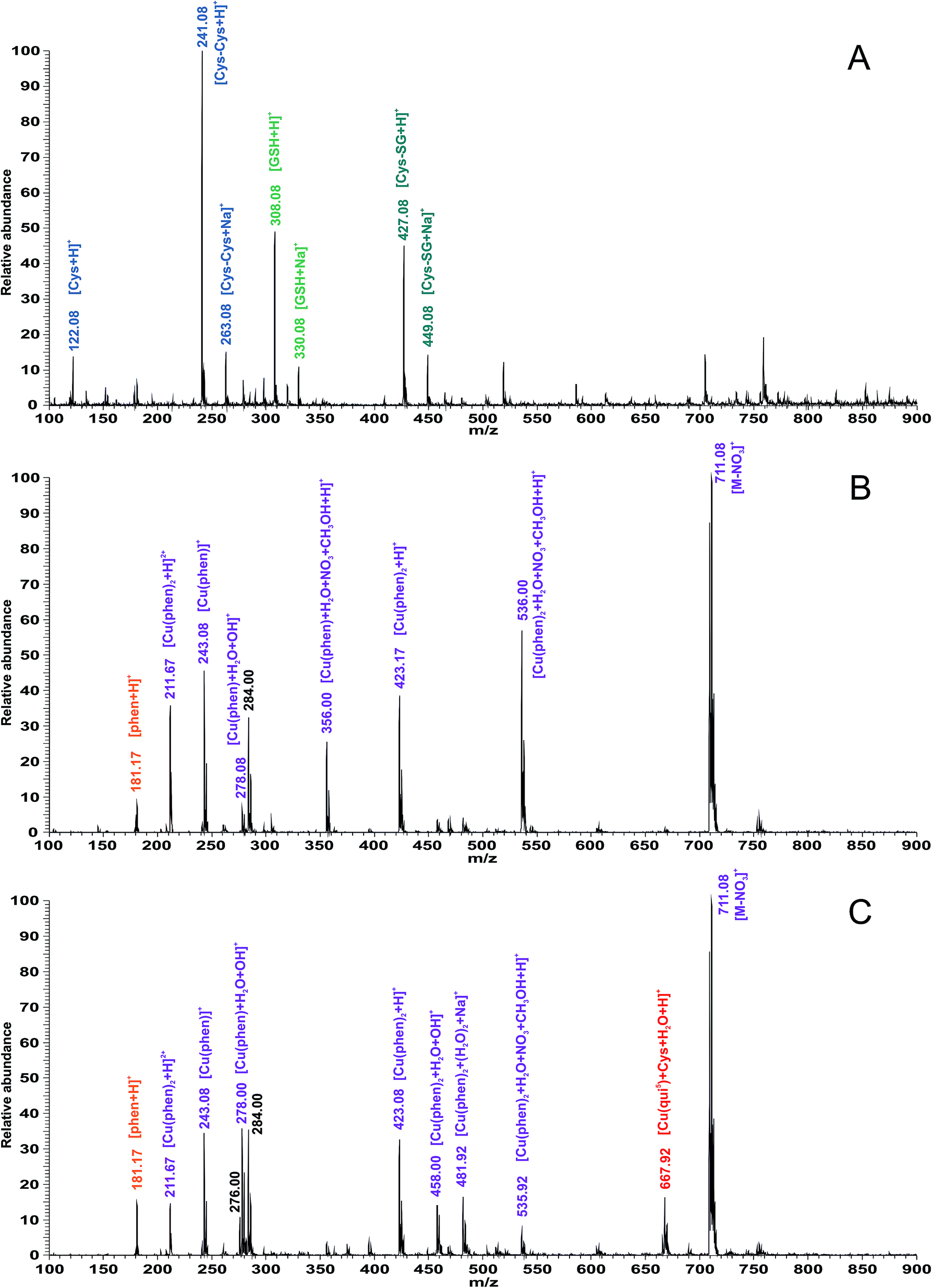 | ||
| Fig. 5 Results of the ESI-MS studies of the interactions of complex 5 with physiological concentrations of L-cysteine and L-glutathione. ESI-MS spectra of the mixture containing physiological concentrations of L-cysteine and L-glutathione (A), a 10 μM solution of complex 5 in methanol:water (1:1, v/v) 24 h after preparation (B), and the interacting system containing complex 5 (at a concentration of 10 μM), L-cysteine (at a concentration of 290 μM) and reduced glutathione (at a concentration of 6 μM) (C). The identified ionic species for the selected peaks are indicated. | ||
The described set of interactions in solution may contribute significantly to the behaviour of the prepared complexes in biological systems, leading to their higher affinity in the context of DNA binding in the form of the species containing {Cu(phen)}. These findings are in agreement with those reported in our previous study.20–22
4. Conclusions
Five copper(II) mixed-ligand complexes with the general composition [Cu(quix)(phen)]NO3·yH2O (1–5), where Hquix stands for variously N-substituted 2-(4-amino-3,5-dichlorophenyl)-3-hydroxy-4(1H)-quinolinone-7-carboxamides; phen = 1,10-phenanthroline and y = 0 or 1, were prepared, thoroughly characterized and screened for their in vitro cytotoxicity against a panel of human cancer cell lines (HOS, MCF7, HeLa, G361, A2780, and A2780R). In addition, their toxicity to healthy cells was evaluated on a non-malignant lung fibroblast cell line, MRC-5. The presented complexes, except for 4, were found to be potent in vitro anticancer agents; complex 5 was found to have the best IC50 value, i.e. 0.6 ± 0.1 μM against A2780. Moreover, complex 5 showed a very favourable selectivity index, calculated as the ratio of IC50(MRC-5)/IC50(A2780), reaching a value higher than ca. 80. Complexes 1–5 were able to bind to CT-DNA, as evidenced by significant florescence quenching of the ethidium bromide–DNA system, with binding constants comparable to that of ethidium bromide. Furthermore, pUC19 plasmid DNA cleavage experiments showed the ability of the prepared complexes to act as chemical nucleases in the presence of hydrogen peroxide, and the addition of reducing agent (L-ascorbic acid) significantly increased their pro-oxidative effects. The results of the mass spectrometric evaluation of the interactions of the representative complex 5 with sulphur-containing biomolecules confirmed the ability of the complex to interact with physiological levels of L-cysteine and simultaneously suggested that the species responsible for the biological activities of the prepared complexes may be [Cu(qui)(phen)]+ or similar cationic species containing the {Cu(phen)} moiety.The current study unambiguously demonstrated the importance of suitable structural modifications of the O,O-ligands in Casiopeínas®-like copper(II) complexes. The results of the most promising complex, 5, suggested that the introduced 7-(p-xylyl)carboxamido-substitution on the qui skeleton resulted in a more beneficial cytotoxic profile, with comparable anticancer activity and significantly increased selectivity towards cancer cells over non-malignant cells. In conclusion, it can be pointed out that the presented results represent a successful next step in the optimization of the composition of Cu-quinolinonato complexes, leading to potentiation of their anticancer activity and, concurrently, suppression of their negative side-effects.
Acknowledgements
The authors would like to acknowledge the financial support from the National Program of Sustainability I (LO1305) of the Ministry of Education, Youth and Sports of the Czech Republic and Palacký University (PrF_2015_019). We are also grateful to Ms Kateřina Kubešová for help with cytotoxicity testing, Dr Bohuslav Drahoš for help with mass spectrometry experiments and Mr Jakub Hutyra for performing DNA cleavage experiments.References
- L. R. Kelland and N. P. Farrell, Platinum Based Drugs in Cancer Therapy, Humana Press, Totowa, NJ, USA, 2000 Search PubMed.
- M. Gielen and E. R. T. Tiekink, Metallotherapeutic Drugs and Metal-Based Diagnostic Agents, John Wiley & Sons, Ltd., Chichester, UK, 2005 Search PubMed.
- E. Alessio, Bioinorganic Medicinal Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2011 Search PubMed.
- P. Heffeter, U. Jungwirth, M. Jakupec, C. Hartinger, M. Galanski, L. Elbling, M. Micksche, B. Keppler and W. Berger, Drug Resist. Updates, 2008, 11, 1 CrossRef CAS PubMed.
- X. Y. Wang and Z. J. Guo, New Trends and Future Developments of Platinum-Based Antitumor Drugs in Bioinorganic Medicinal Chemistry, ed. E. Alessio, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, 2011 Search PubMed.
- C. X. Zhang and S. J. Lippard, Curr. Opin. Chem. Biol., 2003, 7, 481 CrossRef.
- M. C. Linder, Biochemistry of Copper, Plenum Press, New York, 1991 Search PubMed.
- J. M. C. Gutteridge, Med. Biol., 1985, 63, 41–42 CAS.
- W. Zhou, X. Wang, M. Hu, C. Zhu and Z. Guo, Chem. Sci., 2014, 5, 2761 RSC.
- S. Tardito, A. Barilli, I. Bassanetti, M. Tegoni, O. Bussolati, R. Franchi-Gazzola, C. Mucchino and L. Marchio, J. Med. Chem., 2012, 55, 10448 CrossRef CAS PubMed.
- D. Palanimuthu, S. V. Shinde, K. Somasundaram and A. G. Samuelson, J. Med. Chem., 2013, 5, 722 CrossRef PubMed.
- C. Marzano, M. Pellei, F. Tisato and C. Santini, Anti-Cancer Agents Med. Chem., 2009, 9, 185 CrossRef CAS.
- F. P. Dwyer, E. Mayhew, E. M. Roe and A. Shulman, Br. J. Cancer, 1965, 19, 195 CrossRef CAS PubMed.
- C. Mejia and L. Ruiz-Azuara, Pathol. Oncol. Res., 2008, 14, 467 CrossRef PubMed.
- M. E. Bravo-Gomez, J. C. Garcia-Ramos, I. Garcia-Mora and L. Ruiz-Azuara, J. Inorg. Biochem., 2009, 103, 299 CrossRef CAS PubMed.
- L. Becco, A. Rodriguez, M. E. Bravo, M. J. Prieto, L. Ruiz-Azuara, B. Garat, V. Moreno and D. Gambino, J. Inorg. Biochem., 2012, 109, 49 CrossRef CAS PubMed.
- A. G. Gutierrez, A. Vazquez-Aguirre, J. C. Garcia-Ramos, M. Flores-Alamo, E. Hernandez-Lemus, L. Ruiz-Azuara and C. Mejia, J. Inorg. Biochem., 2013, 126, 17 CrossRef CAS PubMed.
- G. Vértiz, L. E. García-Ortuno, J. P. Bernal, M. E. Bravo-Gómez, E. Lounejeva, A. Huerta and L. Ruiz-Azuara, Fundam. Clin. Pharmacol., 2014, 28, 78 CrossRef PubMed.
- L. Becco, J. C. García-Ramos, L. Ruiz Azuara, D. Gambino and B. Garat, Biol. Trace Elem. Res., 2014, 161, 210 CrossRef CAS PubMed.
- R. Buchtík, Z. Trávníček, J. Vančo, R. Herchel and Z. Dvořák, Dalton Trans., 2011, 40, 9404 RSC.
- R. Buchtík, Z. Trávníček and J. Vančo, J. Inorg. Biochem., 2012, 116, 163 CrossRef PubMed.
- Z. Trávníček, J. Vančo, J. Hošek, R. Buchtík and Z. Dvořák, Chem. Cent. J., 2012, 6, 160 CrossRef PubMed.
- M. Soural, J. Hlaváč, P. Funk, P. Džubák and M. Hajdúch, ACS Comb. Sci., 2011, 13, 39 CrossRef CAS.
- M. Soural, J. Hlaváč, P. Hradil, I. Fryšová, M. Hajdúch, V. Bertolasi and M. Maloň, Eur. J. Med. Chem., 2006, 41, 467 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372 CrossRef CAS.
- V. Rassolov, J. A. Pople, M. Ratner, P. C. Redfern and L. A. Curtiss, J. Comput. Chem., 2001, 22, 976 CrossRef CAS.
- V. Shao, L. F. Molnar, Y. Jung, J. Kussmann, C. Ochsenfeld, S. T. Brown, A. T. B. Gilbert, L. V. Slipchenko, S. V. Levchenko, D. P. O'Neill, R. A. DiStasio Jr, R. C. Lochan, T. Wang, G. J. O. Beran, N. A. Besley, J. M. Herbert, C. Y. Lin, T. van Voorhis, S. H. Chien, A. Sodt, R. P. Steele, V. A. Rassolov, P. E. Maslen, P. P. Korambath, R. D. Adamson, B. Austin, J. Baker, E. F. C. Byrd, H. Dachsel, R. J. Doerksen, A. Dreuw, B. D. Dunietz, A. D. Dutoi, T. R. Furlani, S. R. Gwaltney, A. Heyden, S. Hirata, C. P. Hsu, G. Kedziora, R. Z. Khalliulin, P. Klunzinger, A. M. Lee, M. S. Lee, W. Z. Liang, I. Lotan, N. Nair, B. Peters, E. I. Proynov, P. A. Pieniazek, Y. M. Rhee, J. Ritchie, E. Rosta, C. D. Sherrill, A. C. Simmonett, J. E. Subotnik, H. L. Woodcock III, W. Zhang, A. T. Bell, A. K. Chakraborty, D. M. Chipman, F. J. Keil, A. Warshel, W. J. Hehre, H. F. Schaefer, J. Kong, A. I. Krylov, P. M. W. Gill and M. Head-Gordon, Phys. Chem. Chem. Phys., 2006, 8, 3172 RSC.
- QC Expert 3.2, Statistical software, TriloByte Ltd., Pardubice, Czech Republic, 2009 Search PubMed.
- A. Banerjee, J. Singh and D. Dasgupta, J. Fluoresc., 2013, 23, 745 CrossRef CAS PubMed.
- G. Salemi, M. C. Gueli, M. D'Amelio, V. Saia, P. Mangiapane, P. Aridon, P. Ragonese and I. Lupo, Neurol. Sci., 2009, 30, 361 CrossRef PubMed.
- E. I. Solomon and A. B. P. Lever, Inorganic Electronic Structure and Spectroscopy, Applications and Case Studies, Wiley, New York, 1999, vol. 2 Search PubMed.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, Part B: Applications in Coordination Organometallic and Bioinorganic Chemistry, 5th edn, Wiley, New York, 1997 Search PubMed.
- A. Barve, A. Kumbhar, M. Bhat, B. Joshi, R. Butcher, U. Sonawane and R. Joshi, Inorg. Chem., 2009, 48, 9120 CrossRef CAS PubMed.
- L. Gasque, G. Medina, L. Ruiz-Ramírez and R. Moreno-Esparza, Inorg. Chim. Acta, 1999, 288, 106 CrossRef CAS.
- D. Suh and J. B. Chaires, Bioorg. Med. Chem., 1995, 3, 723 CrossRef CAS PubMed.
- G. Zhao, H. Lin, S. Zhu, H. Sun and Y. Chen, J. Inorg. Biochem., 1998, 70, 219 CrossRef CAS.
- R. F. Pasternack, M. Caccam, B. Keogh, T. A. Stephenson, A. P. Williams and E. J. Gibbs, J. Am. Chem. Soc., 1991, 113, 6835 CrossRef CAS.
- J. C. Tung, J. M. Barnes, S. R. Desai, C. Sistrunk, M. W. Conklin, P. Schedin, K. W. Eliceiri, P. J. Keely, V. L. Seewaldt and V. M. Weaver, Free Radical Biol. Med., 2015, 79, 269 CrossRef CAS PubMed.
- M. L. Kremer, Phys. Chem. Chem. Phys., 1999, 1, 3595 RSC.
- P. C. Dedon, Chem. Res. Toxicol., 2008, 21, 206 CrossRef PubMed.
- A. S. Kumbhar, S. G. Damle, S. T. Dasgupta, S. Y. Rane and A. S. Kumbhar, J. Chem. Res., Synop., 1999, 98 RSC.
- A. Silvestri, G. Barone, G. Ruisi, M. T. Lo Giudice and S. Tumminello, J. Inorg. Biochem., 2004, 98, 589 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig.S1: 1H and 13C NMR spectra of Hqui4. Fig. S2: mid-FTIR spectra of Hqui4 (up) and complex 4 (down). Fig. S3: the electrophoreogram depicting the interactions of complexes 1 (lanes 1–3), 2 (lanes 4–6), 3 (lanes 7–9), and 5 (lanes 10–12) applied at 200 μM concentration with pUC19 plasmid DNA. Blank sample contained only the native form of pUC19 plasmid DNA (lane 13). Fig. S4: dose-viability curves for complexes 1–3 and 5. See DOI: 10.1039/c5ra22141b |
| This journal is © The Royal Society of Chemistry 2016 |