
Promotional effect of Fe on the performance of supported Cu catalyst for ambient pressure hydrogenation of furfural†
Abstract
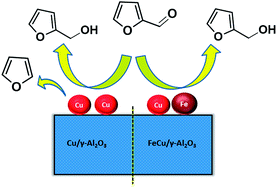
A noble-metal free FeCu based bimetallic catalyst system prepared by facile co-impregnation method was found to be highly admirable for vapour phase selective hydrogenation of furfural to furfuryl alcohol at ambient pressure. Monometallic Cu/γ-Al2O3, Fe/γ-Al2O3 and bimetallic FeCu/γ-Al2O3 catalysts with different Fe loadings were prepared. Structural and morphological features of the catalysts were thoroughly investigated by several physico-chemical characterization techniques. The influence of various reaction parameters, such as Fe loading, reaction temperature and flow of reactants was examined with respect to furfural conversion and furfuryl alcohol yield. The results clearly showed that an optimum amount of Fe is necessary to enhance the catalytic activity of monometallic Cu/γ-Al2O3 for the selective hydrogenation of furfural. The catalyst FC-10 with 10 wt% Fe exhibited excellent activity which led to high furfural conversion (>93%) and furfuryl alcohol selectivity (>98%) under mild reaction conditions. The higher activity of bimetallic FeCu/γ-Al2O3 compared to monometallic Cu/γ-Al2O3 is ascribed to the formation of FeCu bimetallic particles and the existence of oxygen vacancies in the Fe oxide system. The superior activity after Fe loading on the Cu-based catalyst was attributed to the synergy between Cu and Fe. A plausible mechanism is proposed to explain the promoting effect of Fe, which involves synergism between Fe sites with strong oxophilic nature and Cu sites with a high ability for hydrogen activation. Based on the activity results, prolonged catalytic activity and spent catalyst analysis, the developed FeCu/γ-Al2O3 catalyst is inexpensive, eco-benign and robust, which makes it a promising candidate for the efficient conversion of biomass-derived substrates to fine chemicals and drop-in biofuels.
 Please wait while we load your content...
Please wait while we load your content...

