DOI:
10.1039/C5RA21779B
(Paper)
RSC Adv., 2016,
6, 11870-11879
Lipase-catalyzed synthesis of oxidation-responsive poly(ethylene glycol)-b-poly(β-thioether ester) amphiphilic block copolymers†
Received
5th November 2015
, Accepted 19th January 2016
First published on 22nd January 2016
Abstract
A series of novel oxidation-responsive amphiphilic diblock copolymers with β-thioether ester groups were synthesized via the one-step lipase-catalyzed polycondensation of methyl 3-((2-hydroxyethyl)thio)propanoate (MHETP) with monomethoxy poly(ethylene glycol) (mPEG) using immobilized lipase B from Candida antarctica (CALB) as the catalyst. The amphiphilic poly(ethylene glycol)-b-poly(β-thioether ester) (mPEG-b-PTE) diblock copolymers could self-assemble to form nanosized micelles in aqueous solution with low critical micelle concentration (CMC). The micelles were able to undergo a H2O2-triggered disassembly due to the oxidizable thioether groups and degradable ester groups in the hydrophobic PTE core. The oxidation-responsive behaviour of the micelles was further investigated by dynamic light scattering (DLS) and transmission electron microscopy (TEM). Nile Red, a hydrophobic model drug, was encapsulated into the polymeric micelles and showed fast release upon the addition of H2O2. Cell cytotoxicity tests indicated that the micelles had low cytotoxicity toward HeLa cells. The oxidation-insensitive poly(ethylene glycol)-b-poly(ε-caprolactone) (mPEG-b-PCL) diblock copolymer was also prepared for comparison. All these findings demonstrated the potential of mPEG-b-PTE diblock copolymers as a promising oxidation-responsive nanocarrier for controlled drug delivery.
Introduction
Over the past few decades, stimuli-responsive polymeric materials have been widely used for target-specific therapeutic delivery, sensors, and tissue engineering scaffolds in biomedical fields.1–3 These materials can undergo physical or chemical changes in response to various external stimuli, such as pH,4 redox species,5,6 enzymes,7 temperature,8 light,9 or ultrasound.10 Among these stimuli, reactive oxygen species (ROS) including hydrogen peroxides (H2O2), hydroxyl radicals, and superoxide anions play an important role in cell signaling pathways and contribute greatly to the cellular redox state.11 However, overproduction of ROS can cause oxidative stress and damage DNA, proteins, and lipids in the cell, which is associated with many diseases, such as inflammatory pathologies, tumors, cardiovascular and degenerative diseases.12,13 It has been demonstrated that mucosal ROS concentrations are 10–100 times higher in patients suffering from inflammatory bowel disease or colon cancer than the healthy person.14,15 Large amounts of H2O2 could be produced in many different human cancer cell lines.16,17 Therefore, the abnormal level of ROS in tumor and inflammatory tissues can be considered as a target or an indicator to develop oxidation-responsive materials for site-specific delivery of therapeutic and imaging agents.18
In recent years, various oxidation-responsive polymeric materials have been prepared and studied as potential carriers for drug/gene delivery or bioimaging,18–20 including polyoxalate,21 polythioketal,22,23 thioether-, selenium- or tellurium-containing polymers,24–26 arylboronic ester-containing polymers,27,28 ferrocene-based polymers,29 and oligoproline-crosslinked polymers.30 Among them, thioether-containing polymers have attracted significant attention for drug delivery systems because of their unique oxidation properties.31 Hydrophobic thioethers are known to undergo facile oxidation to more hydrophilic sulfoxides or sulfones upon exposure to H2O2.18,31 In 2004, Tirelli et al.32 synthesized a thioether-containing polymer, poly(propylene sulfide) (PPS), by the living anionic ring-opening polymerization of episulfides, which was the first example of the oxidation-responsive polymeric materials. A variety of oxidation-responsive microspheres,33 nanoparticles,34 micelles,35 vesicles,32 and hydrogels36 based on PPS have also been reported. Nevertheless, the non-degradable property of PPS limits their in vivo application in a certain extent. To address this issue, biodegradable thioether-containing polypeptide and polyester materials were gradually developed. Deming et al.37 and Li et al.38 prepared oxidation-responsive poly(L-lysine) with the side-chain thioether linkages by ring-opening polymerization. Leroux et al.39 synthesized a library of redox-responsive thioether-containing polyester stabilizers by postpolymerization modification using the thiol–yne reaction. Recently, Chen et al.40 demonstrated a kind of thermal and oxidation dual responsive PEG-based poly(β-thioether ester) by thiol–ene polymerization of PEG diacrylate and 1,2-ethanedithiol monomers and a triblock copolymer was obtained by modifications of terminal groups with mPEG. However, most of biodegradable thioether-containing polymers were prepared by chemical polymerization and complex chemical modification, which often require toxic chemical catalysts. Green methods for efficient synthesis of novel oxidation-responsive polymeric materials are highly desirable.
Lipase-catalyzed polymerization provides a new strategy for producing biodegradable functional polymers with many advantages including mild reaction conditions, high catalytic activity, and the use of nontoxic biocatalysts.41–43 Jiang et al.44,45 successfully prepared pH-responsive and reduction-degradable amphiphilic block copolymers by enzymatic polymerization. Amphiphilic block copolymers could form nanosized micelles with a core–shell architecture in a selective solvent and have attracted increasing attention for drug delivery.46 The hydrophobic core serves as a natural carrier environment for hydrophobic drugs, and the hydrophilic shell stabilizes the particles in aqueous solution.47 To the best of our knowledge, lipase-catalyzed synthesis of oxidation-responsive amphiphilic block copolymers have never been achieved before. We have recently prepared a linear thioether-containing polymer, poly(β-thioether ester) (PTE), by the lipase-catalyzed polycondensation of methyl 3-((2-hydroxyethyl)thio)propanoate (MHETP),48 which can be used as the hydrophobic segment of the amphiphilic block copolymers.
In this work, we wish to report the lipase-catalyzed, one-step synthesis of a new type of oxidation-responsive poly(ethylene glycol)-b-poly(β-thioether ester) (mPEG-b-PTE) amphiphilic block copolymers via polycondensation of MHETP with mPEG. The hydrophobicity of PTE segment was readily adjusted by varying the feed ratio of mPEG and MHETP. As shown in Scheme 1, these amphiphilic block copolymers were able to self-assemble into micellar aggregates in aqueous solution. In addition, the micelles were disassembled upon addition of H2O2 because the hydrophobic PTE cores became hydrophilic after oxidation and the ester groups further underwent hydrolysis, which could be developed as a potential oxidation-responsive biodegradable material for controlled release systems.
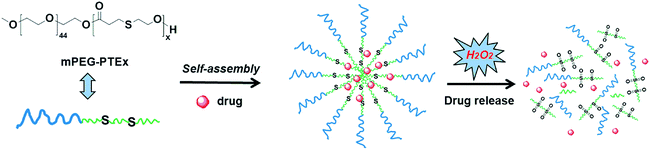 |
| | Scheme 1 Self-assembled micelles of mPEG-b-PTE block copolymers and their oxidation-responsive disassembly for drug release. | |
Experimental
Materials
Methyl acrylate (98%), 2-mercaptoethanol (99%), trimethylamine (99%), ε-caprolactone (99%), diphenyl ether (99%), Nile Red (99%) and 30% H2O2 were obtained from commercial suppliers and used without further purification. Monomethoxy poly(ethylene glycol) 2000 (mPEG, Mn = 2000 Da) were purchased from Sigma-Aldrich and used as received. Novozym-435 (immobilized lipase B from Candida antarctica, CALB) was purchased from Novozymes (Bagsvaerd, Denmark) and used as received. All solvents and other chemicals were of analytical grade and were used without further purification. Methyl 3-((2-hydroxyethyl)thio)propanoate (MHETP) and methyl 6-hydroxyhexanoate (MHH) were synthesized according to the literature.49,50
Lipase-catalyzed synthesis of poly(ethylene glycol)-b-poly(β-thioether ester) (mPEG-b-PTE) and poly(ethylene glycol)-b-poly(ε-caprolactone) (mPEG-b-PCL) block copolymers
In a typical procedure, monomethoxy poly(ethylene glycol) 2000 (600 mg, 0.3 mmol), methyl 3-((2-hydroxyethyl)thio)propanoate (MHETP) and the solvent diphenyl ether (200 wt% vs. total substrates) was placed into a Schlenk tube. The feed molar ratios of mPEG/MHETP were 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10, 1:15, 1:20 and 1:30. Novozym-435 (10 wt% vs. total substrates) was transferred into the tube under magnetic stirring. The tube was heated at 80 °C for 48 h under reduced pressure (350 mbar) controlled by a Pfeiffer vacuum pump. The reaction was quenched with chloroform after 48 h, and the enzyme was removed by filtration. The concentrated filtrate was washed with a large amount of hexane (30 mL × 5) to extract and remove the diphenyl ether solvent and subsequently precipitated three times using a mixture of chloroform and hexane (3 mL/15 mL). The products were dried under vacuum. Control reactions without addition of enzyme were also performed with the feed molar ratios of mPEG/MHETP 1:20 under the same reaction condition and the product was directly analysed by GPC. mPEG-b-PCL block copolymer was synthesized through the same polymerization of mPEG and methyl 6-hydroxyhexanoate (MHH) with the feed molar ratios of mPEG/MHH 1:20.
:10, 1:15, 1:20 and 1:30. Novozym-435 (10 wt% vs. total substrates) was transferred into the tube under magnetic stirring. The tube was heated at 80 °C for 48 h under reduced pressure (350 mbar) controlled by a Pfeiffer vacuum pump. The reaction was quenched with chloroform after 48 h, and the enzyme was removed by filtration. The concentrated filtrate was washed with a large amount of hexane (30 mL × 5) to extract and remove the diphenyl ether solvent and subsequently precipitated three times using a mixture of chloroform and hexane (3 mL/15 mL). The products were dried under vacuum. Control reactions without addition of enzyme were also performed with the feed molar ratios of mPEG/MHETP 1:20 under the same reaction condition and the product was directly analysed by GPC. mPEG-b-PCL block copolymer was synthesized through the same polymerization of mPEG and methyl 6-hydroxyhexanoate (MHH) with the feed molar ratios of mPEG/MHH 1:20.
mPEG-b-PTE20, 1H NMR (400 MHz, CDCl3), δ 2.65 (t, J = 7.2 Hz, SCH2CH2CO), 2.77 (t, J = 6.2 Hz, SCH2CH2O), 2.84 (t, J = 7.2 Hz, SCH2CH2CO), 3.38 (s, CH3O–PEG), 3.54–3.82 (br, OCH2CH2O, SCH2CH2OH end group), 4.26 (t, J = 7.0 Hz, SCH2CH2OCO, OCH2CH2OCO). 13C NMR (100 MHz, CDCl3), δ 171.46, 71.93, 70.56, 69.05, 63.86, 63.70, 63.60, 60.67, 59.02, 35.37, 34.74, 30.57, 27.15, 26.67.
mPEG-b-PCL20, 1H NMR (400 MHz, CDCl3), δ 1.38 (m, CH2CH2CH2O), 1.64 (m, CH2CH2CH2CH2O), 2.31 (t, J = 7.6 Hz, CH2CO), 3.38 (s, CH3O–PEG), 3.54–3.82 (br, OCH2CH2O, CH2CH2OH end group), 4.06 (t, J = 6.8 Hz, CH2CH2CH2OCO), 4.23 (t, J = 4.8 Hz, OCH2CH2OCO). 13C NMR (100 MHz, CDCl3), δ 173.54, 71.91, 70.54, 69.15, 64.13, 63.43, 62.53, 59.02, 34.22, 34.10, 32.31, 28.33, 25.51, 25.30, 24.68, 24.56.
Characterizations
1H NMR and 13C NMR spectra were obtained on a Bruker DMX 400 Spectrometer in CDCl3 and referenced to an internal TMS standard. 1H, 1H-COSY, and 13C, 1H-HSQC spectra were recorded using the standard pulse sequence provided by Bruker. Gel permeation chromatography (GPC) to obtain the polymer molecular weights and their distributions was performed on a Waters HPLC system equipped with a model 1515 isocratic pump, a 717 plus autosampler, and a 2414 refractive index (RI) detector with Waters Styragel® HT3 and HT4 columns in series. The eluting solvent was THF at a flow rate of 1.0 mL min−1 at 45 °C. The retention times were calibrated against poly(ethylene glycol) standards with molecular weight range 400–80000 Da.
Preparation of polymeric micelles by self-assembly
mPEG-b-PTE and mPEG-b-PCL block copolymers were directly dissolved in deionized water to form the micelles. The average size and size distribution of micelles were determined by DLS after filtration through a 0.45 μm Millipore filter with polymer solution concentration of 1 mg mL−1. Dynamic light scattering (DLS) measurements were carried out at 25 °C using a Zetasizer Nano-ZS90 system from Malvern Instruments equipped with a 633 nm He–Ne laser using backscattering detection with a fixed detector angle of 90°. The size and morphology of the polymeric micelles were further investigated by transmission electron microscopy (TEM, Hitachi H-600) at an acceleration voltage of 100 kV. The samples were prepared by dropping 2.5 mg mL−1 of the micellar solution onto a copper grid followed by negatively staining with a 1 wt% aqueous solution of phosphotungstic acid.
Critical micelle concentration (CMC) measurements
The critical micelle concentration was determined using pyrene as a fluorescence probe. The block copolymer concentration varied from 1.0 × 10−6 mg mL−1 to 1 mg mL−1, and the pyrene concentration was fixed at 6.0 × 10−7 M. The fluorescence spectra were recorded using a F-7000 fluorescence spectrometer (HITACHI) at room temperature. Both the emission and excitation slit widths were 5 nm. The samples were excited at 335 nm, and the emission spectra were recorded from 350 to 500 nm. The intensity values of fluorescence emission, I372 and I382 at 372 nm and 382 nm, respectively, were used for the subsequent calculations. The CMC was determined from the plots of the I382/I372 ratio versus the logarithm of the polymer concentration using the intersection of the linear regression lines as the CMC value.
Oxidation of mPEG-b-PTE copolymer
The oxidation experiments were performed according to a reported similar procedure.38 mPEG-b-PTE20 (50 mg) was dissolved in 1 mL of deionized water. Next, 1 mL of 2% (w/v) H2O2 was added. The mixture was performed in a shaking incubator with 200 rpm at 37 °C. At appropriate intervals, samples were withdrawn and 1 mL of sodium sulfite (1 M) was added to reduce unreacted H2O2. The solution mixture was then transferred into a dialysis tubing (MWCO 1000 Da), and dialyzed against 800 mL deionized water for 24 h with water changing four times. The resulting solution was finally lyophilized to yield oxidized products. The molecular weights of products were determined using GPC, and the structures were investigated by 1H NMR and FTIR spectra. FTIR spectra were recorded on a Shimadzu FTIR-4200 spectrometer. mPEG-b-PCL20 was investigated in the same method.
Oxidation-responsive properties of mPEG-b-PTE micelles
The size change of the micelles oxidized by H2O2 was monitored in time using DLS. The micelle solution of mPEG-b-PTE20 (1 mg mL−1) with different concentration H2O2 was performed in a shaking incubator with 200 rpm at 37 °C. At appropriate intervals, 1 mL of the mixture solution were withdrawn and determined by DLS. The morphology of the micelles treated with 5% (w/v) H2O2 at 37 °C for 24 h was investigated by TEM.
Encapsulation and H2O2-triggered release of Nile Red
Nile Red-loaded micelles were prepared by a film hydration method.51 50 mg of the mPEG-b-PTE20 block copolymer and 0.25 mg of Nile Red were co-dissolved in 2.5 mL of THF. Then, the solvent was removed using a rotary evaporator at room temperature to form a solid thin film. Subsequently, the solid thin film was dissolved in 10 mL of deionized water by using water bath heating at 60 °C for 5 min and vortexed for 2 min. After cooling to room temperature, the solution was transferred into a 25 mL volumetric flask and diluted with deionized water to a polymer concentration of 2 mg mL−1. The resulting solution was filtered through 0.45 μm membrane filters to remove non-encapsulated Nile Red and stored at 4 °C as the stock solution of Nile Red-loaded micelles. The size and morphology of the blank micelles of mPEG-b-PTE20 prepared by the film hydration method without Nile Red were further investigated by DLS and TEM, respectively. UV-Vis absorption spectra of Nile Red-loaded micelles were recorded on a Hitachi PharmaSpec UV-1900 UV-Visible spectrophotometer and their fluorescent emission spectra were recorded using a F-7000 fluorescence spectrometer (HITACHI). The loading amount and encapsulation efficiency of Nile Red were determined by a fluorescence-based method.35 1 mL of the stock solution was diluted with DMF into 90% DMF aqueous solution in 10 mL volumetric flask. The obtained solution was measured by a fluorescence spectrometer with the excitation wavelength of 557 nm. The intensity value of fluorescence emission at 630 nm was used to quantify the Nile Red content, according to the standard calibration curve of Nile Red measured in 90% DMF. The loading amount was calculated as the weight percent of dye relative to polymer and encapsulation efficiency was defined as the weight percent of the dye loaded versus what was added to the micelle solution.
The Nile Red release was performed by mixing 2 mL of the stock solution with 2 mL of different concentration H2O2 and incubating at 37 °C. The fluorescence spectra were recorded at different time intervals with the excitation wavelength of 557 nm. The intensity value of fluorescence emission at 637 nm was used for the subsequent calculations. The initial fluorescence intensity of each was measured as 100% intensity, respectively. Nile Red-loaded micelles of mPEG-b-PCL20 were investigated in the same method and used 626 nm as the emission wavelength for calculations.
In vitro cytotoxicity study
The cytotoxicity of mPEG-b-PTE20 and mPEG-b-PCL20 toward HeLa cells was evaluated by using a Cell Counting Kit-8. HeLa cells (8000 cells per well) were seeded into 96-well plates and cultured overnight for 70–80% cell confluence. The cells were then incubated in a culture medium containing polymer with a particular concentration for 24 h. After that, polymer solutions were removed, 100 μL of sterile filtered CCK-8 (0.1 mg mL−1) stock solution in PBS was added to each well for additional 1 h incubation at 37 °C. Then, the absorbance of each sample was measured using an ELISA plate reader (model 680, BioRad) at a wavelength of 450 nm. The cell survival was expressed as follows: cell viability = (ODtreated/ODcontrol) × 100%.
Results and discussion
Lipase-catalyzed synthesis of amphiphilic block copolymers
The synthesis of mPEG-b-PTE diblock copolymers was carried out in one step via enzymatic polymerization using CALB as the catalyst. The amphiphilic block copolymers consist of both hydrophilic monomethoxy poly(ethylene glycol) (mPEG) blocks and hydrophobic poly(β-thioether ester) (PTE) blocks. Scheme 2 illustrates a general self-condensation of MHETP with mPEG2000 (Mn 2000 Da) as a chain-terminating agent in diphenyl ether at 80 °C for 2 days under reduced pressure. The use of reduced pressure during the polymerization could efficiently remove the methanol byproduct to increase the molecular weights of the products. The molar feed ratios of mPEG and MHETP were varied from 1:10, 1:15, 1:20 and 1:30. As shown in Table 1, both mPEG and PTE contents in the copolymers were readily controlled by adjusting the feed ratios. All the mPEG-b-PTE copolymers demonstrated good yields and relatively narrow polydispersities ranging from 1.14–1.35. The Mn values of the copolymers increased from 2700 Da to 4560 Da as the PTE content increased. For mPEG-b-PTE block copolymers, the GPC results show smaller molecular weight of the polymer than NMR results. That difference may be caused by the different chemical structure between mPEG-b-PTE block copolymers and linear PEG polymer standards. mPEG-b-PTE block copolymers represent smaller hydrodynamic volume than linear PEG. The GPC traces for the polymers in Fig. 1 demonstrate a monomodal distribution, indicating that mPEG successfully linked to the end of PTE chain. The control reaction in the absence of enzyme produced material with molecular weights below 2000 Da by GPC (data not shown), indicating that no polymerization occurred without CALB. Poly(ethylene glycol)-b-poly(ε-caprolactone) (mPEG-b-PCL) diblock copolymer, one of the most studied biocompatible and biodegradable drug delivery carriers,52 was used to compare with mPEG-b-PTE in view of their similar polymer structures. The mPEG-b-PCL20 block copolymer was prepared by polymerization of mPEG2000 and methyl 6-hydroxyhexanoate (MHH) with the molar ratio of 1:20 under the same reaction condition.
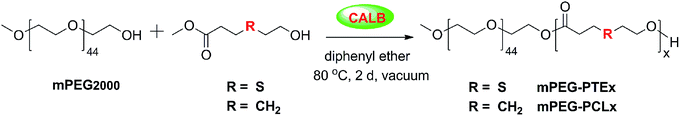 |
| | Scheme 2 Lipase-catalyzed synthesis of mPEG-b-PTE and mPEG-b-PCL block copolymers. | |
Table 1 Characterization of mPEG-b-PTE and mPEG-b-PCL copolymers
| Name |
mPEG/monomer feed ratioa |
Composition ratiob |
Yield (%) |
Mnc (Da) |
Mw/Mnc |
Mnd (Da) |
fw,PEGd (%) |
CMCf (mg L−1) |
Sizeg (nm) |
PdIg |
| Molar ratio of mPEG to monomer in feed. Molar ratio of mPEG to TE or CL units from copolymers determined by 1H NMR. Determined by GPC. Molecular weight and weight percent of mPEG in copolymer determined by 1H NMR. Nominal value from supplier. Determined using pyrene as a fluorescence probe. Size and PdI (size polydispersity index) of micelles (1 mg mL−1) determined by DLS. |
| mPEG2000 |
— |
— |
— |
1980 |
1.04 |
2000e |
— |
— |
— |
— |
| mPEG-b-PTE11 |
1:10 |
1:11 |
79 |
2700 |
1.14 |
3450 |
57.9 |
6.34 |
29.6 ± 4.3 |
0.238 ± 0.006 |
| mPEG-b-PTE15 |
1:15 |
1:15 |
86 |
3450 |
1.22 |
3980 |
50.2 |
3.84 |
17.2 ± 0.3 |
0.067 ± 0.009 |
| mPEG-b-PTE20 |
1:20 |
1:20 |
83 |
3700 |
1.29 |
4640 |
43.1 |
3.36 |
21.3 ± 0.8 |
0.257 ± 0.007 |
| mPEG-b-PTE31 |
1:30 |
1:31 |
86 |
4560 |
1.35 |
6100 |
32.8 |
2.37 |
27.9 ± 1.2 |
0.252 ± 0.031 |
| mPEG-b-PCL20 |
1:20 |
1:20 |
74 |
4360 |
1.42 |
4280 |
46.7 |
1.19 |
20.0 ± 1.8 |
0.155 ± 0.010 |
 |
| | Fig. 1 GPC traces of mPEG2000, mPEG-b-PTE and mPEG-b-PCL20 copolymers. | |
Structural characterization of block copolymers
The chemical structures of the mPEG-b-PTE block copolymers were analysed and confirmed by NMR spectroscopy. Fig. 2A presents the 1H NMR spectra of mPEG-b-PTE20. The signal at 3.64 ppm with strong intensity was attributed to the ethylene oxide protons CH2CH2O of mPEG segment. The terminal methyl CH3O and methylene CH2OH protons of mPEG-b-PTE20 chains resonated at 3.38 and 3.75 ppm, respectively. The proton signals at 2.65, 2.77 and 2.84 ppm were ascribed to the methylene protons of PTE units. The new proton signals at 4.26 ppm were ascribed to the methylene protons CH2OCO of PTE and mPEG, indicating that the monomer MHETP reacted and was incorporated into the block copolymer backbone. The composition of mPEG-b-PTE block copolymers was analysed by 1H NMR spectra using the ratio between the relative intensities of the CH3O protons of mPEG units at 3.38 ppm and the CH2CO protons of PTE units at 2.65 ppm. The methyl peak of mPEG units at 3.38 ppm and the methylene peak of PCL units at 1.6 ppm are chosen to determine the molar composition of mPEG-b-PCL copolymer. Good agreement between the feed ratio and the experimental values in Table 1 indicated good control over the block copolymer composition. Fig. 2B provides the 13C NMR spectra of mPEG-b-PTE20. The resonance of the carbonyls of PTE units appeared at 172.38 ppm, while the resonance of the carbonyls of MHETP at 171.46 ppm completely disappeared. The terminal CH3O and CH2OH carbon atoms of mPEG-b-PTE20 resonated at 59.02 and 60.67 ppm, respectively. The 2D COSY NMR spectra of mPEG-b-PTE20 were obtained to further confirm the signal assignments and structures in Fig. S3 (ESI†). The methylene protons a and e of PTE units were coupled, as were the methylene protons d and f. All the above data provided strong evidence for the successful synthesis of mPEG-b-PTE diblock copolymers.
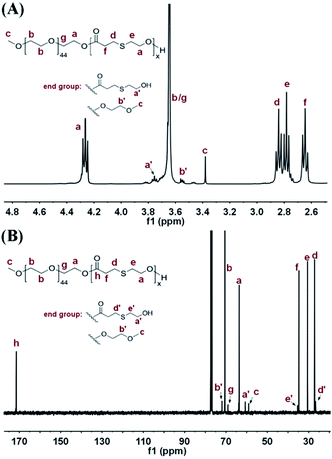 |
| | Fig. 2 (A) 1H NMR and (B) 13C NMR spectra of mPEG-b-PTE20 in CDCl3. | |
Self-assembly behaviour of mPEG-b-PTE block copolymers
To study the self-assembly behavior of amphiphilic mPEG-b-PTE copolymers, the critical micelle concentration (CMC) was measured by the fluorescent probe method using pyrene as the probe.53 Fig. 3 shows the intensity ratios (I382/I372) obtained from the fluorescence emission spectra of the pyrene against the logarithms of the copolymer concentrations. The CMC values of the four different copolymers, mPEG-b-PTE11, mPEG-b-PTE15, mPEG-b-PTE20 and mPEG-b-PTE31, were 2.37, 3.36, 3.84 and 6.34 mg L−1, respectively. As presented in Table 1, the CMC values gradually decreased with increasing PTE chain lengths due to the enhanced hydrophobicity of the micelle inner cores. Furthermore, the low CMC values obtained for mPEG-b-PTE ensure high stability of these micelles resulting in less disruption upon in vivo dilution, which is essential for in vivo drug delivery.54 The CMC value of mPEG-b-PTE20 was higher than mPEG-b-PCL20, which may be because the polarity of thioether in the hydrophobic PTE segment is stronger than that of alkane in the PCL segment.
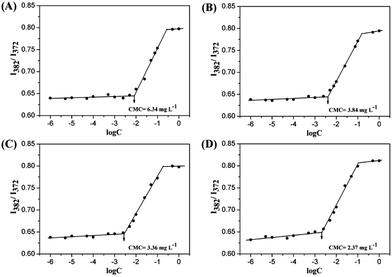 |
| | Fig. 3 Plots of the intensity ratio I382/I372 from the pyrene emission spectra versus the logarithm of the concentration for self-assembling micelles in aqueous media from (A) mPEG-b-PTE11, (B) mPEG-b-PTE15, (C) mPEG-b-PTE20 and (D) mPEG-b-PTE31. | |
The size and morphology of mPEG-b-PTE micelles prepared by the direct dissolution method were confirmed by the DLS and TEM analysis. Table 1 summarizes the average size of the micelles formed from the mPEG-b-PTE copolymers, which ranged from 17 to 30 nm. The size of the micelles for mPEG-b-PTE15, mPEG-b-PTE20 and mPEG-b-PTE31 gradually increased with increasing the lengths of the hydrophobic PTE chain. However, the mPEG-b-PTE11 micelles showed a larger particle size, which may be due to the formation of more loose aggregates resulting from the very short hydrophobic PTE segment. The mPEG-b-PCL20 micelles displayed almost the same size as the mPEG-b-PTE20 micelles by reason of their same degree of polymerization. The size distribution values of all the micelles were moderate in the range 0.067–0.257 determined by DLS. Fig. 4 displays the TEM micrographs of the mPEG-b-PTE and mPEG-b-PCL micelles. All the micelles adopted spherical morphologies. The sizes of the mPEG-b-PTE20 and mPEG-b-PCL20 micelles were approximately 20 nm according to the TEM observations (Fig. 8C and E), which were consistent with the average size obtained from DLS. mPEG-b-PTE block copolymers at the mPEG content in the range of 32.8–57.9 wt% (Table 1) could readily form spherical small-sized micelles in aqueous solution.
 |
| | Fig. 4 TEM of (A) mPEG-b-PTE11 (B) mPEG-b-PTE15 (C) mPEG-b-PTE20 (D) mPEG-b-PTE31 (E) mPEG-b-PCL20 micelles and (F) mPEG-b-PTE20 micelles treated with 5% (w/v) H2O2 at 37 °C for 24 h. | |
Oxidation of mPEG-b-PTE block copolymers
To study the oxidation-responsive behaviour of the mPEG-b-PTE block copolymers, we used H2O2 as a model oxidant. The oxidized products from mPEG-b-PTE20 block copolymer were prepared in the aqueous solution of 1% (w/v) H2O2 at different reaction time. The structure of the oxidized products was analysed by NMR and FTIR spectra. Fig. 5 compares the 1H NMR spectra of mPEG-b-PTE20 block copolymer before and after oxidation. An obvious higher-frequency shift of the peaks corresponding to the four types of methylene protons of PTE around the sulfur atoms was observed after being oxidized for 2 h, suggesting a polarity increase of these functional groups. The oxidation of the thioether groups was also confirmed by the disappearance of a proton signal at 3.75 ppm ascribed to the terminal CH2OH groups of mPEG-b-PTE20 and the appearance of a new proton signal a′′ at 4.15 ppm. Two new multiple peaks a′ at 4.52 and 4.62 ppm presumably ascribed to the methylene CH2OCO groups in the β position of sulfoxides and sulfones, respectively, appeared and rised gradually with the increase of oxidized time. The 2D COSY NMR spectra of the oxidized products of mPEG-b-PTE20 treated with H2O2 for 24 h was provided in Fig. S6 (ESI†). Both the protons a′ at 4.52 and 4.62 ppm were coupled with the protons e′ at 3.08 ppm corresponding to the methylene in the α position to –SOx– moieties, indicating the existence of sulfoxide and sulfone groups in the oxidized products. Additionally, the oxidized products were further investigated by FTIR spectra. As shown in Fig. 6, the stretching vibration band of sulfoxide groups at 1039 cm−1 and two stretching vibrations of sulfone groups at 1176 and 1360 cm−1 were clearly observed and strengthened gradually with the increase of oxidized time, which verified that the thioether groups of mPEG-b-PTE20 were converted to sulfoxide and sulfone groups. The similar phenomenon was also described in some other publications.40,55 Moreover, there were no changes of the 1H NMR and FTIR spectra of mPEG-b-PTE20 without H2O2 treatment for 24 h.
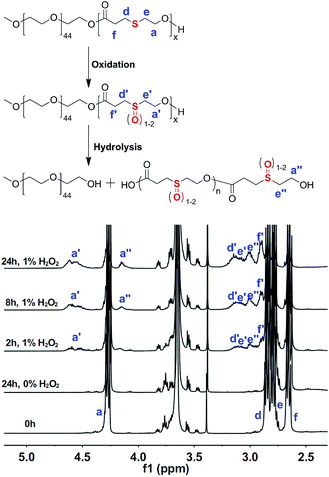 |
| | Fig. 5 1H NMR spectra of mPEG-b-PTE20 oxidized by H2O2 as a function of the reaction time. | |
 |
| | Fig. 6 FTIR of (a) mPEG-b-PTE20, (b) mPEG-b-PTE20 treated without H2O2 for 24 h (c) mPEG-b-PTE20 treated with H2O2 for 2 h (d) mPEG-b-PTE20 mPEG-b-PTE20 treated with H2O2 for 8 h and (e) mPEG-b-PTE20 treated with H2O2 for 24 h. | |
Fig. 7 shows the change of GPC traces of the oxidized products from mPEG-b-PTE20 over various time intervals. Their molecular weights were given in Table S1 (ESI†). After oxidation for 2 h, there was a minor shift of the elution band of products toward shorter elution time and the Mw value increased from 4780 Da to 5040 Da, which may be due to the oxidation induced chain volume expansion and increase of molecular weight.43 In the meantime, the GPC peak assigned to mPEG2000 appeared, then increased regularly over time, suggesting the degradation of polymer during oxidation. After oxidation for 24 h, the original copolymer peak disappeared completely and the copolymer degraded into the low molecular weight products and mPEG2000. Chen et al.40 recently also discovered the degradation of poly(β-thioether ester) after oxidation by H2O2, which was probably because of the hydrolysis of the ester bonds. Besides, only a little degradation of mPEG-b-PTE20 without H2O2 treatment for 24 h was observed in Fig. 7. To as a control, mPEG-b-PCL20 had no change of 1H NMR after oxidation for 24 h (Fig. S7†) and the GPC trace showed only a little degradation (Fig. S8†). These results strongly implied that mPEG-b-PTE20 degraded more rapidly under the oxidation condition than mPEG-b-PCL20.
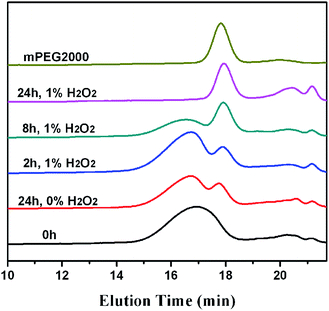 |
| | Fig. 7 GPC traces of mPEG-b-PTE20 oxidized by H2O2 as a function of the reaction time. | |
Oxidation-triggered disruption of the mPEG-b-PTE micelles
The oxidation-sensitive property of mPEG-b-PTE20 micelles was further studied using DLS by monitoring change of micelle sizes in response to H2O2 in aqueous solution. As shown in Fig. 8, the size of mPEG-b-PTE20 micelles gradually decreased to 3.3 nm after oxidation in 5% (w/v) H2O2 for 24 h, demonstrating the disassociation of the self-assembled micellar aggregates, while the size of mPEG-b-PCL20 micelles remained unchanged. Besides, no changes of the sizes for both mPEG-b-PTE20 and mPEG-b-PCL20 micelles were observed without H2O2 treatment for 24 h by DLS (Fig. S9†). Moreover, the TEM results proved that no obvious aggregates were observed in the solution after oxidation in 5% (w/v) H2O2 for 24 h (Fig. 3F), indicating that the micellar structures have been destroyed by the oxidation stimuli. However, the PEG-b-PCL20 micelles were quite stable in an oxidation environment and could maintain their spherical structures (Fig. S10†). The oxidation-responsiveness of mPEG-b-PTE20 micelles could be attributed to two reasons. On the one hand, the hydrophobic thioether groups of PTE chain were transformed into the hydrophilic sulfoxide or sulfone groups in an oxidative environment and the amphiphilic copolymer changed into hydrophilic species, which leaded to the disassembly of the micelles. On the other hand, the ester bonds of β-thioether ester groups in the mPEG-b-PTE20 backbone were more easily hydrolyzed after oxidation of the thioethers,56,57 resulting in the destruction of the micelles.
 |
| | Fig. 8 DLS plots of (A) mPEG-b-PTE20 and (B) mPEG-b-PCL20 micelles treated with 5% (w/v) H2O2 at 37 °C. | |
Nile Red loading and H2O2-triggered release
In view of the good oxidation-responsiveness of mPEG-b-PTE20 block copolymer, it is possible to use the micelles to incorporate hydrophobic drugs. Here, we chose Nile Red as a model hydrophobic drug for the investigation of H2O2-triggered drug release. For encapsulation of Nile Red, the film hydration method was used to prepare nanoparticles. The size of the blank micelles of mPEG-b-PTE20 and PEG-b-PCL20 without Nile Red using the film hydration method were 18.9 nm and 23.6 nm (Fig. S14†), respectively, which were close to the values by the direct dissolution method. According to the TEM images (Fig. S14C†), the micelles of mPEG-b-PTE20 remained the spherical morphologies. However, mPEG-b-PCL20 formed mixed micelles of spheres and cylinders (Fig. S14D†) due to its crystallization-driven self-assembly, which have been previously described by Eisenberg et al.58 The loading amount and encapsulation efficiency of Nile Red for mPEG-b-PTE20 were 0.18 wt% and 35%, respectively. In contrast, PEG-b-PCL20 showed a higher loading amount (0.22 wt%) and encapsulation efficiency (44%), which could be due to the more hydrophobic micellar cores for PEG-b-PCL20. Furthermore, Nile Red, an environment-dependent fluorescent probe, can be also used to ascertain the hydrophobicity of the micelles.59 As shown in Fig. 9, the dye loaded mPEG-b-PTE20 micelles exhibited weaker color intensity than PEG-b-PCL20. In addition, both the UV absorption and fluorescent emission spectra for the dye loaded mPEG-b-PTE20 micelles showed a slight bathochromic shift, suggesting that the Nile Red was encapsulated into a more hydrophilic environment. Fig. 9 provides the in vitro release of Nile Red from the micelles at different concentrations of H2O2. The decrease of fluorescence intensity indicates the release of Nile Red due to its insolubility in aqueous solution.60 The results showed clearly a H2O2-dependent release behavior from mPEG-b-PTE20 micelles, in which the fluorescence of Nile Red decreased more than 99% at 5% (w/v) H2O2 in 8 h, while only about 14% of Nile Red was released for the sample in the absence of H2O2 (Fig. 9A). The higher concentration of H2O2 would cause quicker release of Nile Red from mPEG-b-PTE20 micelles. In contrast, no loss of Nile Red fluorescence intensity from mPEG-b-PCL20 micelles was observed with or without H2O2 (Fig. 9B). The rapid release of drug payload from mPEG-b-PTE20 micelles in H2O2 could be ascribed to the destruction of micellar structure upon oxidation, which has been previously demonstrated. These results suggested that oxidation-responsive mPEG-b-PTE20 micelles may achieve site-specific drug delivery in the presence of H2O2. We preliminary try to load a hydrophobic anti-cancer drug paclitaxel into the mPEG-b-PTE block copolymer micelles using the same method of encapsulation of Nile Red (ESI†). Unfortunately, we didn't observe a H2O2-dependent release behavior. As shown in Fig. S15,† within 26 h, 19.4% of paclitaxel was released from micelles without H2O2, while only about 3.3% and 1.9% of paclitaxel were detected by HPLC from micelles at 1% and 5% w/v H2O2, respectively. We detected the residual paclitaxel in dialysis bags and found that only 49.4% and 31.9% of paclitaxel remained at 1% and 5% w/v H2O2, respectively, which indicated that paclitaxel may be oxidized under high concentration of H2O2 conditions. Besides, the thioether-containing polymer micelles couldn't be destroyed under low concentration of H2O2 conditions, which limits their further application at biologically relevant concentrations of H2O2 (50–100 μM) in a certain extent.61 To overcome this problem, we are developing more oxidation-sensitive polymer materials using a selenium-containing polymeric monomer instead of the thioether-containing one by enzyme-catalyzed polymerization.
 |
| | Fig. 9 Nile Red fluorescence in micelles as a function of time at different H2O2 at 37 °C of (A) mPEG-b-PTE20 and (B) mPEG-b-PCL20 copolymers. | |
In vitro cell cytotoxicity
Cytotoxicity is a very critical factor for a drug delivery carrier, especially for in vivo application. Fig. 10 provides the in vitro cytotoxicity results for both mPEG-b-PTE20 and mPEG-b-PCL20 copolymers using a Cell Counting Kit-8. As shown in Fig. 10, mPEG-b-PTE20 exhibited very low cytotoxicity toward HeLa cells (>95% cell viability), even at high polymer concentration (800 mg L−1), as well as mPEG-b-PCL20. These results indicated that the amphiphilic mPEG-b-PTE20 block copolymer is highly biocompatible and nontoxic.
 |
| | Fig. 10 In vitro cell cytotoxicity of mPEG-b-PTE20 and mPEG-b-PCL20 copolymers in HeLa cells. | |
Conclusions
We have successfully developed a new lipase-catalyzed polymerization method for the synthesis of novel oxidation-responsive biodegradable mPEG-b-PTE diblock copolymers by the polycondensation of MHETP with mPEG2000 using a CALB catalyst. The molecular weights and compositions of the copolymers were well-controlled by varying the feed ratio. These amphiphilic block copolymers efficiently formed spherical small-sized micelles in aqueous solution with low CMC values. In vitro oxidation studies indicated that the mPEG-b-PTE micelles could be destroyed by the oxidation stimuli due to the oxidized thioether groups of hydrophobic PTE segment and further hydrolysis of the ester bonds. In vitro release experiments revealed that the Nile Red loaded mPEG-b-PTE20 micelles more rapidly released dye than mPEG-b-PCL20 in the presence of H2O2. Moreover, mPEG-b-PTE20 exhibited low cytotoxicity toward HeLa cells. These biocompatible and biodegradable amphiphilic block copolymers have the potential as a new type of oxidation-responsive polymeric materials.
Acknowledgements
This work was financially supported by the National Program on Key Basic Research Project of China (973 Program, 2013CB328900) and the National Natural Science Foundation of China (No. 21232005 and 21321061), the Program for Changjiang Scholars in China. We also thank the Sichuan University Analytical & Testing Center for NMR analysis.
Notes and references
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Müller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef PubMed.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS PubMed.
- Y. Wang, M. S. Shim, N. S. Levinson, H. W. Sung and Y. Xia, Adv. Funct. Mater., 2014, 24, 4206–4220 CrossRef CAS PubMed.
- J. Liu, Y. Huang, A. Kumar, A. Tan, S. Jin, A. Mozhi and X.-J. Lia, Biotechnol. Adv., 2014, 32, 693–710 CrossRef CAS PubMed.
- M. Huo, J. Yuan, L. Tao and Y. Wei, Polym. Chem., 2014, 5, 1519–1528 RSC.
- F. Meng, W. E. Hennink and Z. Zhong, Biomaterials, 2009, 30, 2180–2198 CrossRef CAS PubMed.
- J. Hu, G. Zhang and S. Liu, Chem. Soc. Rev., 2012, 41, 5933–5949 RSC.
- C. Webera, R. Hoogenboomd and U. S. Schubert, Prog. Polym. Sci., 2012, 37, 686–714 CrossRef.
- Y. Zhao, Macromolecules, 2012, 45, 3647–3657 CrossRef CAS.
- T. Yin, P. Wang, J. Li, Y. Wang, B. Zheng, R. Zheng, D. Cheng and X. Shuai, Biomaterials, 2014, 35, 5932–5943 CrossRef CAS PubMed.
- S. Papa and V. P. Skulachev, Mol. Cell. Biochem., 1997, 174, 305–319 CrossRef CAS PubMed.
- C. C. Winterbourn, Nat. Chem. Biol., 2008, 4, 278–286 CrossRef CAS PubMed.
- B. P. Yu, Physiol. Rev., 1994, 74, 139–162 CAS.
- N. J. Simmonds, R. E. Allen, T. R. Stevens, R. N. V. Someren, D. R. Blake and D. S. Rampton, Gastroenterology, 1992, 103, 186–196 CAS.
- Y. C. Peng, C. L. Hsu, C. F. Tung, W. K. Chou, L. R. Huang, D. Z. Hung, W. H. Hu and D. Y. Yang, Hepatogastroenterology, 2008, 55, 770–773 CAS.
- S. D. Lim, C. Sun, J. D. Lambeth, F. Marshall, M. Amin, L. Chung, J. A. Petros and R. S. Arnold, Prostate, 2005, 62, 200–207 CrossRef CAS PubMed.
- T. P. Szatrowski and C. F. Nathan, Cancer Res., 1991, 51, 794–798 CAS.
- S. H. Lee, M. K. Gupta, J. B. Bang, H. Bae and H.-J. Sung, Adv. Healthcare Mater., 2013, 2, 908–915 CrossRef CAS PubMed.
- S. Joshi-Barr, C. D. G. Lux, E. Mahmoud and A. Almutairi, Antioxid. Redox Signaling, 2014, 21, 730–754 CrossRef CAS PubMed.
- C.-C. Song, F.-S. Du and Z.-C. Li, J. Mater. Chem. B, 2014, 2, 3413–3426 RSC.
- D. Lee, S. Bae, Q. Ke, J. Lee, B. Song, S. A. Karumanchi, G. Khang, H. S. Choi and P. M. Kang, J. Controlled Release, 2013, 172, 1102–1110 CrossRef CAS PubMed.
- D. S. Wilson, G. Dalmasso, L. Wang, S. V. Sitaraman, D. Merlin and N. Murthy, Nat. Mater., 2010, 9, 923–928 CrossRef CAS PubMed.
- M. S. Shim and Y. Xia, Angew. Chem., Int. Ed., 2013, 52, 6926–6929 CrossRef CAS PubMed.
- C. D. Vo, G. Kilcher and N. Tirelli, Macromol. Rapid Commun., 2009, 30, 299–315 CrossRef CAS PubMed.
- H. Xu, W. Cao and X. Zhang, Acc. Chem. Res., 2013, 46, 1647–1658 CrossRef CAS PubMed.
- R. Fang, H. Xu, W. Cao, L. Yang and X. Zhang, Polym. Chem., 2015, 6, 2817–2821 RSC.
- K. E. Broaders, S. Grandhe and J. M. Fréchet, J. Am. Chem. Soc., 2011, 133, 756–758 CrossRef CAS PubMed.
- Y. Zhang, Q. Yin, L. Yin, L. Ma, L. Tang and J. Cheng, Angew. Chem., Int. Ed., 2013, 52, 6435–6439 CrossRef CAS PubMed.
- L. Liu, L. Rui, Y. Gao and W. Zhang, Polym. Chem., 2015, 6, 1817–1829 RSC.
- S. H. Lee, T. C. Boire, J. B. Lee, M. K. Gupta, A. L. Zachman, R. Rath and H.-J. Sung, J. Mater. Chem. B, 2014, 2, 7109–7113 RSC.
- C. D. Vo, G. Kilcher and N. Tirelli, Macromol. Rapid Commun., 2009, 30, 299–315 CrossRef CAS PubMed.
- A. Napoli, M. Valentini, N. Tirelli, M. Muller and J. A. Hubbell, Nat. Mater., 2004, 3, 183–189 CrossRef CAS PubMed.
- K. M. Poole, C. E. Nelson, R. V. Joshi, J. R. Martin, M. K. Gupta, S. C. Haws, T. E. Kavanaugh, M. C. Skala and C. L. Duvall, Biomaterials, 2015, 41, 166–175 CrossRef CAS PubMed.
- D. Jeanmaire, J. Laliturai, A. Almalik, P. Carampin, R. d'Arcy, E. Lallana, R. Evans, R. E. P. Winpennyd and N. Tirelli, Polym. Chem., 2014, 5, 1393–1404 RSC.
- M. K. Gupta, T. A. Meyer, C. E. Nelson and C. L. Duvall, J. Controlled Release, 2012, 162, 591–598 CrossRef CAS PubMed.
- M. K. Gupta, J. R. Martin, T. A. Werfel, T. Shen, J. M. Page and C. L. Duvall, J. Am. Chem. Soc., 2014, 136, 14896–14902 CrossRef CAS PubMed.
- J. R. Kramer and T. J. Deming, J. Am. Chem. Soc., 2012, 134, 4112–4115 CrossRef CAS PubMed.
- X. Fu, Y. Ma, Y. Shen, W. Fu and Z. Li, Biomacromolecules, 2014, 15, 1055–1061 CrossRef CAS PubMed.
- K. Fuhrmann, A. Pozomska, C. Aeberli, B. Castagner, M. A. Gauthier and J.-C. Leroux, ACS Nano, 2013, 7, 8243–8250 CrossRef CAS PubMed.
- C. Xiao, J. Ding, L. Ma, C. Yang, X. Zhuang and X. Chen, Polym. Chem., 2015, 6, 738–747 RSC.
- S. Kobayashi and A. Makino, Chem. Rev., 2009, 109, 5288–5353 CrossRef CAS PubMed.
- R. A. Gross, M. Ganesh and W. Lu, Trends Biotechnol., 2010, 28, 435–443 CrossRef CAS PubMed.
- W.-X. Wu, N. Wang, B.-Y. Liu, Q.-F. Deng and X.-Q. Yu, Soft Matter, 2014, 10, 1199–1213 RSC.
- X. Zhang, B. Liu, Z. Yang, C. Zhang, H. Li, X. Luo, H. Luo, D. Gao, Q. Jiang, J. Liu and Z. Jiang, Colloids Surf., B, 2014, 115, 349–358 CrossRef CAS PubMed.
- B. Liu, X. Zhang, Y. Chen, Z. Yao, Z. Yang, D. Gao, Q. Jiang, J. Liu and Z. Jiang, Polym. Chem., 2015, 6, 1997–2010 RSC.
- K. Kataoka, A. Harada and Y. Nagasaki, Adv. Drug Delivery Rev., 2012, 64, 37–48 CrossRef.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- W.-X. Wu, L. Qu, B.-Y. Liu, W.-W. Zhang, N. Wang and X.-Q. Yu, Polymer, 2015, 59, 187–193 CrossRef CAS.
- J. Liu, F. Zhan, Q. Fu, X. Zhu and W. Shi, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7543–7555 CrossRef CAS.
- S. E. Choi and M. K. H. Pflum, Bioorg. Med. Chem. Lett., 2012, 22, 7084–7086 CrossRef CAS PubMed.
- X. Ma, X. Huang, G. Huang, L. Li, Y. Wang, X. Luo, D. A. Boothman and J. Gao, Adv. Healthcare Mater., 2014, 3, 1210–1246 CrossRef CAS PubMed.
- X. Wei, C. Gong, M. Gou, S. Fu, Q. Guo, S. Shi, F. Luo, G. Guo, L. Qiu and Z. Qian, Int. J. Pharm., 2009, 381, 1–18 CrossRef CAS PubMed.
- H. Zhang, P. Ni, J. He and C. Liu, Langmuir, 2008, 24, 4647–4654 CrossRef CAS PubMed.
- L. Y. Qiu and Y. H. Bae, Pharm. Res., 2006, 23, 1–30 CrossRef CAS PubMed.
- P. Carampin, E. Lallana, J. Laliturai, S. C. Carroccio, C. Puglisi and N. Tirelli, Macromol. Chem. Phys., 2012, 213, 2052–2061 CrossRef CAS.
- J. Wang, X. Sun, W. Mao, W. Sun, J. Tang, M. Sui, Y. Shen and Z. Gu, Adv. Mater., 2013, 25, 3670–3676 CrossRef CAS PubMed.
- B. Liu, D. Wang, Y. Liu, Q. Zhang, L. Meng, H. Chi, J. Shi, G. Li, J. Li and X. Zhu, Polym. Chem., 2015, 6, 3460–3471 RSC.
- G. Rizis, T. G. M. van de Ven and A. Eisenberg, Soft Matter, 2014, 10, 2825–2835 RSC.
- J. Wu, L. Zhao, X. Xu, N. Bertrand, W. Choi, B. Yameen, J. Shi, V. Shah, M. Mulvale, J. L. MacLean and O. C. Farokhzad, Angew. Chem., Int. Ed., 2015, 54, 9218–9223 CrossRef CAS PubMed.
- J.-H. Ryu, S. Jiwpanich, R. Chacko, S. Bickerton and S. Thayumanavan, J. Am. Chem. Soc., 2010, 132, 8246–8247 CrossRef CAS PubMed.
- M.-F. Chung, W.-T. Chia, W.-L. Wan, Y.-J. Lin and H.-W. Sung, J. Am. Chem. Soc., 2015, 137, 12462–12465 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra21779b |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 











