DOI:
10.1039/C5RA21565J
(Paper)
RSC Adv., 2016,
6, 9132-9138
An investigation of the role increasing π-conjugation has on the efficiency of dye-sensitized solar cells fabricated from ferrocene-based dyes†
Received
16th October 2015
, Accepted 5th January 2016
First published on 22nd January 2016
Abstract
In this article, we report the synthesis of new ferrocene incorporating dyes and their evaluation as sensitizers for dye-sensitized solar cells. We have shown that the sequential addition of alkyne units as π-spacers plays an important role in modulating their solution optical and redox properties. Likewise, the most extensively conjugated dye (Fc-D3) provided the highest power conversion efficiency (η) compared to the other less-conjugated dyes in the series. We have also demonstrated that the choice of electrolyte is important in determining η for this series of dyes.
Introduction
The advantages that dye-sensitized solar cells (DSSCs)1 have over silicon-based photovoltaic devices, such as their ability to operate under low light conditions and their lower cost, has ensured that devices of this type continue to receive significant attention.2 Ruthenium-based dyes dominated the early progress in this field, however, metal-free organic dyes have become increasingly attractive due to their higher molar extinction coefficients, convenient synthesis, purification and structural modification.3 More recently, porphyrin-based dyes have revitalised research into metal-incorporating dyes and to date have produced headlining power conversion efficiencies of around 13%.4
The ability of ferrocene (Fc) to undergo reversible one-electron chemical and electrochemical oxidation at low potential has allowed this unit to become a ubiquitous building block in a range of disparate applications including medicine and materials science.5 Although Fc has proved to be an effective redox electrolyte for DSSCs,6 its incorporation as a donor unit in photosensitizers has received surprisingly little attention. For example, Chauhan et al. have reported DSSCs fabricated from Fc-based dyes which indicated that both the dye anchoring group and the electrolyte play an important role in determining the power conversion efficiency (η).7 More recently, Sirbu et al. reported DSSCs fabricated using a trisubstituted ferrocene-based porphyrin derivative with a cobalt(II/III) electrolyte, however, DSSC performance was rather modest (η = 0.0081%).8 In this study, we describe the synthesis and characterisation of new Fc-based dyes featuring benzothiadiazole and cyanoacrylic acid residues9 as acceptor units and alkyne residues as π-linker units.10 We report the significance of increasing the number of alkyne linker units has on the optical and redox properties of the dyes and the key parameters of their resulting DSSCs. We have also shown that the choice of electrolyte is important in terms of controlling η.
Results and discussion
Synthesis
The three dyes were prepared according to the synthetic pathway shown in Scheme 1. Ferrocene boronic acid 1 was coupled with 4,7-dibromobenzothiadiazole 2,11 via Suzuki cross-coupling reaction, affording 3 in 46% yield. Compound 3 was further reacted with 4-formylphenylboronic acid, affording compound 4 in 84% yield. Compound 6 was obtained, in a 89% yield, via a Sonogashira cross-coupling reaction of ethynylferrocene 5 and 2.12 This compound was then reacted with 4-formylphenylboronic acid and 4-ethynylbenzaldehyde, via Suzuki and Sonogashira coupling reactions, to give compound 7 and 8, in 85% and 45% yield, respectively. The three aldehydes 4, 7 and 8 were reacted with cyanoacetic acid to afford the corresponding cyanoacrylates Fc-D1 (77% yield), Fc-D2 (98% yield) and Fc-D3 (44% yield).
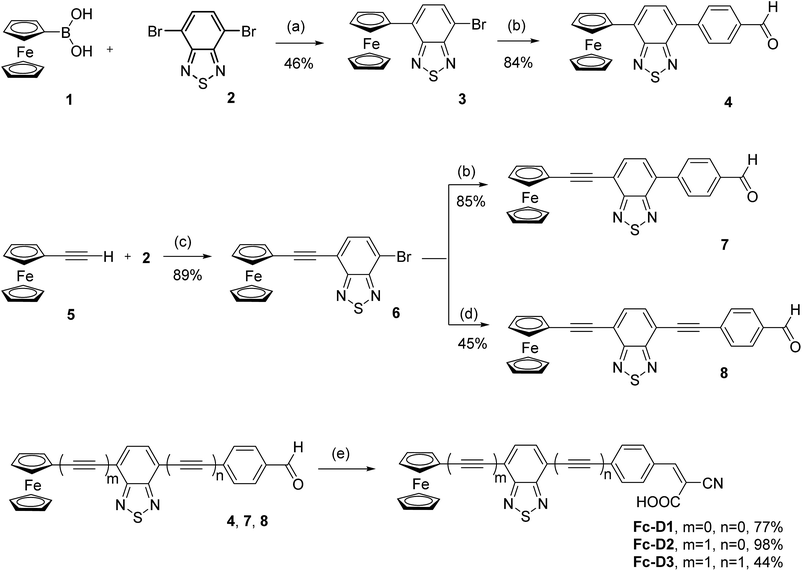 |
| | Scheme 1 Synthesis of ferrocene dyes Fc-D1–Fc-D3. Reagents and conditions: (a) Pd(dppf)Cl2, K2CO3, dioxane, H2O, reflux, 14 h; (b) 4-formylphenylboronic acid, Pd(PPh3)4, K2CO3, THF, H2O, reflux, 48 h; (c) PdCl2(PPh3)4, CuI, Et3N, THF, 60 °C, 6 h; (d) 4-ethynylbenzaldehyde, PdCl2(PPh3)4, CuI, Et3N, THF, r.t., 6 h; (e) cyanoacetic acid, piperidine, acetic acid, MgSO4, toluene, 100 °C, 6 h. | |
Characterization
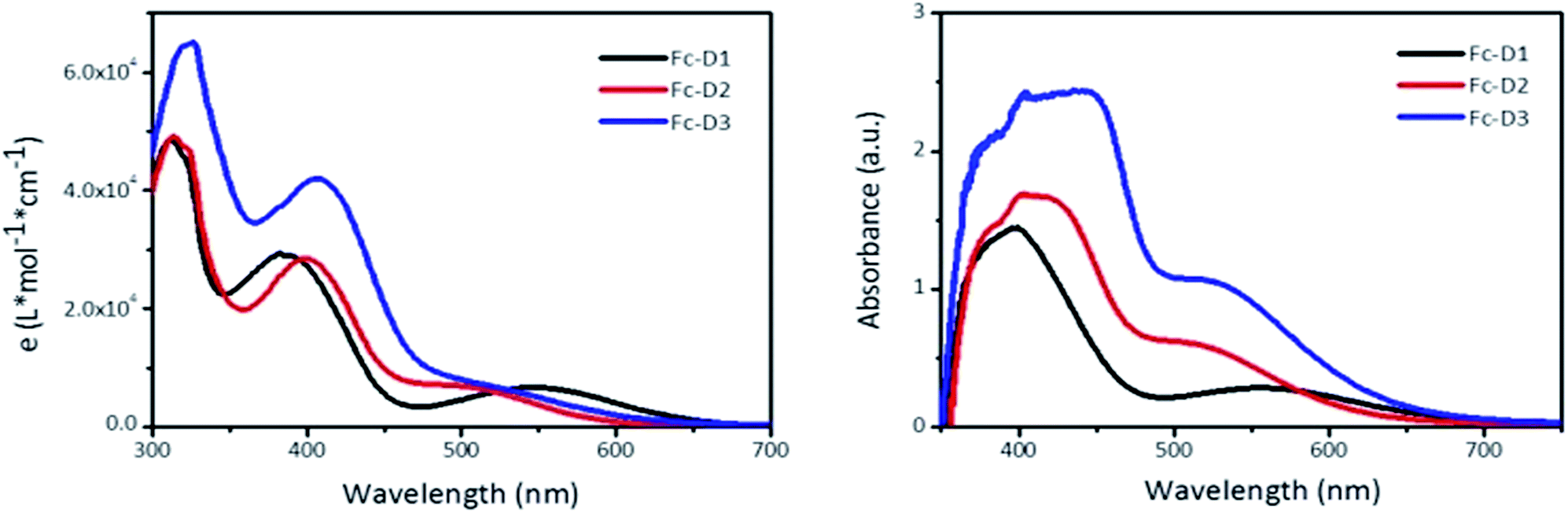
The UV-vis spectra of the three compounds recorded in DMF are provided in Fig. 1. The absorption band around 300 nm is likely due to n–π* transition of the benzothiadiazole (BTD) unit or π–π* transition of the conjugated aromatic moieties,13 whilst the absorption around 400 nm corresponds to an intramolecular charge-transfer (ICT) between the donor and acceptor groups. It is clear that the increase in the conjugation length results in the red shift of the two absorption maxima with higher molar extinction coefficients. The distinct absorption band around 550 nm for Fc-D1 is attributed to a d–π* charge transfer (CT). However, d–π* CT bands are sensitive to adjacent acceptor strength, and are not as pronounced in the spectra of the alkyne containing dyes Fc-D2 and Fc-D3. Compared with the spectra in DMF solution, the ICT absorption band of these dyes on TiO2 films exhibits significant spectral broadening and red-shift because of the interaction of the anchoring groups with the surface of TiO2 and J-aggregation of photosensitizer molecules. Unlike the spectra in DMF solution, the absorption related to the metal-acceptor charge transfer is observable in all dyes on TiO2 films. From the cut-off wavelengths of the spectra in DMF solution, the estimated values of the optical gaps (Eopt) are listed in Table 1.
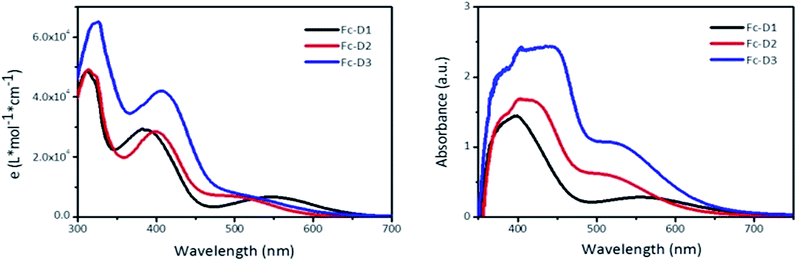 |
| | Fig. 1 Absorption spectra of Fc-D1–Fc-D3 in DMF (left), and deposited on TiO2 film (right). | |
Table 1 Summary of optical and electrochemical properties of the dyes. Eopt = optical gap; Efund = IP − EA
| Dye |
λmax, nm (ε, 103 M−1 cm−1) |
Eopt (eV) |
IP (eV) |
EA (eV) |
Efund (eV) |
| Fc-D1 |
312(48.7), 387(29.1), 550(6.7) |
1.62 |
−5.11 |
−3.28 |
1.83 |
| Fc-D2 |
314(49.2), 399(28.5), 483(7.0) |
1.70 |
−5.18 |
−3.44 |
1.74 |
| Fc-D3 |
323(65.6), 407(42.1), 487(6.6) |
1.61 |
−5.13 |
−3.48 |
1.75 |
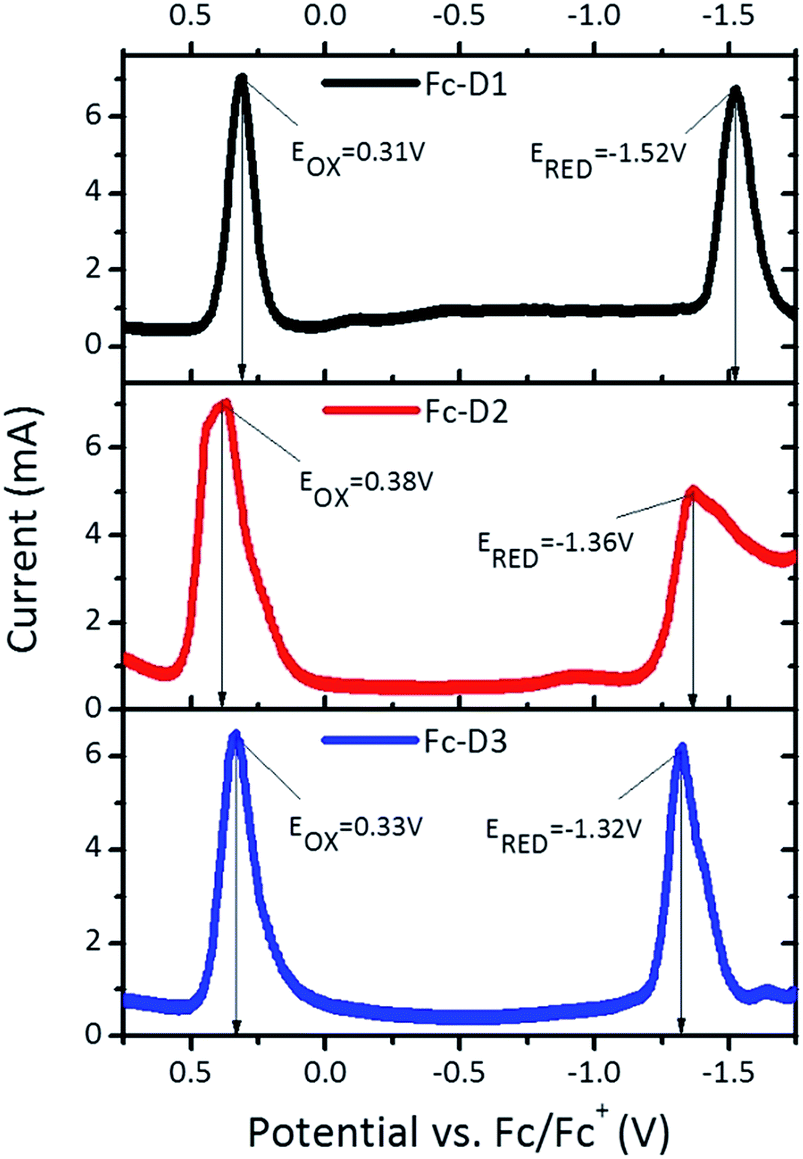
The electrochemical behaviour of the three dyes in DMF solution was explored by cyclic voltammetry (CV) and square wave voltammetry (SWV) (Fig. 2). The redox-active ferrocenyl group exhibits a single oxidation wave whereas a one electron reduction wave attributed to the BTD acceptor unit is observed. Addition of alkyne π-linker units on going from Fc-D1 to Fc-D3, results in the reduction potentials being shifted to more positive values indicating that the benzothiadiazole unit is easier to reduce. The estimated electron affinities (EAs) and ionization potentials (IPs) are presented in Table 1.14 As shown in Fig. 3, all the IPs are slightly lower in energy than the I−/I3− redox couple, ensuring that the generation of all the dyes is energetically favourable. On the other hand, the positions of the EAs are found to be almost 1 eV above the TiO2 conduction band.
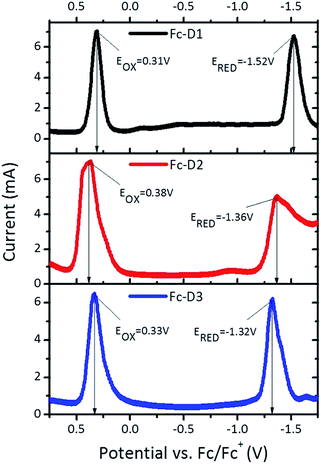 |
| | Fig. 2 SWV plots for three dyes recorded in DMF (1 × 10−3 M). | |
 |
| | Fig. 3 Energy level diagram showing the IPs and EAs of each dye, and their position relative to the TiO2 conduction band and I−/I3− redox potential. | |
Theoretical calculations
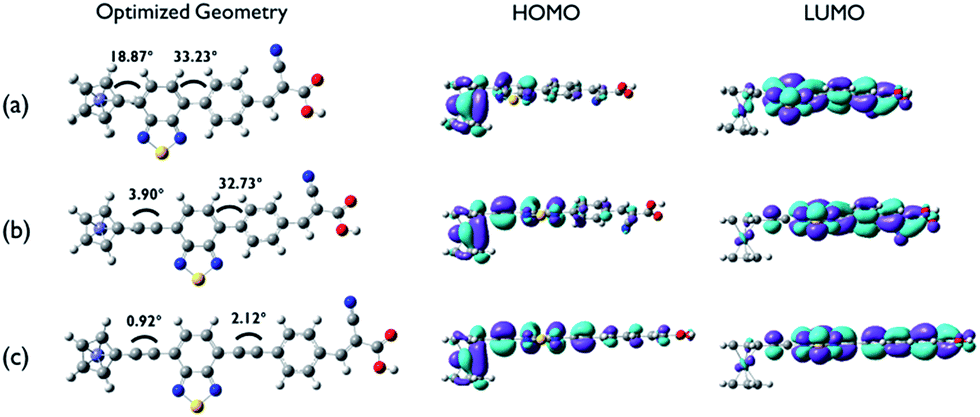
To gain insight on the electronic structure and optical properties of the ferrocene-based dyes, density functional theory (DFT) and time dependent DFT calculations were performed. Fig. 4 shows ground state optimized geometries and electronic density distributions of the HOMO and LUMO of the dyes, which clearly show the role of the acetylene linker on maintaining planarity and facilitating ICT separation. In particular, on going form FC-D1 to FC-D3 the sequential addition of the acetylene linker moiety makes the dihedral angles between the ferrocenyl and benzothiadiazole moieties and between the benzothiazole and phenyl ring moieties significantly decrease from 18.87° to 0.92° and from 33.23° to 2.12°, respectively. This indicates that acetylene linkers eliminate the steric hindrance between the hydrogen atoms of these rings. The ferrocenyl groups in all dyes adopt an eclipsed D5h conformation rather than the staggered conformation.15,16 The electron densities of the HOMOs for all dyes are mainly localized on the ferrocenyl moiety and are extended along to the benzothiadiazole moiety to the phenyl ring. The LUMOs for all dyes are localised on the benzothiadiazole and cyanoacrylate residues. In all dyes, significant overlap of the frontier orbitals occurs suggesting that efficient ICT likely occurs from the donor to the terminal acceptor group which anchors to the TiO2 surface.
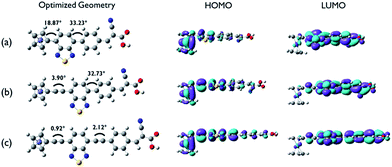 |
| | Fig. 4 Ground state optimized geometry of the dyes, and electron density distribution representations: (a) Fc-D1, (b) Fc-D2, and (c) Fc-D3. | |
Photovoltaic measurements
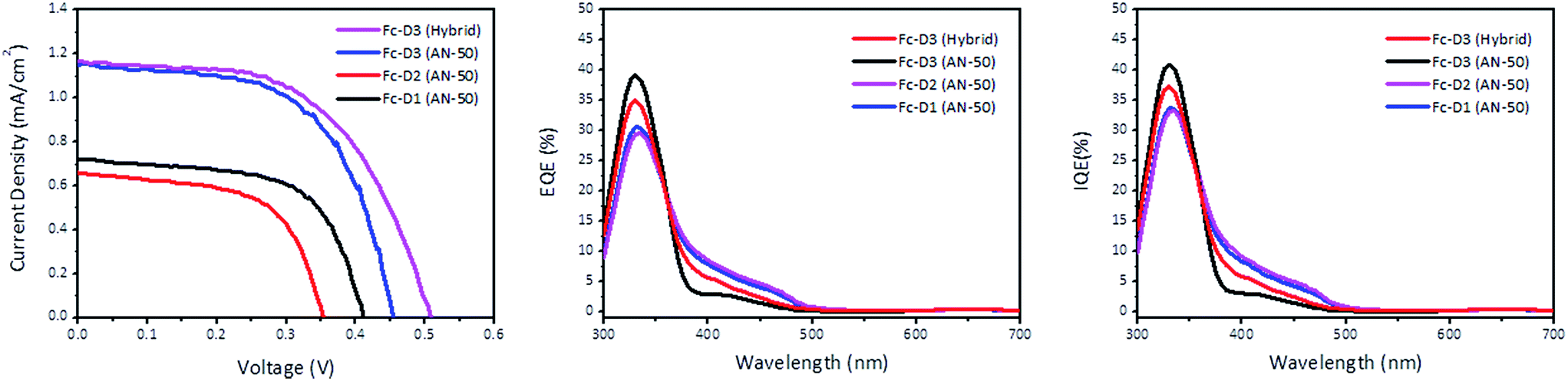
Fig. 5 shows J–V characteristics of DSSCs sensitized with the three dyes. The photovoltaic parameters are summarized in Table 2. The addition of the two acetylene bridging units in Fc-D3 resulted in better photovoltaic performance with the iodide/triiodide redox couple than Fc-D1: Voc from 0.407 V to 0.434 V, Jsc from 0.730 mA cm−2 to 1.070 mA cm−2 and η from 0.180% to 0.279%. The η value of Fc-D3 compared to Fc-D1 is more than doubled which may be ascribed to the improvement of light harvesting efficiency or the electron injection efficiency of Fc-D3. Fig. 5 also shows both external quantum efficiency (EQE) and internal quantum efficiency (IQE) spectra of the DSSCs. The EQE and IQE spectra of DSSCs featuring Fc-D3 extend to the longer wavelength region and exhibit higher values in the shorter wavelength region when compared to those using Fc-D1. Interestingly, our hybrid electrolyte containing both iodide/triiodide (I−/I3−) and organic redox couples allowed for better DSSC performance. The organic redox couple consists of the thiolate form (McMT−) and disulfide dimer (BMT) derived from 2-mercapto-5-methyl-1,3,4-thiadiazole (McMT). Although the redox potential of McMT−/BMT (0.155 V vs. NHE) is more negative than that of I−/I3− (0.4 V vs. NHE),17 the addition of the organic redox couple resulted in the increase in Voc. We think that variations in the recombination kinetics or shifts in the TiO2 conduction band position could lead to the higher Voc. For example, different cations (i.e. Li+ and tetra-n-butylammonium (TBA+) in the hybrid electrolyte) can give different potentials for the conduction band edge (Ecb) of the TiO2 nanoporous film. An increase of a cation radius or a less-adsorptive cation shifts Ecb negatively and thus the increase of Eredox − Ecb leads to the increase of the photovoltage.18,19
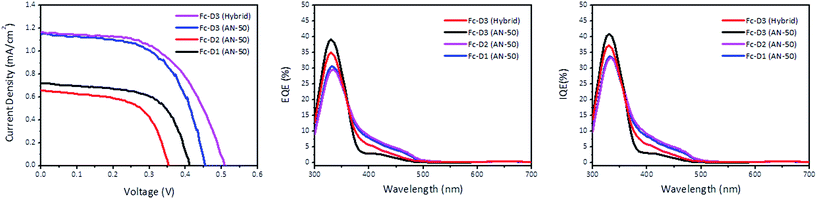 |
| | Fig. 5 J–V curve of the DSSC devices (left), EQE plots (middle), and IQE plots (right). | |
Table 2 Summary of the photovoltaic parameters of the DSSC devices
| |
Voc (V) |
Jsc (mA cm−2) |
FF (%) |
η (%) |
| Fc-D1 (AN-50) |
0.407 ± 0.004 |
0.730 ± 0.049 |
58.4 ± 2.1 |
0.180 ± 0.009 |
| Fc-D1 (Hybrid) |
0.405 ± 0.021 |
0.610 ± 0.046 |
61.2 ± 1.2 |
0.160 ± 0.019 |
| Fc-D2 (AN-50) |
0.337 ± 0.014 |
0.590 ± 0.056 |
57.9 ± 2.0 |
0.115 ± 0.018 |
| Fc-D2 (Hybrid) |
0.380 ± 0.010 |
0.770 ± 0.050 |
60.3 ± 1.9 |
0.190 ± 0.004 |
| Fc-D3 (AN-50) |
0.434 ± 0.005 |
1.070 ± 0.052 |
57.5 ± 1.3 |
0.279 ± 0.023 |
| Fc-D3 (Hybrid) |
0.494 ± 0.008 |
1.190 ± 0.057 |
54.1 ± 2.3 |
0.325 ± 0.005 |
As electrochemical impedance spectroscopy (EIS) can offer very valuable insight into interfacial charge-transfer processes, we have used this technique to explore DSSCs sensitized with Fc-D3. The semicircle in the intermediate frequency region in Fig. 6a represents the electron transfer process at the TiO2/dye/electrolyte interface and its radius is related to the charge recombination rate, i.e. a larger radius indicates a slower charge recombination. The semicircle in the low frequency region in Fig. 6a is associated with the Warburg diffusion of the redox couple in the electrolyte. The electron lifetime corresponding to the TiO2/dye/electrolyte interface, can be estimated from the peak frequency (fpeak) in Fig. 6b according to τe = 1/2πfpeak. The calculated values of Fc-D3 (AN-50) and Fc-D3 (Hybrid) are 0.36 ms and 0.71 ms, respectively. The addition of the organic redox couple suppresses the back charge recombination at the TiO2/dye/electrolyte and thus this resulted in the increase in Jsc.
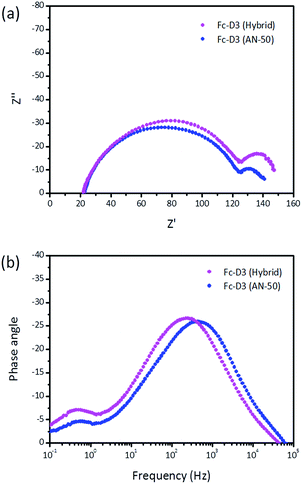 |
| | Fig. 6 EIS plots of the DSSC devices using Fc-D3. | |
Experimental
Materials and methods
All reactions were undertaken under a nitrogen atmosphere. Solvents were purified using a PureSolv solvent purifier system. Reagents were purchased from Sigma-Aldrich, and were used without further purification. NMR spectra were obtained with either a Bruker AVIII 400 MHz or a Bruker AVIII 500 MHz spectrometer and all reported chemical shifts are relative to TMS. UV-vis spectra were recorded on a Perkin-Elmer Lamda 25 instrument. Optically determined band gaps (Eopt) were estimated using the absorption edge of the longest wavelength absorption (λ) using Eopt (eV) = (1240/λ (nm)). Cyclic voltammetry measurements were undertaken using a CH Instruments 440A electrochemical analyzer using a platinum working electrode, a platinum wire counter electrode and a silver wire pseudo-reference electrode. Ferrocene was used as an internal standard and all redox couples are reported versus the ferrocene/ferrocenium (Fc/Fc+) redox couple, adjusted to 0.0 V. The solutions were prepared using dry dimethylformamide (DMF) containing electrochemical grade tetrabutylammonium hexafluorophosphate (0.1 M) as the supporting electrolyte. The solutions were purged with nitrogen gas for 3 min prior to recording the electrochemical data.
Theoretical calculations
Density functional theory (DFT) calculations were performed using Gaussian 09 to gain insight electronic properties of the dye molecules.20 Global minimum states were confirmed by absence from imaginary frequencies under ground-state geometry optimization followed by vibrational frequency calculations. All calculations were conducted with Becke's three-parameter hybrid and Lee–Yang–Parr's gradient corrected correlation (B3LYP) functional and 6-311G(d,p) basis set for H, C, N, O, S and Lanl2DZ functional for Fe, under vacuum.
Fabrication of DSSCs and photovoltaic measurements
The TiO2 photoanodes were screen-printed onto transparent fluorine-doped SnO2 (FTO)-coated conducting glass (TEC 8, Pilkington, 2.2 mm-thick, sheet resistance = 8 Ω sq.−1).21 The resulting layer was placed in a muffle furnace and gradually heated to 300 °C over a 30 min period, then heated at 300 °C for 1 hour, and then heated to 575 °C for 1 hour, and then cooled to room temperature. The screen printing and calcination processes were repeated until a thickness of approximately 20 μm was obtained. The active area of the electrodes was 0.25 cm2. The prepared photoanodes were immersed in a 0.04 M solution of TiCl4 at 75 °C for 1 hour, rinsed with deionized water, and then sintered at 500 °C for 30 min. They were exposed to O2 plasma for 10 min and then immersed for 24 hours in one of three dye-containing ethanol solutions (0.5 mM). The Pt counter electrodes were prepared on the FTO-coated glass with magnetron sputtering and two holes were drilled in the glass. Both the dye-sensitized TiO2 electrode and Pt counter electrode were sealed with a 60 μm-thick layer of Surlyn (Solaronix, Switzerland). An iodide based redox electrolyte (Iodolyte AN-50, Solaronix) was injected into the rear side of the counter electrode. A hybrid electrolyte, a mixture of the I−/I3− and McMT−/BMT couples was also used. The photovoltaic characteristics of the DSSCs under AM 1.5 illumination (equivalent to one sun, 1 kW m−2) were investigated with a solar cell current–voltage (I–V) measurement system (K3000 LAB, McScience, Korea). The photocurrent density (Jsc), open-circuit voltage (Voc), fill factor (FF), and power-conversion-efficiency (η) were simultaneously measured. EQE and IQE were recorded as a function of excitation wavelength (λ) by a spectral incident photon-to-current efficiency (IPCE) measurement system (K3100, McScience, Korea).
Synthetic procedures
Compound 3. Ferroceneboronic acid 1 (500 mg, 2.17 mmol) and 2 (960 mg, 3.26 mmol) were dissolved in dry dioxane (15 mL). A 2 M K2CO3 aqueous solution (10.9 mL, 21.7 mmol) was added, and the mixture was degassed for 30 minutes with N2. Then Pd(dppf)Cl2 (791 mg, 0.11 mmol) was added and the resulting mixture was left to stir at reflux. After 14 h, the mixture was allowed to cool to room temperature, and was then quenched with water (50 mL) and extracted with DCM (3 × 50 mL). The combined organic extracts were dried over MgSO4, filtered and concentrated under vacuum. The crude compound was purified by column chromatography (SiO2, petroleum ether![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :toluene (1:1)), affording the title compound as a purple solid (393 mg, 46%). mp 165–166 °C. δH (400 MHz, CDCl3, TMS), 7.73 (1H, d, J 7.7), 7.55 (1H, d, J 7.7), 5.24 (2H, t, J 1.9), 4.51 (2H, t, J 1.9), 4.03 (5H, s). δC (100 MHz, CDCl3, TMS), 154.2, 153.1, 134.1, 132.4, 125.2, 110.5, 80.4, 70.3, 70.0, 68.8. HRMS (ESI, m/z): [M + H]+ found, 397.9155; calc. for (C16H11BrFeN2S), 397.9170.
:toluene (1:1)), affording the title compound as a purple solid (393 mg, 46%). mp 165–166 °C. δH (400 MHz, CDCl3, TMS), 7.73 (1H, d, J 7.7), 7.55 (1H, d, J 7.7), 5.24 (2H, t, J 1.9), 4.51 (2H, t, J 1.9), 4.03 (5H, s). δC (100 MHz, CDCl3, TMS), 154.2, 153.1, 134.1, 132.4, 125.2, 110.5, 80.4, 70.3, 70.0, 68.8. HRMS (ESI, m/z): [M + H]+ found, 397.9155; calc. for (C16H11BrFeN2S), 397.9170.
Compound 4. Compound 3 (390 mg, 0.98 mmol) and 4-formylphenylboronic acid (177 mg, 1.18 mmol) were dissolved in THF (15 mL). A 2 M K2CO3 aqueous solution (4.9 mL, 9.8 mmol) was added, and the solution was degassed for 30 minutes with N2. Then, Pd(PPh3)4 (57 mg, 0.05 mmol) was added and the solution was left to stir at reflux. After 48 h, the mixture was allowed to cool to room temperature, and was then quenched with water (50 mL) and extracted with DCM (3 × 50 mL). The combined organic extracts were dried over MgSO4, filtered and concentrated under vacuum. The crude compound was purified by column chromatography (SiO2, toluene), affording the title compound as a purple solid (355 mg, 84%). mp > 300 °C. δH (400 MHz, CDCl3, TMS), 10.11 (1H, s), 8.17 (2H, d, J 8.2), 8.05 (2H, d, J 8.2), 7.80 (1H, d, J 7.5), 7.70 (1H, d, J 7.5), 5.34 (2H, t, J 1.9), 4.55 (2H, t, J 1.9), 4.07 (5H, s). δC (100 MHz, CDCl3, TMS) 192.1, 154.1, 153.9, 143.9, 135.7, 134.8, 130.1, 129.7, 129.5, 129.1, 125.0, 80.8, 70.3, 70.1, 69.0. HRMS (EI, m/z): [M]+ found, 424.0331; calc. for (C23H16FeN2OS), 424.0333.
Dye Fc-D1. Compound 4 (332 mg, 0.78 mmol), cyanoacetic acid (80 mg, 0.94 mmol) and MgSO4 (40 mg, 0.16 mmol) were dissolved/suspended in toluene (50 mL). Piperidine (8 μL, 0.08 mmol) and acetic acid (27 μL, 0.47 mmol) were added, and the mixture was then left to stir at 100 °C. After 6 h, the reaction was stopped and the solvent was removed under vacuum. The crude compound was purified by column chromatography (SiO2, DCM:methanol (9:1)), affording the title compound as a green solid (270 mg, 71%). mp > 300 °C. δH (500 MHz, d6-DMSO, TMS), 8.18 (2H, d, J 8.4), 8.05 (3H, d, J 8.4), 8.01 (1H, d, J 7.5), 7.90 (1H, d, J 7.5), 5.41 (2H, t, J 1.9), 4.57 (2H, t, J 1.9), 4.06 (5H, s). HRMS (ESI, m/z): [M − H]− found, 490.0315; calc. for (C26H16FeN3O2S), 490.0318.
Compound 612. Ethynylferrocene 5 (157 mg, 0.75 mmol), and 2 (200 mg, 0.68 mmol) were dissolved in a mixture of THF (10 mL) and Et3N (10 mL). The solution was degassed for 30 minutes, with N2, then Pd(PPh3)2Cl2 (24 mg, 0.03 mmol) and CuI (7 mg, 0.03 mmol) were added. The mixture was left to stir at 60 °C. After 6 h, the mixture was allowed to cool to room temperature and was then quenched with an aqueous solution of HCl (5%, 50 mL). The organic layer was extracted with DCM (3 × 50 mL), washed with water, dried over MgSO4, filtered and concentrated under vacuum. The crude compound was purified by column chromatography (SiO2, petroleum ether:DCM (9:1)), affording the title compound as an orange solid (250 mg, 89%). mp 170–171 °C. δH (500 MHz, CDCl3, TMS), 7.6 (1H, d, J 8.3), 7.2 (1H, d, J 8.3), 4.5 (2H, s), 4.2 (7H, b). δC (125 MHz, CDCl3, TMS) 154.2, 153.1, 132.1, 132.0, 117.4, 113.5, 97.0, 81.1, 71.9, 70.2, 69.4, 63.9.
Compound 7. Compound 6 (200 mg, 0.47 mmol) and 4-formylphenylboronic acid (85 mg, 0.56 mmol) were dissolved in THF (20 mL). A 2 M K2CO3 solution (2.4 mL, 4.7 mmol) was added, and the solution was degassed for 30 minutes with N2. Then Pd(PPh3)4 (27 mg, 0.03 mmol) was added and the solution was then left to stir at reflux. After 6 h, the mixture was allowed to cool to room temperature, and was then quenched with water (50 mL) and extracted with DCM (3 × 50 mL). The combined organic extracts were dried over MgSO4, filtered and concentrated under vacuum. The crude compound was purified by column chromatography (SiO2, petroleum ether:DCM (1:1)), affording the title compound as a brown solid (180 mg, 86%). mp > 300 °C. δH (400 MHz, CDCl3, TMS), 10.11 (1H, s), 8.15 (2H, d, J12 8.5), 8.05 (2H, d, J 8.5), 7.87 (1H, d, J 7.4), 7.77 (1H, d, J 7.4), 4.67 (2H, t, J 1.9), 4.34 (2H, t, J 1.9), 4.32 (5H, s). δC (100 MHz, CDCl3, TMS) 153.8, 116.9, 116.9, 98.4, 95.7, 95.0, 94.1, 92.1, 91.4, 81.5, 77.9, 51.7, 44.4, 34.5, 32.8, 32.1, 26.5. HRMS (EI, m/z): [M]+ found, 448.0331; calc. for (C25H16FeN2OS), 448.0333.
Dye Fc-D2. Compound 7 (130 mg, 0.29 mmol), cyanoacetic acid (30 mg, 0.35 mmol) and MgSO4 (14 mg, 0.06 mmol) were dissolved/suspended in toluene (20 mL). Piperidine (3 μL, 0.03 mmol) and acetic acid (10 μL, 0.17 mmol) were added, and the mixture was then left to stir at 100 °C. After 6 h, the reaction was stopped and the solvent was removed under vacuum. The crude compound was purified by column chromatography (SiO2, DCM:methanol (9:1)), affording the title compound as a dark brown solid (144 mg, 96%). mp > 300 °C. δH (500 MHz, d6-DMSO, TMS), 8.17 (2H, d, J 8.5), 8.05 (2H, d, J 8.5), 8.02 (1H, s), 7.98 (2H, s), 4.70 (2H, t, J 1.9), 4.44 (2H, t, J 1.9), 4.35 (5H, s). HRMS (ESI, m/z): [M − H]− found, 514.0295; calc. for (C28H16FeN3O2S), 514.0318.
Compound 8. Compound 6 (280 mg, 0.66 mmol), and 4-ethynylbenzaldehyde (95 mg, 0.73 mmol) were dissolved in a mixture of THF (10 mL) and Et3N (10 mL). The solution was degassed for 30 minutes, with N2, then Pd(PPh3)2Cl2 (24 mg, 0.03 mmol) and CuI (7 mg, 0.03 mmol) were added. The mixture was left to stir at room temperature. After 6 h, the mixture was quenched with a saturated aqueous solution of NH4Cl (50 mL). The organic layer was extracted with DCM (3 × 50 mL), washed with water, dried over MgSO4, filtered and concentrated under vacuum. The crude compound was purified by column chromatography (SiO2, toluene:diethyl ether (9:1)), affording the title compound as an orange solid (140 mg, 45%). mp > 300 °C. δH (500 MHz, CDCl3, TMS), 10.05 (1H, s), 7.92 (2H, d, J 8.4), 7.82 (2H, d, J 8.4), 7.80 (1H, d, J 7.4), 7.75 (1H, d, J 7.4), 4.66 (2H, t, J 1.9), 4.35 (2H, t, J 1.9), 4.31 (5H, s). δC (125 MHz, CDCl3, TMS) 191.5, 154.5, 136.1, 133.3, 132.6, 131.7, 129.8, 129.0, 119.1, 115.5, 98.8, 95.9, 89.4, 82.1, 72.2, 70.4, 69.8, 64.1. HRMS (EI, m/z): [M]+ found, 472.0328; calc. for (C27H16FeN2OS), 472.0333.
Dye Fc-D3. Compound 8 (140 mg, 0.30 mmol), cyanoacetic acid (30 mg, 0.35 mmol) and MgSO4 (14 mg, 0.06 mmol) were dissolved/suspended in toluene (20 mL). Piperidine (3 μL, 0.03 mmol) and acetic acid (10 μL, 0.17 mmol) were added, and the mixture was then left to stir at 100 °C. After 6 h, the reaction was stopped and the solvent was removed under vacuum. The crude compound was purified by column chromatography (SiO2, DCM:methanol (9:1)), affording the title compound as an orange solid (190 mg, 44%). mp > 300 °C. δH (400 MHz, d6-DMSO, TMS), 8.07 (1H, bs), 8.02 (2H, d, J 8.2), 7.98 (1H, d, J 7.4), 7.90 (1H, d, J 7.4), 7.78 (2H, d, J 8.2), 4.70 (2H, t, J 1.9), 4.45 (2H, t, J 1.9), 4.34 (5H, s). HRMS (ESI, m/z): [M − H]− found, 538.0326; calc. for (C30H16FeN3O2S), 538.0318.
[nBu4N]+McMT−. 2-Mercapto-5-methyl-1,3,4-thiadiazole (McMT) (1.32 g) was neutralized with a 1 M solution of tetra-n-butylammonium (TBA+) hydroxide in 10 mL methanol. The reaction was heated under reflux for 3 hours under a N2 atmosphere. Then, the solvent was evaporated and the resulting thiolate salt was dried under vacuum at 40 °C for 12 hours and placed at room temperature for 12 hours.
1,2-Bis(5-methyl-1,3,4-thiathiazole-2-yl)sulfide (BMT). McMT (1.32 g) was deprotonated with potassium carbonate (0.69 g) in 20 mL methanol. The mixture underwent ultrasonic treatment for 2 hours and then iodine (1.25 g) was added to the solution and the reaction was sonicated for a further 10 minutes until the iodine disappeared completely. The solvent was removed under vacuum and the residue was dissolved in excess CH2Cl2. The solution was washed with water (3 × 30 mL), and finally the organic phase was collected and dried over anhydrous Na2SO4, filtered and the solvent evaporated to yield BMT as a white solid which was dried under vacuum at 40 °C for 12 hours.
Conclusions
In conclusion, a series of new Fc-functionalised dyes have been synthesised and characterised. We have shown that the sequential addition of alkyne units on going from Fc-D1 to Fc-D3 has a significant effect on both the light absorption characteristics and LUMO of the dyes. In addition, DSSCs fabricated using dye Fc-D3 display the highest η values of the series, providing a maximum value of 0.325% when a hybrid electrolyte was used. Currently, we are exploring other n-type metal oxide semiconductors in order to avoid direct electron injection from the HOMO of the Fc dyes to the conduction band (CB) of the metal oxide and back electron transfer from the CB to the HOMO. However, an important problem facing the development of ferrocene-based dyes is the well-documented instability of oxidized state of Fc (ferrocenium) to molecular oxygen,22 which will need to be addressed if efficient and stable DSSCs are to be fabricated in the future.
Acknowledgements
GC and MC thank the EPSRC for funding (EP/E036244/1, EP/J500434/1). MC JH acknowledges a Small and Medium Business Administration (SMBA) grant (No. S212933) and the International Cooperative R&D program through Korea Institute for Advancement of Technology (KIAT) funded by the Ministry of Trade, Industry and Energy (MOTIE) of Korea. Ministry of Trade, Industry and Energy (MOTIE) of Korea. Open access data: http://dx.doi.org/10.5525/gla.researchdata.250
Notes and references
- B. O'Regan and M. Gratzel, Nature, 1991, 353, 737–740 CrossRef.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed.
- For representative reviews see:
(a) A. Mishra, M. K. R. Fischer and P. Bäuerle, Angew. Chem., Int. Ed., 2009, 48, 2474–2499 CrossRef CAS PubMed;
(b) Y. Ooyama and Y. Harima, ChemPhysChem, 2012, 13, 4032–4080 CrossRef CAS PubMed;
(c) R. K. Kanaparthi, J. Kandhadi and L. Giribabu, Tetrahedron, 2012, 68, 8383–8393 CrossRef CAS;
(d) B.-G. Kim, K. Chung and J. Kim, Chem.–Eur. J., 2013, 19, 5220–5230 CrossRef CAS PubMed.
- T. Higashino and H. Imahori, Dalton Trans., 2015, 44, 448–463 RSC.
- Ferrocenes: Ligands, Materials and Biomolecules, ed. P. Stepnicka, John Wiley and Sons Ltd., 2008 Search PubMed.
-
(a) T. Daeneke, T.-H. Kwon, A. B. Holmes, N. W. Duffy, U. J. Bach and L. Spiccia, Nat. Chem., 2011, 3, 211–215 CrossRef CAS PubMed;
(b) T. Daeneke, A. J. Mozer, T.-H. Kwon, W. Duffy, A. B. Holmes, N. U. Bach and L. Spiccia, Energy Environ. Sci., 2012, 5, 790–799 RSC;
(c) S. Sönmezoğlu, C. Akyürek and S. Akin, J. Phys. D: Appl. Phys., 2012, 45, 425101 CrossRef.
- R. Chauhan, M. Trivedi, L. Bahadur and A. Kumar, Chem.–Asian J., 2011, 6, 1525–1532 CrossRef CAS PubMed.
- D. Sirbu, C. Turta, A. C. Benniston, F. Abou-Chahine, H. Lemmetyinen, N. V. Tkachenko, C. Wood and E. Gibson, RSC Adv., 2014, 4, 22733–22742 RSC.
- M. Liang and J. Chen, Chem. Soc. Rev., 2013, 42, 3453–3488 RSC.
-
(a) C. Teng, X. Yang, C. Yang, H. Tian, S. Li, X. Wang, A. Hagfeldt and L. Sun, J. Phys. Chem. C, 2010, 114, 11305–11313 CrossRef CAS;
(b) J.-L. Song, P. Amaladass, S.-H. Wen, K. K. Pasunooti, A. Li, Y.-L. Yu, X. Wang, W.-Q. Deng and X.-W. Liu, New J. Chem., 2011, 35, 127–136 RSC.
- K. Pilgram, M. Zupman and R. Skiles, J. Heterocycl. Chem., 1970, 7, 629–633 CrossRef CAS.
- R. Misra, P. Gautam, T. Jadhav and S. M. Mobin, J. Org. Chem., 2013, 78, 4940–4948 CrossRef CAS PubMed.
- O. F. Mohammed and A. A. O. Sarhan, Chem. Phys., 2010, 372, 17–21 CrossRef CAS.
- J.-L. Bredas, Mater. Horiz., 2014, 1, 17–19 RSC.
- Y. Liao, B. E. Eichinger, K. A. Firestone, M. Haller, J. Luo, W. Kaminsky, J. B. Benedict, P. J. Reid, A. K.-Y. Jen, L. T. Dalton and B. H. Robinson, J. Am. Chem. Soc., 2005, 127, 2758–5766 CrossRef CAS PubMed.
- S.-I. Kato, M. Kivala, W. B. Schweizer, C. Boudon, J.-P. Gisselbrecht and F. Diederich, Chem.–Eur. J., 2009, 15, 8687–8691 CrossRef CAS PubMed.
- H. Tian, X. Jiang, Z. Yu, L. Kloo, A. Hagfeldt and L. Sun, Angew. Chem., Int. Ed., 2010, 49, 7328–7331 CrossRef CAS PubMed.
- Y. Liu, A. Hagfeldt, X.-R. Xiao and S.-E. Lindquist, Sol. Energy Mater. Sol. Cells, 1998, 55, 267–281 CrossRef CAS.
- S. Nakade, T. Kanzaki, W. Kubo, T. Kitamura, Y. Wada and S. Yanagida, J. Phy. Chem. B, 2005, 109, 3480–3487 CrossRef CAS PubMed.
- M. J. Frisch, et al., Gaussian 09, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- M. Al-Eid, S. Lim, K.-W. Park, B. Fitzpatrick, C.-H. Han, K. Kwak, J. Hong and G. Cooke, Dyes Pigm., 2014, 104, 197–203 CrossRef CAS.
- For examples see:
(a) M. Sato, T. Yamada and A. Nishimura, Chem. Lett., 1980, 925–926 CrossRef CAS;
(b) G. Zotti, G. Schiavon, S. Zecchin and D. Favretto, J. Electroanal. Chem., 1998, 456, 217–221 CrossRef CAS;
(c) J. P. Hurvois and C. Moinet, J. Organomet. Chem., 2005, 690, 1829–1839 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: NMR spectra of new compounds and cyclic voltammetry of the dyes. See DOI: 10.1039/c5ra21565j |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a