
Fabrication and performance evaluation of a novel membrane electrode assembly for DMFCs†
Abstract
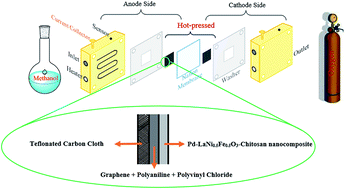
In the current study, a nano-scale perovskite LaNi0.5Fe0.5O3 (LaNi0.5Fe0.5O3NPs) was synthesized by a rapid ultrasonic-assisted co-precipitation method and characterized by various techniques. A modified glassy carbon electrode with Pd nanoparticles (PdNPs) and LaNi0.5Fe0.5O3NPs dispersed into chitosan (CH) polymer was prepared and its catalytic activity toward methanol oxidation was investigated. Based on the electrochemical studies, the PdNPs-LaNi0.5Fe0.5O3NPs-CH nanocomposite showed considerable activity for methanol oxidation in comparison to PdNPs-CH. Then a gas diffusion layer (GDL) was prepared with the ratio of 40% wt graphene carbon and 60% wt polymer mixture (consisting of 35% wt polyvinyl chloride and 65% wt polyaniline) for the micro-porous layer and a teflonated carbon cloth as the macro-porous substrate. A direct methanol fuel cell was designed, assembled and tested with the suggested PdNPs-LaNi0.5Fe0.5O3NPs-CH nanocomposite as anodic catalyst and the synthetic GDL under several different conditions.
 Please wait while we load your content...
Please wait while we load your content...

