DOI:
10.1039/C5RA16732A
(Paper)
RSC Adv., 2016,
6, 575-585
Supramolecular complexes involving non-symmetric viologen cations and hexacyanoferrate(II) anions. A spectroscopic, crystallographic and computational study†
Received
19th August 2015
, Accepted 14th December 2015
First published on 17th December 2015
Abstract
We have investigated spectrally, crystallographically as well as computationally the charge transfer complexes involving newly synthesized N-aryl-N′-methyl non-symmetric viologens (AMVs) and hexacyanoferrate(II) (HCF) anions. The supramolecular binding of AMVs and HCF was studied in solution and in the crystal state for one of the obtained complexes. Substituent effects on the electron affinities of the dicationic AMVs, determined using Mulliken's theory [R. S. Mulliken, J. Am. Chem. Soc., 1952, 74, 811–824] were quantified. The structure of one of the AMV//FeII(CN)6 pairs solved through Single-Crystal X-ray Diffraction (SCXRD), provided insights for the supramolecular binding of the anionic and cationic counterparts and the role of lattice water molecules. Supramolecular binding in solution, studied with the use of NMR spectroscopy, is in agreement with the results obtained in the solid state.
Introduction
Viologens are a well-known family of heterocyclic compounds with numerous applications in various research areas.1 Their strong electron accepting aptitude along with their ability to undergo one-electron reversible reductions, both chemically and electrochemically, resulting in relatively stable, intensely coluored radical-cations has attracted much interest for uses in printing and display technologies.2,3 Additionally 4,4′-bipyridines can serve as building blocks in photoactive molecular shuttles,4 electrochromic devices,3,5 biochemical sensors,6,7 small viologen-based medium responsive molecules,8 monoquat-ligated pentacyanoferrate(II) solvatochromic complexes9–12 as well as important components in photoconductive interlocked molecules.13 The use of viologen halide salts in photochromic applications has also attracted much interest.14 Charge transfer is a very important feature for the aforementioned photochemical activity of viologens, and it can also occur between the organic counterparts in viologen/electron donors charge transfer complexes.15–17 Utilization of hexacyanoferrate(II) anions and 4,4′-dipyridinium dications to build charge transfer complexes (CTCs) has attracted much attention as well. The first CTCs of that type were reported as early as 1963 by Nakahara and Wang, bearing the symmetric methylviologen18 (also known as paraquat). Ever since, several studies in solution19–21 as well as some crystallographic studies were reported.22–24 Noteworthy, the number of crystallographically analyzed CTCs with charged N-heterocycles remains small up to date. The interionic charge transfer (IICT) of the viologen/hexacyanoferrate(II) CTCs resembles the intervalence charge transfer (IVCT) of Prussian blue, responsible for its characteristic deep blue colour. IICT between the [FeII(CN)6]4− electron rich and the viologen electron deficient counterparts is of great interest in these complexes. The electronic spectra of such viologen-based CTCs can be readily affected by different factors, such as pressure,25 temperature,24,26 and light14,27 therefore opening new routes towards novel chromic systems. Hitherto, a big number of CTCs bearing variously substituted dipyridinium salts, such as various paraquats,14–27 lucigenin,28 and several diquats (diquaternized 2,2′-bipyridinium salts)29 acting as the electron deficient (cationic) parts of these CTCs, have been reported. However, to the best of our knowledge only CTCs containing symmetric viologens have been reported up to now. In this work we report the synthesis, in-solution, crystallographic and computational study of new CTCs involving hexacyanoferrate(II) and non-symmetric viologens. The effect of different substituents on the two nitrogen atoms of 4,4′-bipyridine on the 1H-NMR and UV-vis spectra of these new CTCs obtained in solution, in combination with the data collected crystallographically, are used in order to rationalize the structure–properties relation.
Results and discussion
Synthesis and characterization
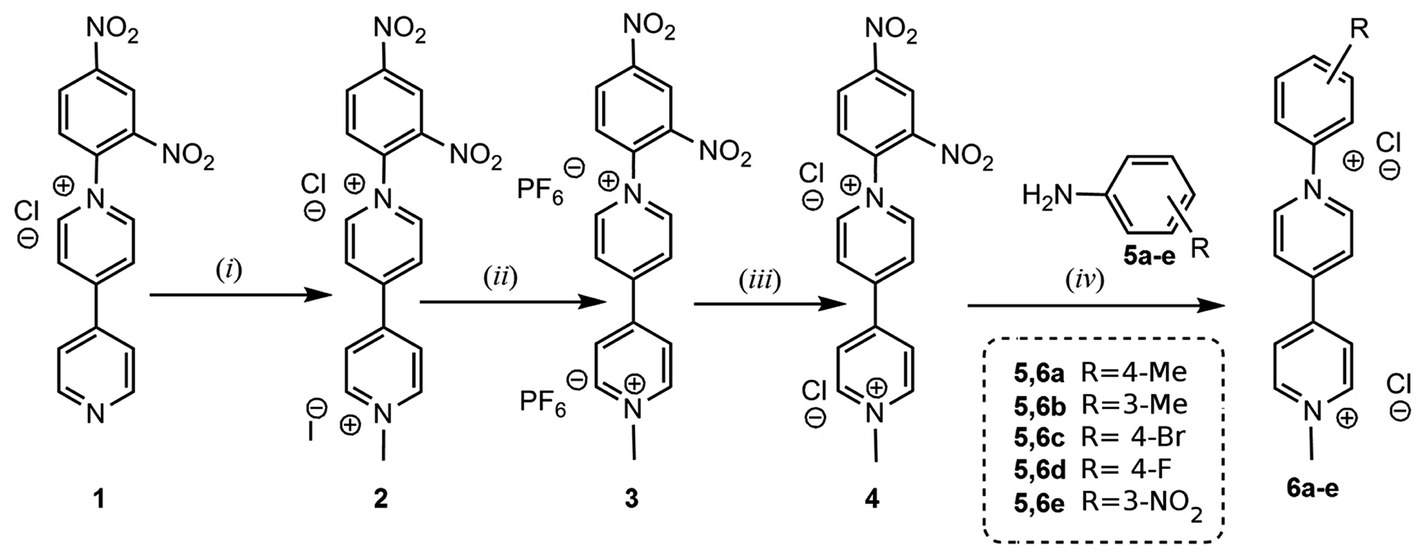
In this work a group of six non-symmetric viologens (N-aryl-N′-methyl viologens: AMVs) were synthesized and fully characterized using various spectroscopic techniques. The general synthetic route followed for the synthesis of viologens 4 and 6a–e is depicted in Scheme 1. The “monoquat” precursor 1 which was synthesized according to a previously described method,10 gave through quaternization of the second N atom, via a reaction with MeI, product 2 in high yield. The latter compound was purified through two sequential anion exchanges yielding viologen 4 as a dichloride salt. This compound was further used both for the study of its supramolecular complexation with FeII(CN)6, as well as for the synthesis of the group of AMVs 6a–e. The latter were prepared through Zincke reactions of 4 with suitable anilines in MeOH. The reaction times significantly depended on the aryl substituent, varying within 4–96 h, and the AMVs in all cases were isolated in yields higher than 68%.
 |
| | Scheme 1 Synthetic route followed for the preparation of viologens 4 and 6a–e. (i) Δ, MeI in EtOH(ii) Δ, aqueous NH4PF6. (iii) Excess of Et4N+Cl− in MeCN. (iv) Zincke reaction. Refluxing MeOH, 2,4-dinitraniline. | |
Compounds 4 and 6a–e were preferably isolated as dichloride salts, because of their higher solubility in water as compared to the mixed Cl−, I− salts. Solubility in water is very important for the formation of the AMV//HCF ion pairs.
The AMVs were characterized using standard analytical methods including 1H and 13C NMR spectroscopy, electrospray high resolution mass spectrometry (ESI HRMS) as well as UV-vis spectrophotometry. The ESI HRMS of all AMV chloride salts (4 and 6a–e) exhibited peaks corresponding to the radical cations (see ESI† data file). All measured m/z of [AMV–2Cl−] radical cation species are listed in the Experimental section.
On the structure of CTCs
Mulliken's theory accounts CTCs as resonance hybrids of non-polar and dative structures.30 Assuming the chemical equation of Scheme 2 in case of a CTC between an electron acceptor (A) and an electron donor (D) and Ψ0 and Ψ1 the wave functions describing the non-polar (D⋯A) and dative (D+⋯A−) resonance structures respectively, the wave functions describing the CTC in its electronic ground and excited state (ΨG and ΨE respectively) will be given by the equations shown in Scheme 2. Absorption of light and photoexcitation of the CTC facilitates the charge transfer from D to A, consequently rendering the dative resonance structure more important than the non-polar resonance structure in the excited state. The opposite applies to the electronic ground state. In other words in the electronic ground state it will be: |a| > |b| whereas in the excited state |a*| < |b*|, where a and a* correspond to the coefficients of the wave function Ψ0 in the ground and excited state respectively whereas b and b* correspond to the coefficients of the wave function Ψ1 in the ground and excited state respectively (Scheme 2).
 |
| | Scheme 2 | |
From the many theories on CTCs, Mulliken's theory is probably the most applicable in a wide range of CTCs.30,31 This theory provides a relation of the charge transfer energy (ECT) of a CTC with the electron affinity of the electron withdrawing part (EA) and the ionization potential of the electron donating part (ID) of the CTC, according to the following equation:
In this equation Δ corresponds to the difference between the sum of van der Waals, exchange, repulsion, as well as CT resonance energies, and the excited and ground states of the charge transferred.
Formation of the CTCs in solution
The formation of a CTC of the type AMV//HCF is typically followed by intense colouration (green to blue). Even though the aqueous solutions of an AMV salt and K4FeII(CN)6 are yellow, upon mixing these two solutions, a deep coloured solution is obtained. The appearance of weak visible bands in the electronic spectra of aqueous equimolar mixtures of AMV and FeII(CN)6 is attributed to the transition: d(t2g6)p(π0)* → d(t2g5)p(π1)*. As shown in Fig. 1 the energy of the aforementioned transition (ECT) in water is highly affected by the electron donating or withdrawing nature of the phenyl substituent of the AMVs. A bathochromic shift of the charge transfer visible band maximum, of approx. 60 nm (ΔECT = 0.193 eV) was observed when going from the electron donating group (m-tolyl) to the strong electron accepting DNPh (Table 1). This variation of the charge transfer energy is clearly connected to the tunable electron accepting ability of the AMVs which is strongly dependent on the nature of the phenyl substituent. Benniston et al.32 have recently indicated the importance of the torsion angles around the pyridinium rings of some viologens. Herein, focus will be placed on the substituent effects, which are according to our spectroscopic results the most dominant. As anticipated, increasing the electron accepting character of the aforementioned substituent renders viologen dications more prone to reduction by hexacyanoferrate(II) anions (see ESI† different resonance structures for DNPh). The Hammett plots of the charge transfer energies (ECT) depicted in Fig. 2 shows a very good correlation between ECT and σ. A ρ value of −0.115 (±0.028) eV is obtained through linear regression. The negative ρ value reflects the bathochromic effect observed while increasing the electron withdrawing character of the aryl substituent of the AMVs.
 |
| | Fig. 1 The visible charge transfer band observed in aqueous solutions of 4,6a–e containing [FeII(CN)6]4− at equal concentrations (10 mM). Blue arrow indicates bathochromism while going from electron donating to electron withdrawing substituents. | |
Table 1 Chemical shifts observed for the AMVs 4, 6a–e when codissolved in D2O with K4[Fe(CN)6] in equimolar quantities (assignment of protons according to Fig. 4)
| Compound |
Δδaa (ppm) |
Δδba (ppm) |
Δδca (ppm) |
Δδda (ppm) |
ΔδMea,b (ppm) |
| Δδ are determined as the difference: (δAMV//FC − δAMV) where δAMV//FC corresponds to the chemical shift of a certain type of protons of AMV2+ in the presence of [FeII(CN)6]4− and δAMV corresponds to the chemical shift of the same type of protons of the AMV2+ cation with counter anion Cl−. All measurements were performed in D2O at 25 °C. For the determination of δAMV: [AMVCl2] = 38 mM and for δAMV//FC [AMVCl2] = [K4[Fe(CN)6]] = 38 mM. ΔδMe corresponds to the Δδ of the Me protons attached directly to a nitrogen atom of an AMV. |
| 6a |
0.027 |
0.099 |
0.135 |
0.135 |
0.069 |
| 6b |
0.037 |
0.088 |
0.126 |
0.130 |
0.054 |
| 6c |
0.040 |
0.083 |
0.122 |
0.122 |
0.047 |
| 6d |
0.042 |
0.072 |
0.121 |
0.120 |
0.038 |
| 6e |
0.051 |
0.086 |
0.143 |
0.141 |
0.045 |
| 4 |
0.078 |
0.067 |
0.183 |
0.153 |
0.061 |
 |
| | Fig. 2 Hammett plot of the experimentally determined charge transfer energies (ECT) of the CTCs between the viologen cations 4, 6a–e (AMVs) and hexacyanoferrate(II) anions. ECT were determined in water solutions at 25 °C with [AMV] = [FeIICN6] = 10 mM. Relation obtained through linear regression: ECT = −0.115 (±0.028)σ + 2.129 (±0.018); R = 0.900. | |
In addition to the emergence of the characteristic visible IICT band upon mixing AMVs with HCF anions in water, significant changes in the 1H-NMR spectra of AMVs are also observed upon complexation. This effect was first reported by Haque, Coshow and Johnson, who noticed that because of the electrostatic interactions between methylviologen dications and HCF anions in solution, significant downfield shifts were observed on the proton signals of the aromatic rings of 4,4′-bipyridyl.33 As long as these interactions are electrostatic, they should be drastically affected by the distance between anions and cations. Hence, for a symmetric viologen cation (such as methylviologen) there should be an equal electrostatic effect on both pyridine rings of 4,4′-bipyridyl. However, for non-symmetric viologens (e.g. the AMVs studied in this work) the effect of the FeII(CN)6 anions on the two rings of 4,4′-bipyridyl, is expected to be different, due to different charge distribution along the 4,4′-bipyridyl backbone. Consequently, 1H-NMR spectroscopy becomes an important tool for understanding the local effect of the ion pairing between the AMV cations and HCF anions in solution (dependence of Δδ on Hammett parameter can be found in ESI†).
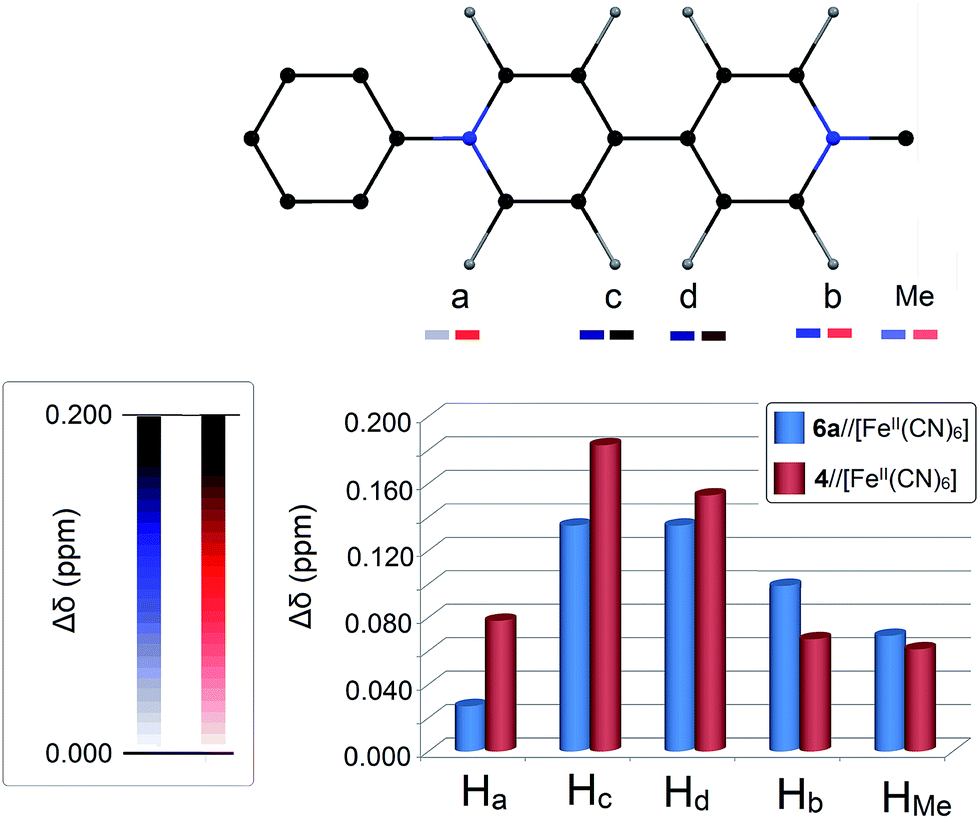
As listed in Table 1, the extent of downfield shifts of the proton signals (both aromatic and methyl protons) induced upon mixing AMV chlorides with K4FeII(CN)6 in equimolar quantities in D2O strongly depends on the nature of the aryl substituents of the AMVs. For the CTC 42+//[FeII(CN)6]4− for instance, all proton signals are downfield shifted when compared to the ones obtained for the dichloride salt of 42+ (Fig. 3 and 4).
 |
| | Fig. 3 1H-NMR spectra of (i) 4 in D2O and (ii) of the CTC: 4//FeII(CN)6 formed in D2O at 25 °C; [42+] = [(FeII(CN)6)4−] = 38 mM. | |
 |
| | Fig. 4 Chart showing the 1H-NMR shifts (Δδ) for 4 and 6a, after mixing these AMVs with K4[Fe(CN)6] in equimolar quantities ([AMV] = [K4[Fe(CN)6]] = 38 mM) in D2O at 25 °C (Δδ values for all AMVs are listed in Table 1). Insets: assignment of 4,4′-bipyridine protons used in this work (top) and colour-scale of Δδ (left); the darker the colour, the higher the Δδ observed. | |
Determination of electron affinities for 4 and 6a–e
According to the theory of Mulliken describing CTCs it is also possible to determine the EA of the AMVs. Eqn (1) connects EA and ECT. In this equation using the value of ID = 3.50 eV obtained by Monk and coworkers34 and letting only EA vary, it is possible to calculate EA according to a previously described methodology.31,34 The values of EA obtained for the AMVs through that procedure are listed in Table 2. The characteristic equation ECT vs. EA (see Fig. 5) obtained for the CTCs involving AMVs and ferrocyanide(II) anions, is the following:
| ECT (eV) = −[0.956 (±0.161)]EA + 3.604 (±0.256); R = 0.947; RSS = 0.003. |
Table 2 Charge transfer energies (ECT) and maxima charge transfer wavelengths (λmax) for CTCs between the 4, 6a–e cations and [FeII(CN)6]4− along with electron affinities (EA), calculated LUMO energies (ELUMO) and HOMO–LUMO energy gaps (EGAP) of 4 and 6a–e
| Viologen |
Substituent |
σx |
λmaxb (nm) |
ECTc (eV) |
EAd (eV) |
ELUMOe (eV) |
EGAPf (eV) |
| For 6b and 6e the σm values were used. For 4, the σ value 1.420 corresponds to the Σ(σp(NO2)). Determined from the first derivatives of Gaussian fitted curves. d(t2g6)p(π0)* → d(t2g5)p(π1)* transition energies. Determination of EA of 4 and 6a–e was based on Mulliken's theory on charge transfer complexes. PCM(H2O)-B3LYP 6-311G(d,p) calculated LUMO energies (for details see ESI). HOMO–LUMO gap based on the calculated HOMO and LUMO energies of 4 and 6a–e by PCM(H2O)-B3LYP 6-311G(d,p) (for details see ESI). |
| 6a |
4-Me |
−0.170 |
574.4 |
2.158(5) |
1.519(9) |
−3.805(8) |
3.671(1) |
| 6b |
3-Me |
−0.069a |
570.6 |
2.172(8) |
1.537(3) |
−3.821(3) |
3.650(7) |
| 6c |
4-F |
0.062 |
588.1 |
2.108(2) |
1.539(9) |
−3.846(6) |
3.854(5) |
| 6d |
4-Br |
0.232 |
606.7 |
2.043(6) |
1.604(0) |
−3.910(5) |
3.544(8) |
| 6e |
3-NO2 |
0.710a |
598.3 |
2.072(3) |
1.596(2) |
−3.937(8) |
4.381(3) |
| 4 |
2,4-DiNO2 |
1.420a |
630.7 |
1.965(8) |
1.727(2) |
−3.942(4) |
4.853(4) |
 |
| | Fig. 5 Plot of the experimentally determined charge transfer energies vs. the electron affinities of the AMVs as determined through Mulliken's theory. | |
This characteristic equation for ferrocyanide(II) anions connects the charge transfer energy which is the energy needed for an electron to be transferred from the iron(II) center to an electron deficient AMV dication with the electron affinity of the AMV. The negative slope obtained indicates that the charge transfer becomes more favorable when going to viologens with higher electron affinity, as anticipated.
We have further validated our results by extrapolation of the obtained linear relation (EA vs. ECT) to methylviologen. The CTC of the aforementioned symmetric viologen with hexacyanoferrate(II) exhibits an ECT value of 2.36 eV (525 nm).34 Thus the value EA = 1.35 eV is obtained for methylviologen which is in good agreement with the reported value 1.24 eV by White.34,35 As anticipated the electron affinity of methylviologen is significantly lower than the obtained EA values of the AMVs studied in this work, apparently as a result of the electron withdrawing character of the substituted phenyl group on one of the two quaternized nitrogens of AMVs. Even with electron donating substituents such as pMe on the phenyl ring (the case of AMV 6a) the electron affinity of the AMV (∼1.52 eV) is approx. 23% higher than that for methylviologen determined by White.35
Moreover, EA strongly depends on the electron donating or accepting nature of the aryl substituent (ρ = 0.121 ± 0.018 eV; R = 0.957. Plot is found in ESI†).
We furthermore calculated the LUMO and HOMO energies for the dications of the viologens 4, 6a–e within Density Functional Theory (DFT). DFT is a very convenient computational tool which has recently been employed in case of viologen/iodide ion pairs, resulting in a very good agreement between experiments and computations.36 Herein, we have calculated the LUMO and HOMO energies for all AMV-dications, employing functionals including dispersion effects (see Computational details section as well as the ESI†) and we have found that EA correlates very well with the calculated LUMO energies (Fig. 6A).37 ELUMO can serve as a good measure of the electron accepting aptitude of a viologen, reflecting its propensity to undergo one-electron reductions. For the viologens studied here, the electron accepting aptitude follows the sequence 4 > 6e > 6d > 6c > 6b > 6a (see Table 2 and Fig. 7) and varies linearly with the Hammett parameter σ (see the ESI†).
 |
| | Fig. 6 Plots showing the linear correlation of the charge transfer energies of 4 and 6a–e with (A) the B3LYP 6-311G(d,p) LUMO energies and (B) the calculated HOMO–LUMO energy gap for 4 and 6a–e at B3LYP 6-311G(d,p). | |
 |
| | Fig. 7 Representations of LUMOs for the dications of the viologens 4 and 6a–e calculated at PCM(H2O)-B3LYP 6-311G(d,p). | |
We have furthermore observed a good correlation between the calculated HOMO–LUMO energy gaps (EGAP) and the electron affinities of 4 and 6a–e (see Fig. 6B). EGAP reflects the inherent capacity of a compound to undergo intramolecular charge transfer. In case of the viologens studied here, it further indicates that intramolecular charge transfer would be energetically more feasible with electron donating substituents on the phenyl ring attached to one of the quaternized nitrogen atoms of the viologen. The tunable character of EGAP is reflected by the very good correlation of the calculated EGAP with the Hammett σ parameter (see ESI†). Now, an increased intramolecular CT aptitude should be connected to a decreased intermolecular CT capacity as long as the latter is associated with the reduced electron accepting character of the 4,4′-bipyridine backbone which is more electron rich when electron donor substituents are present. This is why a negative slope is obtained for the linear correlation ECT vs. EGAP (Fig. 6B).
Crystal structure of complex 7
The CTC 7 was formed in water by mixing (6a2+)Cl2 and K4FeII(CN)6 (see experimental details). 7 was self-precipitated shortly after mixing the solutions of 6a2+ and [FeII(CN)6]4− as a greenish-blue powder, sparingly soluble in cool water but soluble in warm water and several polar organic solvents (e.g. DMSO). 7 was recrystallized from water by heating an aqueous suspension of the obtained green powder of 7 (not exceeding 70 °C). When 7 is fully dissolved in water, the solution has a deep blue colour. After letting the solution slowly cool down to r.t. rhombic crystals of 7 were formed which were suitable for SCXRD (Fig. 8). Unfortunately, in our hands the rest of the ion pairs of 4 and 6b–e with [FeII(CN)6]4− formed in-solution did not give crystals. Especially the CTC 42+//[FeII(CN)6]4− was surprisingly soluble in cool water, presumably because of the strong hydrogen bonding interactions with water molecules.
 |
| | Fig. 8 Crystals of 7 as viewed by scanning electron microscopy (top) and optical microscopy (bottom). | |
The structure of 7 was solved by SCXRD (see experimental section and Table 3) and it was shown that it corresponds to: {(6a2+)4[FeII(CN)6]4-2}·8.75H2O (Fig. 9). The asymmetric unit of 7 is composed of two [FeII(CN)6]4− anions, four viologen cations 6a2+ and disordered water molecules. Among the viologen cations, three of them are in general positions and two 6a2+ moieties are lying on crystallographic two-fold axis, thus accounting for a half unit each. Several interesting features can be highlighted with regard to the packing of the structure (Fig. 9 and 10).
Table 3 Crystallographic data for 7
| w = 1/[σ2(Fo2) + (0.1892P)2 + 10.293P] where P = (Fo2 + 2Fc2)/3. |
| Formula |
C84H89.5Fe2N2008.75 |
| Mw |
1630.95 |
| Crystal system |
Orthorhombic |
| Measurement temperature/K |
293 |
| Space group |
A ea2 |
| a/Å |
14.6547(5) |
| b/Å |
34.5808(13) |
| c/Å |
35.0669(11) |
| V/Å3 |
17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 770.9(11) 770.9(11) |
| Z |
8 |
| Dc/g cm−3 |
1.219 |
| Crystal colour |
Green |
| Crystal size/mm3 |
0.3 × 0.2 × 0.04 |
| μ(Mo-Kα)/mm−1 |
0.39 |
| No. of refl. measured |
87803 |
| No. of unique refl. |
15088 |
| No. of observed refl.[F2 > 4σF2] |
11405 |
| No. parameters refined |
1107 |
| R1 [F2 > 4σF2] |
0.0875 |
| wR1 [F2 > 4σF2] |
0.2420a |
| R2 [all refl.] |
0.1107 |
| wR2 [all refl.] |
0.2694 |
| Goodness of fit [all refl.] |
1.038 |
| Residual Fourier/e Å−3 |
−0.942; 0.897 |
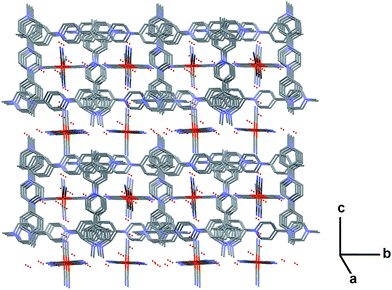 |
| | Fig. 9 View of the crystal packing of 7 along a axis showing the alternance of channels of AMV 6a cations embedding FeII(CN)6 and water, and the “open” FeII(CN)6 – water sheets along b. Water molecules are represented as red small spheres. Atom colours: Fe: orange; C: grey; N: blue; O: red (all H atoms have been omitted for clarity). | |
 |
| | Fig. 10 The crystal structure of the supramolecular complex {(6a2+)4[FeII(CN)6]4-2}·8.75H2O (7) as viewed along the axis (A) a, (B) b and (C) c. Atom colours: Fe: orange; C: black; N: blue; H: grey (all lattice water molecules have been omitted for clarity). | |
The viologen cations pack in alternate orientation of 90° along crystallographic axis a (Fig. 11). They thus form successive layers of “closed tubular channels” with square section embedding ferrocyanide and water, and “open sheets” of ferrocyanide and water along crystallographic axis b (Fig. 11). The structure of 7 also reveals the presence of hydrogen bonds, some of them being involved into a strong infinite 3-dimensional network along a, b and c of H-bonds connecting together the FeII(CN)6 anions through water molecules (Fig. 11), and thus linking the “closed channels” together as well as with the “open sheet”.
 |
| | Fig. 11 Infinite zigzag chain consisting of hydrogen bonded H2O molecules and [FeII(CN)6]4− anions viewed along the axis c. | |
Interestingly despite the fact that the viologen moieties are not planar (the angles between their cyclic units range from 25° to 40°) their terminal aromatic rings stack parallel to each other along axis a and form an infinite network of π–π interactions: the dihedral angles between the terminal rings range from 1.8 to 8.3° and the perpendicular distances range from 3.41 to 3.69 Å. These moieties therefore build-up the edge of the closed channels.
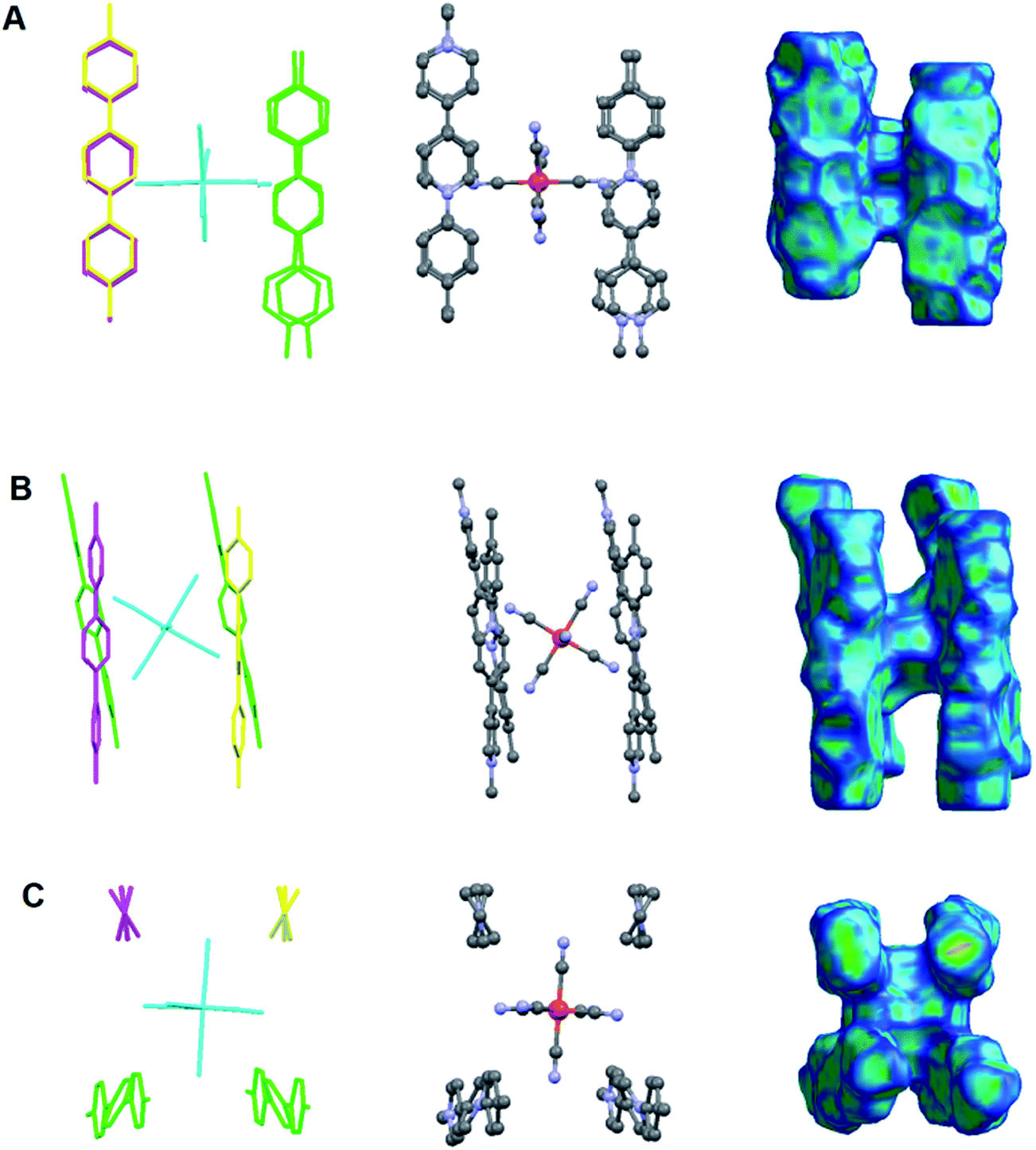
Moreover, viologen/[FeII(CN)6]4− clusters can be also observed. As shown in Fig. 12, four 6a2+ cations can enclose a [FeII(CN)6]4− anion thus forming a charged cluster. Hirshfeld surface reveals the packing of a hexacyanoferrate(II) anion and the four 6a2+ dications in such a cluster (Fig. 12, right column).
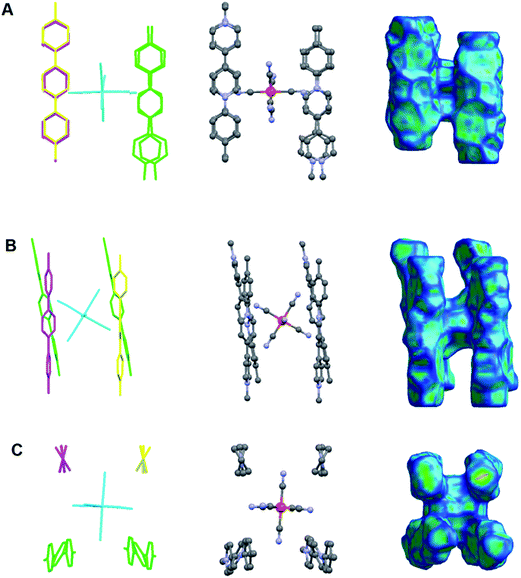 |
| | Fig. 12 A hexacyanoferrate(II) anion surrounded by four viologen cations: 6a2+ forming a charged cluster of the type: [(6a)4FeII(CN)6]4+ observed in the crystal structure of the CTC 7 (A) along axis a, (B) along axis b and (C) along axis c. Left column: capped stick model of the cluster with different colours indicating viologen cations of different symmetries (H atoms are omitted for clarity); middle column: ball and stick model of the cluster (H atoms are omitted for clarity); right column: Hirshfeld surface38 of the cluster mapped with curvedness.39,40 | |
What is interesting is that the distance between the iron center and the centroids of the pyridine rings of 6a2+ is not equal. The ring which is closer to FeII is the aryl substituted with a distance of 5.6–5.7 Å. The corresponding distance for the methyl substituted pyridine ring is 6.4–6.6 Å (ESI†). This finding is consistent with what is observed in solution according to the 1H-NMR study. In all cases of CTCs downfield shifts were greater for the ortho-protons of the aryl-substituted pyridine rings. In other words both in solution and in a crystal, hexacyanoferrate(II) shows some preference to lie closer to the aryl-substituted nitrogen atom of the viologen, and that is an effect caused by the non-symmetric structure of the AMVs. This is not observed in case of CTCs of symmetric viologens reported so far, and reveals the role of the viologen substitution on the charge transfer.
Concluding remarks
The supramolecular binding of a group of synthesized aryl-methyl substituted viologens with hexacyanoferrate anions was studied in solution and in the solid state. 1H-NMR studies showed that the ferrocyanide(II) anions preferentially lie closer to the aryl-substituted pyridine ring of the viologens in all cases. The structure of one of the isolated CTCs was solved and it was shown that the same trend also exists in the solid crystal state. Through the determination of electron affinities of the viologen cations, it was concluded that the aryl substituent readily affects the electron withdrawing aptitude of the viologens and furthermore their propensity to form CTCs with hexacyanoferrate anions. Moreover, the linear dependence of the electron affinities of the viologen cations and the charge transfer energies is in agreement with Mulliken's theory on CTCs. Of high importance is the stabilizing role of hydrogen bonding between water molecules and hexacyanoferrates in 3-dimensional polymeric chains observed in the structure of one of the CTCs, solved through SCXRD. Such information is of high significance as it can help on better understanding solvent–ion interactions in solution.
Computational details
All calculations were performed using GAUSSIAN 0941 program package within the DFT framework. The hybrid functional B3LYP42 was employed in combination with two polarized triple-zeta split-valence basis sets from Pople 6-311G(d,p) and Ahlrichs def2-TZVP. As charge transfer processes usually involve long range interactions, we decided to employ two other functionals including dispersion effects. For that purpose, the WB97XD43 and CAM-B3LYP44 were used with the same basis set as employed before. All geometry optimizations were performed with the default optimization threshold of 3 × 10−4 for the RMS Force criterion. Since all spectroscopic data were collected in water media, the Polarizable Continuum Model (PCM) was used for all calculations (ε = 78.355300). Frequency calculations were systematically carried out to ensure that the resulting structures converged to a local minimum on the potential energy surface.
Experimental section
Physical measurements
NMR spectra were obtained using a Varian Gemini 300 spectrometer (300 MHz 1H, 75 MHz 13C). Both 1H and 13C NMR spectra were recorded in deuterium oxide (D2O) at 25 ± 1 °C (compound 7 studied at 50 ± 1 °C due to low solubility at r.t.). For the 13C NMR measurements, a few drops of [D6]DMSO were added in solutions of the AMV chlorides in D2O and the signal of [D6]DMSO was used as a reference (39.39 ppm).45 The 1H NMR spectra were calibrated by using residual undeuterated solvent as an internal reference (4.79 ppm).45 For the 1H NMR spectroscopic study of CTCs, solutions containing AMV cations and hexacyanoferrate anions both in concentrations of 38 mM were used. IR spectra were recorded on a Perkin Elmer Spectrum 1 FTIR spectrophotometer in the solid state (without any preparation of the samples) using the attenuated total reflectance technique (ATR) in the wavenumber region 600–4000 cm−1. UV-visible spectra were recorded using a Varian CARY 1E UV-visible spectrophotometer at 25 ± 1 °C. The concentrations of the solutions used were 10 mM and they were prepared right before each measurement. For the UV-vis spectroscopic study of CTCs, solutions containing AMV cations and hexacyanoferrate anions both in concentrations of 10 mM were used. SEM images and EDX analyses were obtained using a Quanta 200 FEI Scanning Electron Microscope. Single-crystal XRD analyses were performed on an Agilent SuperNova diffractometer at 293 K at the MoKα radiation (λ = 0.71073 Å). Data collection reduction and multiscan ABSPACK correction were performed with CrysAlisPro (Agilent Technologies). The structures were solved by direct methods with SHELXS46 and SHELXL46 was used for full matrix least squares refinement. All H-atoms were introduced at idealized positions and their coordinates and Uiso parameters were constraint to 1.2 U eq (parent atom) for the aromatics and 1.5 U eq (parent atoms) for the methyls and waters. Electrospray ionization (ESI) HRMS spectra were obtained on a Waters, Inc. Q-TOF Premier Mass Spectrometer (the HRMS facility of Iowa University, USA). All samples 4 and 6a–e were dissolved in water, whereas 7 was dissolved in DMSO for the HRMS measurements. The reported m/z values correspond to the average of three repetitions per sample. The elemental analyses were performed on a Thermo Finnigan elemental analyzer EA 1112 (Spectropole, Aix-Marseille Université, Marseille, France). The results are provided with an absolute accuracy of ±0.3% and are validated for two trials (the reported results correspond to the average of two trials in any case).
Materials and methods
Chloro-2,4-dinitrobenzene, 4,4′-bipyridine, and the substituted anilines (5a–e) were purchased by ACROS Organics. Potassium ferrocyanide trihydrate and methyl iodide were purchased by FLUKA. All solvents used were purified according to literature47 before use. Water was purified with a Barnstead EASYpure RF compact ultrapure water system and then distilled twice. Compound 1 was prepared according to a method already described in the past.10
Synthesis of [1-(2,4-dinitrophenyl)-1′-methyl-4,4′-bipyridine-1,1′-diium chloride iodide] (2). In a 100 mL round bottom flask 1.00 g of the compound 1 (2.79 mmol) was dissolved as possible in 30 mL of absolute EtOH (with mild heating) and methyl iodide (2 mL, 32.1 mmol) was added to the resulting solution. The flask was equipped with a CaCl2 tube and the solution was stirred at r.t. overnight. The precipitate was filtered off with suction, and then washed several times with absolute EtOH and diethyl ether. Finally the red solid was dried in vacuo for several hours. Red powder, 1.38 g (2.75 mmol) 98%. This compound was further transformed to compound 3 by ion exchange.
Synthesis of [1-(2,4-dinitrophenyl)-1′-methyl-4,4′-bipyridine-1,1′-diium hexafluorophosphate] (3). 1.20 g of the compound 2 (2.4 mmol) was dissolved in a mixture of 15 mL of H2O and 15 mL of MeOH, with mild heating and sonication. To the warm solution an aqueous solution of NH4PF6 (3.60 g: 22 mmol in 5 mL) was added. Immediately a shiny yellow solid precipitated. The solid was filtered off with suction and then it was washed with warm H2O, EtOH, and finally diethylether. Finally the solid was dried for several hours under reduced pressure. Yellow powder 0.980 g (1.56 mmol) 65%. Compound 3 was used directly for the next step without any purification.
Synthesis of [1-(2,4-dinitrophenyl)-1′-methyl-4,4′-bipyridine-1,1′-diium chloride] (4). The synthesis of this compound has been reported before.48,49 0.980 g (1.56 mmol) of the compound 3 were easily dissolved in 6 mL of dry acetonitrile. To this solution, a solution of 4.20 g tetraethylammonium chloride (25.3 mmol) in 12 mL dry acetonitrile, was added. Immediately a yellowish precipitate was formed, which was filtered by suction, washed with dry acetonitrile and diethylether. Caution was taken during the whole procedure since compound (4) is hygroscopic and forms sticky solids when exposed to moisture. The solid was dried in dessicator in vacuo over fresh P2O5 overnight (for the needs of synthesis of 6a–e, two batches of 4 were prepared according to the above procedure). Yellow powder, 0.590 g, 92.5% (1.44 mmol); 1H NMR (300 MHz, [D6]DMSO): δ = 9.70 (d, J = 5.7 Hz, 2H; C5H5N), 9.35 (d, J = 5.1 Hz, 2H; C5H5N), 9.18 (s, 1H, DNPh), 9.07 (m, 3H), 8.88 (d, J = 5.1 Hz, 2H; C5H5N), 8.43 (d, J = 8.7 Hz, 1H; DNPh), 4.48 (s, 3H, Me); 13C NMR (75 MHz, [D6]DMSO): δ = 151.45, 149.39, 147.70, 147.21, 146.84, 143.01, 138.36, 131.96, 130.33, 126.49, 126.44, 121.55, 48.19 (Me). HRMS-ESI (m/z): [M − 2Cl−]+, calcd for C17H14N4O4, 338.1015; found, 338.1003.
General procedure for the synthesis of 6a–e. 0.200 g of the compound 4 (0.488 mmol) were dissolved in MeOH (5 mL). To this solution a 4:1 excess of the appropriate substituted aniline (5a–e) was added. The solution then became violet, in all cases. The solution was refluxed for a period varying from 4 to 96 h and then was cooled and 30 mL of H2O were added. 2,4-Dinitroaniline was this way precipitated which was filtered, and the filtrate was condensed to dryness using a rotary evaporator. The solid was dissolved in 3 mL of MeOH and reprecipitated by adding diethyl ether, and it was collected by in vacuo filtration. The solids were washed with EtOH and diethyl ether, and dried in vacuo for several hours.
[1-(4-Methylphenyl)-1′-methyl-4,4′-bipyridine-1,1′-diium chloride] (6a). The synthesis of this compound has been reported before.50 Yellow powder, 4 h reflux, 123 mg (0.369 mmol) 76%; 1H NMR (300 MHz, D2O): δ = 9.36 (d, J = 6.0 Hz, 2H; C5H5N), 9.10 (d, J = 6.3 Hz, 2H; C5H5N), 8.71 (d, J = 6.3 Hz, 2H; C5H5N), 8.62 (d, J = 6.0 Hz, 2H; C5H5N), 7.72 (d, J = 7.5 Hz, 2H; Ph), 7.60 (d, J = 7.5 Hz, 2H; Ph), 4.53 (s, 3H, Me), 2.50 (s, 3H, Me-Ph); 13C NMR (75 MHz, D2O/[D6]DMSO): δ = 149.90, 149.11, 145.78, 144.68, 142.59, 139.34, 130.48, 126.34, 126.20, 123.18, 47.83 (Me), 19.79 (Me-Ph); UV-vis (H2O) λnm(logεmax): 314(3.795). HRMS-ESI (m/z): [M − 2Cl−]+, calcd for C18H18N2, 262.1470; found, 262.1461.
[1-(3-Methylphenyl)-1′-methyl-4,4′-bipyridine-1,1′-diium chloride] (6b). Pink coloured powder, 4 h reflux, 120 mg (0.360 mmol) 74%; 1H NMR (300 MHz, D2O): δ = 9.39 (d, J = 6.9 Hz, 2H; C5H5N), 9.13 (d, J = 6.6 Hz, 2H; C5H5N), 8.74 (d, J = 6.6 Hz, 2H; C5H5N), 8.64 (d, J = 6.6 Hz, 2H; C5H5N), 7.69 (m, 4H, Ph), 4.56 (s, 3H, Me), 2.54 (s, 3H, m-Me-Ph); 13C NMR (75 MHz, D2O/[D6]DMSO): δ = 149.28, 147.72, 146.82, 145.80, 142.22, 140.22, 132.12, 130.08, 126.55, 126.36, 125.16, 121.87, 47.99 (Me), 20.86 (m-Me-Ph); UV-vis (H2O) λnm(logεmax): 316(3.786). HRMS-ESI (m/z): [M − 2Cl−]+, calcd for C18H18N2, 262.1470; found, 262.1463.
[1-(4-Fluorophenyl)-1′-methyl-4,4′-bipyridine-1,1′-diium chloride] (6c). Yellow powder, 16 h reflux, 123 mg (0.364 mmol) 75%; 1H NMR (300 MHz, D2O): δ = 9.41 (d, J = 6.6 Hz, 2H; C5H5N), 9.14 (d, J = 5.7 Hz, 2H; C5H5N), 8.76 (d, J = 6.3 Hz, 2H; C5H5N), 8.65 (d, J = 6.3 Hz, 2H; C5H5N), 7.902 (m, 2H; Ph), 7.55 (m, 2H; Ph), 4.57 (s, 3H, Me); 13C NMR (75 MHz, D2O/[D6]DMSO): δ = 152.29, 150.97, 147.84, 146.98, 128.41, 128.24, 128.08, 127.95, 119.28, 118.97, 49.86 (Me); UV-vis (H2O) λnm(logεmax): 316(3.812). HRMS-ESI (m/z): [M − 2Cl−]+, calcd for C17H15N2F, 266.1219; found, 266.1221.
[1-(4-Bromophenyl)-1′-methyl-4,4′-bipyridine-1,1′-diium chloride] (6d). Yellow powder, 24 h reflux, 133 mg (0.334 mmol) 68%; 1H NMR (300 MHz, D2O): δ = 9.38 (d, J = 6.0 Hz, 2H; C5H5N), 9.09 (d, J = 5.7 Hz, 2H; C5H5N), 8.73 (d, J = 5.4 Hz, 2H; C5H5N), 8.61 (d, J = 5.1 Hz, 2H; C5H5N), 7.96 (d, J = 8.1 Hz, 2H; Ph), 7.75 (d, J = 7.8 Hz, 2H; Ph), 4.52 (s, 3H, Me); 13C NMR (75 MHz, D2O/[D6]DMSO): δ = 150.55, 148.84, 146.98, 145.97, 141.81, 133.84, 127.19, 127.21, 126.93, 125.82, 48.70 (Me); UV-vis (H2O) λnm(logεmax): 322(3.781). HRMS-ESI (m/z): [M − 2Cl−]+, calcd for C17H15N2Br, 326.0409.; found, 326.0419.
[1-(3-Nitrophenyl)-1′-methyl-4,4′-bipyridine-1,1′-diium chloride] (6e). Yellow powder, 96 h reflux, 125 mg (0.343 mmol) 70%; 1H NMR (300 MHz, D2O): δ = 9.53 (d, J = 6.9 Hz, 2H; C5H5N), 9.15 (d, J = 6.0 Hz, 2H; C5H5N), 8.84 (d, J = 6.9 Hz, 2H; C5H5N), 8.72 (s, 1H, Ph), 8.68 (d, J = 6.6 Hz, 2H; C5H5N), 8.33 (d, J = 8.1 Hz, 1H, Ph), 8.08 (t, J = 8.1 Hz, 1H; Ph); 13C NMR (75 MHz, D2O/[D6]DMSO): δ = 153.32, 150.86, 150.17, 147.94, 147.20, 143.90, 133.60, 132.16, 128.71, 128.39, 128.24, 121.63, 49.95 (Me); UV-vis (H2O) λnm(logεmax): 313(3.698). HRMS-ESI (m/z): [M − 2Cl−]+, calcd for C17H15N3O2, 293.1164; found, 293.1154.
Di-(1-methyl-1′-p-tolyl-4,4′-bipyridine-1,1′-diium)(hexacyanoferrate(II)) (7). In an aqueous solution of 6a (70 mg: 0.210 mmol in 400 μL H2O), an aqueous solution of potassium ferrocyanide containing half the equimolar quantity of [FeII(CN)6]·3H2O (36 mg: 0.085 mmol in 400 μL H2O) was added. The formed solution was left overnight in an open vessel resulting in a green-blue semicrystalline solid. Alternatively DMSO was added (approx. 1 mL) resulting again in a green-blue semicrystalline solid. In any case, the solid was filtered by suction and washed carefully with small quantities of H2O, EtOH and Et2O. Finally the solid was dried under reduced pressure. The obtained solid was purified with recrystallization from hot water (60 °C), resulting in green-blue rhombic crystals. The crystals after filtration with suction were washed with cool water, EtOH and Et2O, and dried overnight in a high vacuum dessicator over fresh P2O5 until it was brought to constant weight. Green blue rhombic crystals, 105 mg (0.064 mmol) 76%. 1H NMR (300 MHz, D2O): δ = 9.62 (δ, J = 6.6 Hz, 2H; C5H5N), 9.40 (δ, J = 5.4 Hz, 2H; C5H5N), 9.03 (δ, J = 6.6 Hz, 2H; C5H5N), 8.94 (δ, J = 5.4 Hz, 2H; C5H5N), 8.01 (δ, J = 8.4 Hz, 2H; Ph), 7.88 (δ, J = 7.2 Hz, 2H; Ph), 4.76 (s, 3H, Me), 2.77 (s, 3H, Me–Ph); FTIR ν(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N):2028 cm−1, ν(C
N):2028 cm−1, ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N, pyr): 1636 cm−1, 1499 cm−1, 1438 cm−1; elemental analysis calc. for {(6a2+)2[FeII(CN)6]4−}·8H2O: C42H52N10FeO8 (880.77): C, 57.27; H, 5.95; N, 15.90; found: C, 57.60; H, 5.39; N, 15.15%.
N, pyr): 1636 cm−1, 1499 cm−1, 1438 cm−1; elemental analysis calc. for {(6a2+)2[FeII(CN)6]4−}·8H2O: C42H52N10FeO8 (880.77): C, 57.27; H, 5.95; N, 15.90; found: C, 57.60; H, 5.39; N, 15.15%.
Acknowledgements
Part of this work was supported financially from the IKY foundation (Greek state scholarship foundation).
References
- P. M. S. Monk, The viologens: physicochemical properties, synthesis and applications of the salts of 4,4′-Bipyridine, John Wiley & Sons Ltd, Chichester, 1998 Search PubMed.
- C. L. Bird and A. T. Kuhn, Chem. Soc. Rev., 1981, 10, 49–82 RSC.
- M. Möller, S. Asaftei, D. Corr, M. Ryan and L. Walder, Adv. Mater., 2004, 16, 1558–1562 CrossRef.
- I. Deligkiozi, R. Papadakis and A. Tsolomitis, Supramol. Chem., 2012, 24, 333–343 CrossRef CAS.
- P. Monk, R. Mortimer and D. Rosseinsky, Electrochromism and Electrochromic Devices, Cambridge University Press, Cambridge, 2007 Search PubMed.
- A. L. Delacey, M. T. Bes, C. Gomezmoreno and V. M. Fernandez, J. Electroanal. Chem., 1995, 390, 69–76 CrossRef.
- Z. Sharrett, S. Gamsey, P. Levine, D. Cunningham-Bryant, B. Vilozny and A. Schiller, et al., Tetrahedron Lett., 2008, 49, 300–304 CrossRef CAS.
- R. Papadakis, I. Deligkiozi and A. Tsolomitis, Dyes Pigm., 2012, 95, 478–484 CrossRef CAS.
- I. Deligkiozi, E. Voyiatzis, A. Tsolomitis and R. Papadakis, Dyes Pigm., 2015, 113, 709–722 CrossRef CAS.
- R. Papadakis and A. Tsolomitis, J. Phys. Org. Chem., 2009, 22, 515–521 CrossRef CAS.
- R. Papadakis, Chem. Phys., 2014, 430, 29–39 CrossRef CAS.
- R. Papadakis and A. Tsolomitis, J. Solution Chem., 2011, 40, 1108–1125 CrossRef CAS.
- I. Deligkiozi, R. Papadakis and A. Tsolomitis, Phys. Chem. Chem. Phys., 2013, 15, 3497–3503 RSC.
- Organic photochromic and thermochromics compounds. Main photochromic families, ed. J. C. Crano and R. J. Guglielmetti, Kluwer Academic Publishers, New York, 2002, vol. 1, pp. 341–367 Search PubMed.
- K. B. Simonsen, N. Thorup, M. P. Cavac and J. Becher, Chem. Commun., 1998, 901–902 RSC.
- A. Ponnu, J. Sung and K. G. Spears, J. Phys. Chem. A, 2006, 110, 12372–12384 CrossRef CAS PubMed.
- H. J. Hwang, S. K. Lee, S. Lee and J. W. Park, J. Chem. Soc., Perkin Trans. 2, 1999, 1081–1086 RSC.
- A. Nakahara and J. H. Wang, J. Phys. Chem., 1963, 67, 496–498 CrossRef CAS.
- H. E. Toma, Can. J. Chem., 1979, 57, 2079–2084 CrossRef CAS.
- A. S. N. Murthy and A. P. Bhardwaj, Spectrochim. Acta, Part A, 1982, 38, 207–212 CrossRef.
- S. Aditya, Spectrochim. Acta, Part A, 1988, 44, 941–942 CrossRef.
- V.-S. Eller, M. Adam and R. D. Fischer, Angew. Chem., 1990, 102, 1157–1159 CrossRef.
- S. A. Kostina, A. B. Ilyukhin, B. V. Lokshin and V. Y. Kotov, Mendeleev Commun., 2001, 11, 12–13 CrossRef.
- A. S. Abouelwafa, V. Mereacre, T. S. Balaban, C. E. Anson and A. K. Powell, CrystEngComm, 2010, 12, 94–99 RSC.
- W. S. Hammack, H. G. Drickamer and D. N. Hendrickson, Chem. Phys. Lett., 1988, 151, 469–473 CrossRef CAS.
- T. Kinuta, T. Sato, N. Tajima, R. Kuroda, Y. Matsubara and Y. Imai, J. Mol. Struct., 2010, 982, 45–49 CrossRef CAS.
- M. Nanasawa, M. Kaneko and H. Kamogawa, Bull. Chem. Soc. Jpn., 1993, 66, 1764–1767 CrossRef CAS.
- V. Y. Kotov, A. B. Ilyukhin and A. V. Zenin, Russ. J. Inorg. Chem., 2008, 53, 552–556 CrossRef.
- M. Hofbauer, M. Möbius, F. Knoch and R. Benedix, Inorg. Chim. Acta, 1996, 247, 147–154 CrossRef CAS.
- R. S. Mulliken, J. Am. Chem. Soc., 1952, 74, 811–824 CrossRef CAS.
- C. J. Bender, Chem. Soc. Rev., 1986, 15, 475–502 RSC.
- A. C. Benniston, A. Harriman, P. Li, J. P. Rostron, R. W. Harrington and W. Clegg, Chem.–Eur. J., 2007, 13, 7838–7851 CrossRef CAS PubMed.
- R. Haque, W. R. Coshow and L.-R. F. Johnson, J. Am. Chem. Soc., 1969, 91, 3822–3827 CrossRef CAS.
- P. M. S. Monk, N. M. Hodgkinson and R. D. Partridge, Dyes Pigm., 1999, 43, 241–251 CrossRef CAS.
- B. G. White, Trans. Faraday Soc., 1969, 65, 2000–2015 RSC.
- G. Saielli, J. Phys. Chem. A, 2008, 112, 7987–7995 CrossRef CAS PubMed.
- Similar correlations have been observed by Hofbauer, et al. for diquats through the NDDO formalism29.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC.
- Visualized using GDIS Molecule Modeller40.
- S. Fleming and A. Rohl, Z. Kristallogr., 2005, 220, 580–584 CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- T. Yanai, D. Tew and N. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- H. E. Gottlieb, V. Kotlyar and A. Nudelman, J. Org. Chem., 1997, 62, 7512–7515 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- D. D. Perrin and L. F. Armarego, Purification of laboratory chemicals, Pergamon, Exeter, 1988 Search PubMed.
- V.-A. Constantin, L. Cao, S. Sadaf and L. Walder, Phys. Status Solidi B, 2012, 249, 2395–2398 CrossRef CAS.
- T. Mochida, Y. Funasako, Y. Nezu, K. Hagiwara and R. Horikoshi, Eur. J. Inorg. Chem., 2015, 2330–2337 CrossRef CAS.
- M. Freitag and E. Galoppini, Langmuir, 2010, 26, 8262–8269 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1409467. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra16732a |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.