
QM/MD simulations on the role of SiO2 in polymeric insulation materials†
Abstract
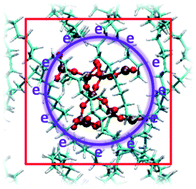
Quantum chemical molecular dynamics (QM/MD) simulations based on a self-consistent charge density functional tight-binding (SCC-DFTB) method on SiO2 filler in polyethylene (PE) showed that: in the absence of SiO2, the PE was quickly charged by high-energy electrons, which resulted in C–C or C–H bonds breaking; on the contrary, in the presence of SiO2 nanoclusters, electron trapping and accumulating were dominated by SiO2 nanoclusters rather than polyethylene, which made polyethylene be preferentially protected and increased the initial time of electrical treeing. In our calculations, we also observed double electric layers around the SiO2 nanocluster, in agreement with recent suggestions from experimental observations. Furthermore, compared with some other investigated nanoclusters, SiO2 was regarded as the most promising candidate attributed to the highest electron affinity. We further observed that once the high-energy electrons were supersaturated in the nanoclusters, the polyethylene chains would be unavoidably charged and C–H bond breaking would occur, which resulted from the interaction between H and O or Si in the nanoclusters. Following that, decomposing and cross-linking of the polyethylene chains were involved in the initial growth of electrical treeing. The current observation can potentially be used in power cable insulation.
 Please wait while we load your content...
Please wait while we load your content...

