
The glass transition temperature measurements of polyethylene: determined by using molecular dynamic method
Qing Yang*,
Xin Chen,
Zhouwen He,
Fengtao Lan and
Hui Liu
State grid smart grid research institute, Department of new electrical materials, Beijing 102211, China. E-mail: yangqingacc@yeah.net
First published on 25th January 2016
Abstract
The unit cell of the polyethylene molecular structure was established to investigate the glass transition temperature (Tg). The properties of interest predicted by the molecular dynamics method were density, free volume, specific volume, radial distribution function, non-bond energy, torsion energy, mean squared displacement and modulus. The Tg of the polyethylene unit cell were determined by means of analyzing these properties. The simulated value of the Tg is about 200 K, which is in good agreement with available data of 195 K in the literature. The method used in this work can be used in studying glass transition of other polymers.
Introduction
The glass transition behavior exists both in amorphous polymers and in the amorphous portion of crystalline polymers when these polymers change from glassy state to rubbery state. It is believed that when polymers experience glass transition, the polymers experience a sudden increase in specific heat capacity, thermal expansivity, motions of molecule chains and other parameters.1–3 The glass transition temperature (Tg) is a key index to evaluate the properties of polymers. The value of this parameter determines the type of use. Values of Tg well below room temperature define the domain of elastomers; values well above room temperature indicate utilization as rigid structural materials. Obviously the ability to predit Tg values would be of great value in the selection and design of new materials and great attention has been given in the characteristic of the Tg of polymers. In order to better understanding of the Tg, experimental research methods such as ellipsometry,4 differential scanning calorimetry (DSC),5 dynamic mechanical test (DMA),6,7 infrared spectroscopy (FTIR),8 thermodilatometry,9 dielectric measurement,10 X-ray reflectivity,11 positron annihilation lifetime spectroscopy (PALS),12 fluorescence intensity13 and other methods have been developed.It is acknowledged that the Tg of polymeric materials is mainly determined by the segment motion of a polymer chain. If this motion can be observed, the polymer chain should have sufficient space (more free volume) and can complete this motion in a short time period (less relaxation time, it mainly depends on rigidity of a polymer chain). Nevertheless, these two factors can not be well discussed in the gas state,14,15 so it is necessary to find an appropriate method to investigate them in the bulk state.
Yet, molecular dynamics (MD) simulation method provides a proper means to study bulk polymer at molecular scale.16–20 Especially, it is showed effective in studying the glass transition temperature of polymers.21–23 In our previous work, the glass transition temperatures of MTB/TDE85 epoxy system and TFMB/TDE85 epoxy system were successfully simulated by using molecular dynamic simulation.24
The relaxation time scale at the Tg is typically around 100 seconds (that value arises from the definition of Tg). MD simulations correspond to much shorter time scales than the typical experiments. Systems are cooled from a higher temperature to a lower one and allowed to equilibrate for a period of time that corresponds to a few picoseconds or nanoseconds. Although there is more than 10 orders of magnitude between these two time values, but according to Jie Han's reports, determination of Tg from MD simulations appears to be a practical procedure.25 Numbers of experimental measurements can be used to analysis the Tg of polymers. However, there were no reports and literature of combining multiple measurements to study the Tg of polymers by using molecular dynamic method. In this study, different measurements of molecular dynamic simulation are used to investigated the Tg of polyethylene. An attempt has been made to establish molecular dynamic measurements–Tg relationships for the polymers. This simulated measurements are especially beneficial for studying the glass transition behavior of complex polymeric structures.
Simulation methodology
Modeling of representative unit cell systems
To investigate the glass transition temperature, one unit cell were prepared. The polyethylene used in the unit cell was made up of two chains of 300 repeat units. For isotropic unit cell, the polyethylene was constructed as an amorphous structure. Periodic boundary conditions were applied to the system to reflect the bulk effect to obtain the macroscopic properties.The COMPASS (Condensed-phase Optimized Molecular Potentials for Atomistic Simulation Studies) forcefield26 was adopted for the interactions between atoms. To minimize the total potential energy of the initial system, the conjugate gradient method was applied until the energy difference between the steps was equal to or less than 10 kcal mol−1. Then, to equilibrate the structures, an NVT (isothermal) ensemble simulation at 300 K for 200 ps was followed by an NPT (isothermal isobaric) ensemble simulation at 300 K and 1 atm for 200 ps. Then the system was simulated from 300 K to 100 K at 10 K/200 ps.
Radial distribution functions
The structural aspects of bulk amorphous PE were examined using radial distribution functions (RDF). The RDF is given as follows:where NA is the amount of atom in group A, NB is the amount of atom in group B, i and j denotes the atom of group A and group B, NAB is the amount of atom both in group A and group B, r denotes the radial distance.27

All atoms and the last 1000 configurations from MD simulations were included in these calculations.
Mean squared displacement
During the cooling-down process, the MSD curves of the unit cell with respect to various temperatures were generated using the initial 100 ps of the NPT simulation at each temperature. The MSD of atoms at time t is given as follows:where
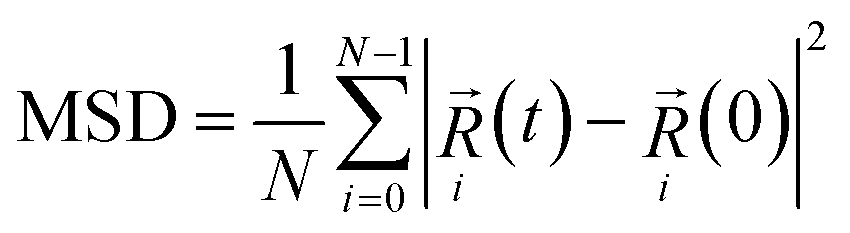
 denotes the current position of the ith atom at time t, N is the total number of the atoms of a given type.28
denotes the current position of the ith atom at time t, N is the total number of the atoms of a given type.28
Modulus
In the modulus calculating process, a total of three loading experiments are performed, in which changes in the total energy of the configurations subjected to deformation are recorded step wise along the x, y, or z directions. After an initial energy minimization, a small strain is applied to the configuration and then a second energy minimization is performed. By definition, the second derivative of the potential energy with respect to strain represents the stiffness matrix. Accordingly, the stiffness matrix, Cij, can be calculated by the following equation:where σ+ and σ− are the ith components of the stress tensor.29

For isotropic amorphous material, the stiffness matrix should always be symmetric, and thus has the following form:



The Young's modulus E is calculated from the following equations:

Results and discussion
The structure of the PE unit cell
The PE unit cell established in this work is amorphous in structure. The packing structure of the simulated PE unit cell was examined by the radial distribution functions (RDF). Fig. 1 shows the radial distribution function, which is calculated for all atoms of the PE unit cell. In the range r < 4 Å, several well pronounced peaks are observed in Fig. 1. These peaks characterize the structure of the PE unit cell. The first peak at around 1.1 Å corresponds to the bond distance between H and other atoms. The second set peaks at around 1.45 Å and 1.56 Å corresponds to the distance between bonded carbon atoms and non-bonded carbon atoms. The subsequent molecular peaks result from distances between atoms two bonds apart, such as hydrogen and carbon in H–C–C sequences (2.16 Å), carbon atoms in C–C–C sequences (2.44 Å).30 Note that any sharp peaks at distances greater than 4 Å are absent and the RDF tend to 1, which is generally regarded as the proof of the amorphous nature of the PE unit cell. | ||
| Fig. 1 The radial distribution function of the PE unit. | ||
The energy of the PE unit cell during the process of MD simulation
It is known that the cooling rate can affect the determination of the glass transition temperature. In fact, both temperature and time could be given to the system to allow the molecules to find their minima. In simulations, enough time is given to the system to reach its minimum energy state and this is verified by monitoring both the kinetic and potential energy of the system.Fig. 2 and 3 show the variation of the potential and kinetic energy versus temperature for one of the simulations. In all simulations, the temperature was set from 300 K to 100 K and it was observed that the graph of potential and kinetic energies of the system reached to a shrinking value. As shown in Fig. 4 and 5, the potential and kinetic energy are plotted as functions of simulation time for the model system at the ten temperatures. All the potential and kinetic energy tend to fluctuate around some well-defined averages, indicating that these model systems have been properly equilibrated.
 | ||
| Fig. 2 Variation of system potential energy versus temperature. | ||
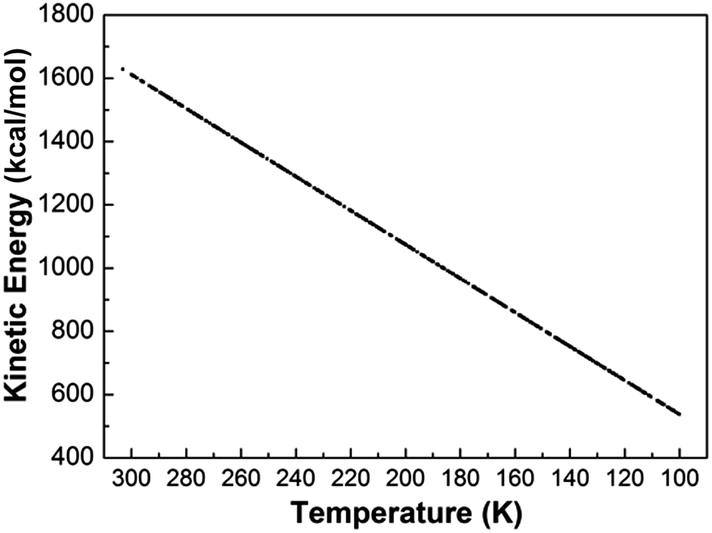 | ||
| Fig. 3 Variation of system kinetic energy versus temperature. | ||
 | ||
| Fig. 4 Variation of system potential energy versus time. | ||
 | ||
| Fig. 5 Variation of system kinetic energy versus time. | ||
The Tg of the PE unit cell obtained from volumetric properties
It is believed that when polymers experience a glass transition, the motion of the molecules can enable the disentanglement of chains; thus, the polymers experience a increase in the thermal expansion. The increase in the thermal expansion appears as a sudden jump in the volumetric properties. Thus, by investigating the change between volumetric properties–temperature curves, an approximate candidate value for the glass transition temperature can be estimated.Fig. 6 depicts the graph of (V − V0)/V versus temperature. Two lines are then fitted to the calculated data, one line to the data before the transition glass temperature and another to the data after Tg. As expected, a kink suggestive of the Tg is observed around 200 °C. This value is about 5 K higher than the experimental value of 195 K for the same system.31
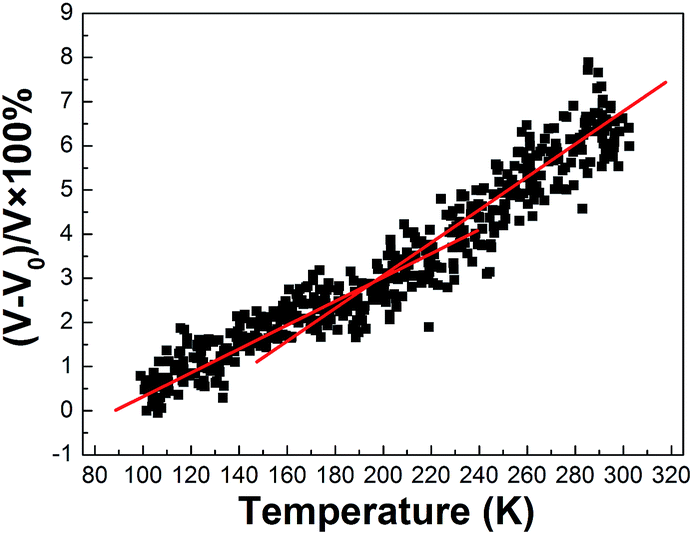 | ||
| Fig. 6 Volume changes versus temperature for the PE unit. | ||
The glass transition of polymers exhibit a free volume dependence relationship. A decrease in free volume that occurs in the glassy state results in inhibition of the molecular chains flow processes. It is believed that the free volume of polymers depends on the packing ability of the molecular structure and the geometric constraints imposed on the segmental packing. To understand the micro structure and its influence on the glass transition, free volume measurements have been carried out using atom volumes & surfaces tools in MS software. Fig. 7 shows the images of free volume of modeling unit cells.
 | ||
| Fig. 7 The free volume of the PE unit. | ||
The dependence of free volume on the temperature for the PE structures is shown in Fig. 8. The data show a non-linear dependence of the free volume on the temperature both above and below the glass transition temperature. Given the uncertainties in the simulated values of free volume, we determined the uncertainty in the Tg by creating a large number of data sets in which free volume at each temperature was sampled from a range around the mean value bound by the uncertainty and then determining the point of intersection of the linear fits in the glassy and rubbery regions. It can be seen that the point of intersection of the curve is about 200 K. The result is according to the one of Fig. 6.
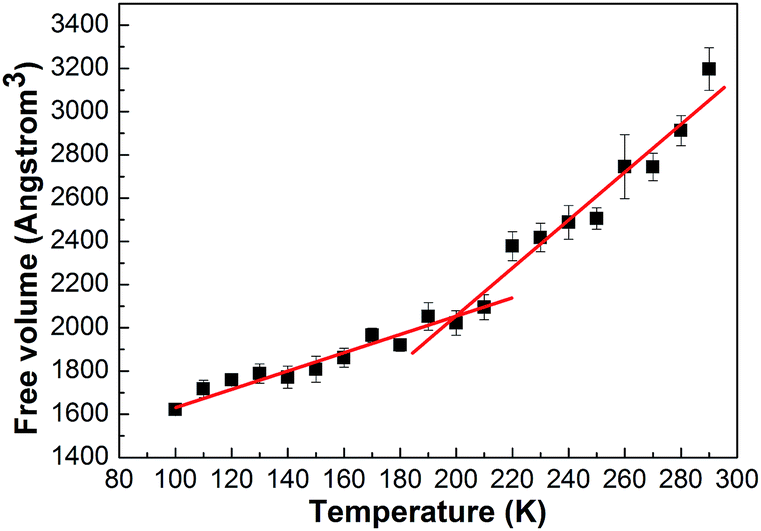 | ||
| Fig. 8 The free volume–temperature curve of the PE unit. | ||
Fig. 9 depicts the density of the PE unit cell versus temperature. Each dot in the graph is a representative for a system state at a specific temperature and density during simulation. The simulation was carried out for several system initial configurations and it was observed that the average results were similar. The idea was to make sure that the results are independent of the system initial configurations. The figure shows that by decreasing the temperature, the density of the PE will increase steadily. A clear change in the slope of the curve is also observable. The change in the slope of the curve is known as the glass transition temperature of the PE. Actually, at this temperature, the PE passes from a glassy state to a rubbery state.
 | ||
| Fig. 9 The density–temperature curve of the PE unit. | ||
Fig. 10 shows the temperature dependence of the density at different simulation times.
 | ||
| Fig. 10 The temperature dependence of the density at different simulation times. | ||
Fig. 10 shows an decrease in Tg (marked by a change in slope of the density–temperature curves) as the simulation time increases. This indicate that the glass transition temperature depends on the quenching rate. A more rapid quenching leads to a higher Tg value.
Fig. 11 shows the temperature dependence of the specific volume of the PE structure. As can be seen from the figure, the specific volume shows a linear relationship with temperature at temperatures both higher (rubbery region) and lower (glassy region) than the glass transition temperature. The glass transition temperature (Tg) can be determined from the point of intersection of the linear fits to these V–T data in the rubbery and glassy regions. The Tg obtained using this procedure for cross-linked epoxy is about 200 K.
 | ||
| Fig. 11 The specific volume–temperature curve of the PE unit. | ||
The Tg obtained from structural properties of the PE unit cell
Shown in Fig. 12 are molecular RDF for the PE unit cell at 100 K, 150 K, 200 K and 300 K. The function give the probability of finding an atom at a distance r from another compared to the ideal gas distribution,32 that is, completely random distribution. Insofar, it give a way to infer structures between atoms or groups and to analyze the structural properties in the polymer systems.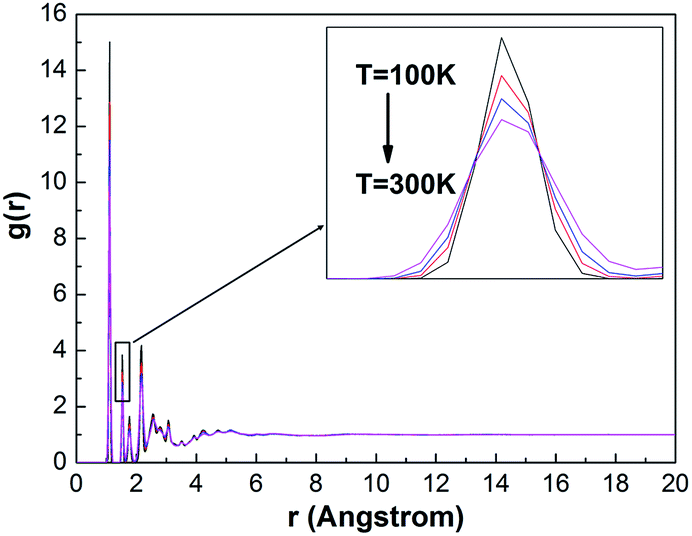 | ||
| Fig. 12 The radial distribution function of the PE unit cell at 100 K, 150 K, 200 K and 300 K. | ||
It can be expected that the correlation function provide the most important information about molecular stacking structure of the polymer chains. Interestingly note from Fig. 12 is that the correlation function at high temperature is lower than those at low temperature, such as g(r = 1.56 Å).
Fig. 13 shows the value of g(r = 1.56 Å) at different temperature. It can be seen that the Tg is determined from the point of intersection of the linear fits to these RDF[g(r = 1.56 Å)]–T data. The peaks at around 1.56 Å corresponds to the distance between non-bonded carbon atoms. It is noted that the distribution of non-bonded carbon atoms is different at lower temperatures and at higher temperatures. With the temperature increasing, the distribution of the non-bonded carbon atoms is changing and it will change suddenly at about 200 K. This temperature is expected as Tg of the PE unit cell.
 | ||
| Fig. 13 The value of g(r = 1.56 Å) at different temperature in the PE unit. | ||
The Tg obtained from mean squared displacement of the PE unit cell
Through the variation of the MSD curve with the temperature, the relative diffusivity of a molecular system that undergoes a temperature change can be estimated. When polymers experience a glassy to rubbery phase transition, the torsional and rotational motions of molecules combined with the local motion can enable the disentanglement of chains; thus, the polymers experience a sudden increase in the diffusivity. The sudden increase in the diffusivity appears as a sudden jump in the MSD curves plotted with respect to the elapsed time. Thus, by investigating the gap between each MSD curve, an approximate candidate range for the glass transition temperature can be estimated to within 10 K.As shown in Fig. 14, the motion of chain segment increases consistently with the increasing temperature. It is interestingly noted that mobility of chain segment is distinctly different for lower temperatures (not higher than 200 K) and higher temperatures (not lower than 210 K). This abrupt change between the two temperature regions can be seen more clearly in Fig. 15, where the MSD is replotted as a function of temperature at 100 ps long time. It can be seen that the Tg is about 200 K. The change in the curve slope of MSD of segments versus T has been employed to identify the Tg of the general polymer surface,33 where the specific volumes are not defined and specific volume versus temperature cannot be used for the same purpose.
 | ||
| Fig. 14 Mean squared displacements (MSD) of the structure as a function of time for the PE unit cell. | ||
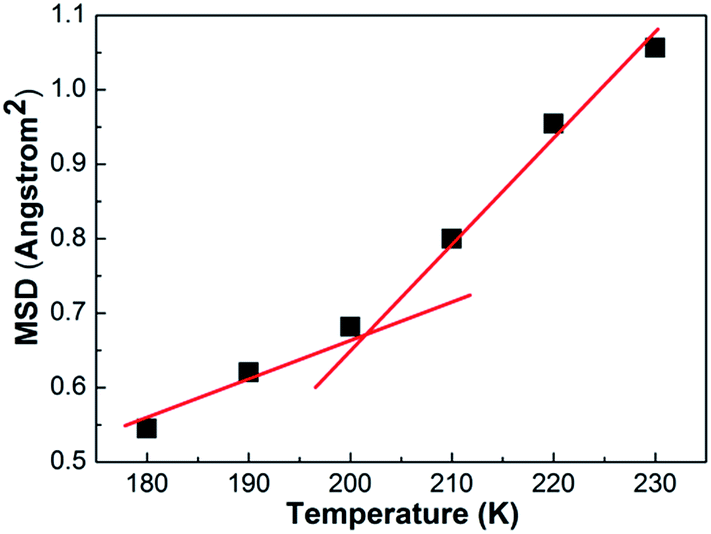 | ||
| Fig. 15 Mean squared displacements (MSD) of the structure as a function of temperature for the PE unit cell. | ||
Previous simulation studies have demonstrated the existence of subdiffusive behavior as deduced by MSD for both unentangled and entangled linear polymer chains.34,35 In order to know if there is some diffusive displacement, we conduct the simulation within 3 ns. Fig. 16 shows that there is some diffusive displacement at a longer times. It also can be seen that at high temperature, the MSD directly crosses over from ballistic motion (t0.25) at short times to subdiffusive motion (tx0 with x0 = 0.5) at intermediate times.
 | ||
| Fig. 16 Mean-squared displacement as a function of time for the atoms of the PE. Lines with slopes of 0.25 and 0.5 are shown as a guide for the eye. | ||
The Tg obtained from energy of the PE unit cell
Various interaction energies can also be used to analyze glass transition occurring in the polymer systems. On this point, Soldera36 has performed energetic analysis of the two PMMA chain tacticities on glass transition. In fact, for our simulations, it is more convenient to analyze the roles of energy components in glass transition. The simulated results of non-bond energy and torsion energy against the temperature are plotted in Fig. 17 and 18. It can be seen that both in the plot of non-bond energy versus temperature and in the plot of torsion energy versus temperature are there a break present, indicating the occurrence of glass transition. At both below and above Tg, non-bond energy and torsion energy increase almost linearly with increasing temperature with a break at Tg. | ||
| Fig. 17 The non bond energy of the PE unit at different temperature. | ||
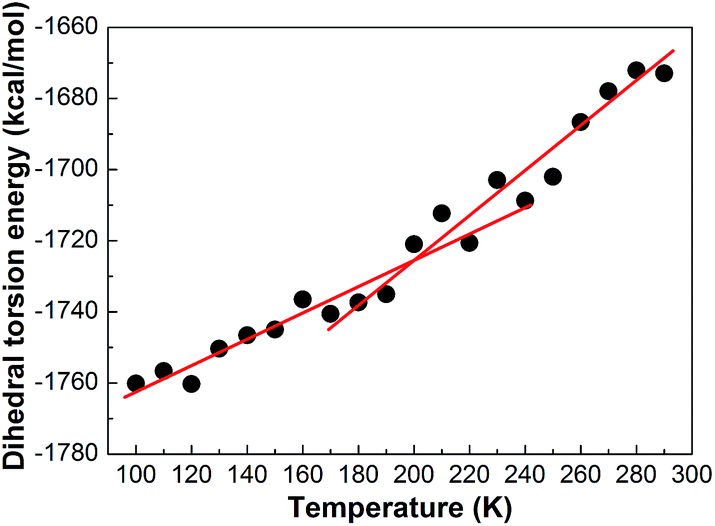 | ||
| Fig. 18 The dihedral torsion energy of the PE unit at different. | ||
These results show that the non-bond energy and torsion energy plays an important role in the glass transition process of the PE system. Li37,38 performed the same analysis on PHB/PEO blend system and drew a similar conclusion with ours: both torsion and non-bond energy played important roles in glass transition.
The Tg obtained from mechanical properties of the PE unit cell
The elastic modulus of PE unit cell at different temperature obtained from the molecular dynamic simulated methods are shown in Fig. 19. In the passage through the glass transition region from the glassy state to the rubbery state, the elastic modulus of the PE dramatically decrease. This is maybe result from that the molecules are easily disentangled as a result of the increased kinetic energy of individual atoms at high temperature.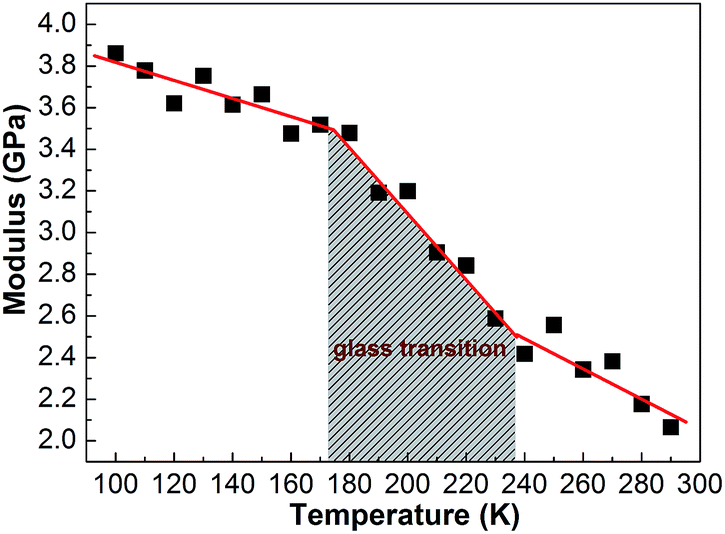 | ||
| Fig. 19 The modulus of the PE unit at different temperature. | ||
Conclusions
The glass transition temperature can be determined by using various molecular dynamic measurements. Through the variation of the MSD curve and the elastic modulus curve with the temperature, the glass transition region of the PE unit cell can be estimated. As expected, a kink suggestive of the Tg can be observed around 200 K from volumetric properties, energy properties and structural properties of the PE unit cell. The Tg of the PE unit cell obtained by molecular dynamic simulated is close to the experimental value of 195 K for the same system. While the illustrative examples we have chosen were relatively simple, they show that the general phenomenology of the glass transition and the properties of the glassy state can be accounted for using molecular simulations, in spite of the relatively small time scale that can be considered in such simulations.Acknowledgements
The authors thank the financial support from the science and technology project of State Grid Corporation of China (project No. 5455DW150009).Notes and references
- A. Djemour, R. Sanctuary and J. Baller, Mobility restrictions and glass transition behaviour of an epoxy resin under confinement, Soft Matter, 2015, 11, 2683–2690 RSC.
- M. L. Huang, B. Tunnicliffe Lewis, G. Thomas Alan and J. C. Busfield James, The glass transition, segmental relaxations and viscoelastic behaviour of particulate-reinforced natural rubber, Eur. Polym. J., 2015, 67, 232–241 CrossRef CAS.
- V. D. Samith and E. Ramos-Moore, Study of glass transition in functionalized poly(itaconate)s by differential scanning calorimetry, Raman spectroscopy and thermogravimetric analysis, J. Non-Cryst. Solids, 2015, 408, 37–42 CrossRef CAS.
- O. K. C. Tsui and H. F. Zhang, Effects of chain ends and chain entanglement on the glass transition temperature of polymer thin films, Macromolecules, 2001, 34, 9139–9142 CrossRef CAS.
- J. Pagacz and K. Pielichowski, PVC/MMT nanocomposites: DSC with stochastic temperature modulation study at glass transition region, J. Therm. Anal. Calorim., 2013, 111, 1571 CrossRef CAS.
- S. Wei, A. P. Vassilopoulos and T. Keller, Effect of thermal lag on glass transition temperature of polymers measured by DMA, Int. J. Adhes. Adhes., 2014, 52, 31–39 CrossRef.
- V. Pistor, L. Puziski and A. J. Zattera, Influence of different concentrations of glycidylisobutyl-POSS on the glass transition of cured epoxy resin, J. Appl. Polym. Sci., 2015, 132, 41453 CrossRef.
- X. F. Liang and D. H. Huang, New Insights into the Effects of Physical Aging on Glass Transition of Polystyrene by FTIR, J. Macromol. Sci., Part B: Phys., 2012, 51, 348–357 CrossRef CAS.
- J. E. Pye and C. B. Roth, Above, below, and in-between the two glass transitions of ultrathin free-standing polystyrene films: Thermal expansion coefficient and physical aging, J. Polym. Sci., Part B: Polym. Phys., 2015, 53, 64–75 CrossRef CAS.
- F. Kremer, M. Tress and E. U. Mapesa, Glassy dynamics and glass transition in nanometric layers and films: a silver lining on the horizon, J. Non-Cryst. Solids, 2015, 407, 277–283 CrossRef CAS.
- O. K. C. Tsui, T. P. Russell and C. J. Hawker, Effect of interfacial interactions on the glass transition of polymer thin films, Macromolecules, 2001, 34, 5535–5539 CrossRef CAS.
- G. B. DeMaggio, W. E. Frieze, D. W. Gidley, M. Zhu, H. A. Hristov and A. F. Yee, Interface and surface effects on the glass transition in thin polystyrene films, Phys. Rev. Lett., 1997, 78, 1524 CrossRef CAS.
- C. J. Ellison and J. M. Torkelson, The distribution of glass-transition temperatures in nanoscopically confined glass formers, Nat. Mater., 2003, 2, 695–700 CrossRef CAS PubMed.
- M. Hasegawa, N. Sensui, Y. Shindo and R. Yokota, Structure and properties of novel asymmetric biphenyl type polyimides. Homo-and copolymers and blends, Macromolecules, 1999, 32, 387–396 CrossRef CAS.
- Y. Tong, W. Huang, J. Luo and M. Ding, Synthesis and properties of aromatic polyimides derived from 2,2′,3,3′-biphenyltetracarboxylic dianhydride, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 1425–1433 CrossRef CAS.
- T. W. Sirk, K. S. Khare, M. Karim, J. L. Lenhart, J. W. Andzelm, G. B. McKenna and R. Khare, High strain rate mechanical properties of a cross-linked epoxy across the glass transition, Polymer, 2013, 54, 7048–7057 CrossRef CAS.
- J. Zhang and D. Jiang, Molecular dynamics simulation of mechanical performance of graphene/graphene oxide paper based polymer composites, Carbon, 2014, 67, 784–791 CrossRef CAS.
- C. Li and A. Strachan, Molecular dynamics predictions of thermal and mechanical properties of thermoset polymer EPON862/DETDA, Polymer, 2011, 52, 2920–2928 CrossRef CAS.
- D. C. Doherty, B. N. Holmes, P. Leung and R. B. Ross, Polymerization molecular dynamics simulations. I. Crosslinked atomistic models for poly(methacrylate) networks, Comput. Theor. Polym. Sci., 1998, 8, 169–178 CrossRef CAS.
- N. B. Shenogina, M. Tsige, S. S. Patnaik and S. M. Mukhopadhyay, Molecular modeling of elastic properties of thermosetting polymers using a dynamic deformation approach, Polymer, 2013, 54, 3370–3376 CrossRef CAS.
- M. Li and X. Y. Liu, et al., Molecular dynamics simulation on glass transition temperature of isomeric polyimide, eXPRESS Polym. Lett., 2009, 3(10), 665 CrossRef CAS.
- A. Solder and N. Metatla, Glass transition phenomena observed in stereoregular PMMAs using molecular modeling, Composites, Part A, 2005, 36, 521 CrossRef.
- J. Pozuelo and J. Baselga, Glass transition temperature of low molecular weight poly(3-aminopropyl methyl siloxane). A molecular dynamics study, Polymer, 2002, 43, 6049 CrossRef CAS.
- Q. Yang, X. P. Yang, X. D. Li, L. Shi and G. Sui, The curing and thermal transition behavior of epoxy resin: a molecular simulation and experimental study, RSC Adv., 2013, 3, 7452–7459 RSC.
- J. Han, H. G. Richard and H. B. Richard, Glass Transition Temperatures of Polymers from Molecular Dynamics Simulations, Macromolecules, 1994, 27, 7781–7784 CrossRef CAS.
- H. Sun, COMPASS: An ab initio force-field optimized for condensed-phase applications overview with details on alkane and benzene compounds, J. Phys. Chem. B, 1998, 102, 7338–7364 CrossRef CAS.
- Amorphous cell and discover modules [Materials Studio 4.0, Visualizer, QSAR], Accelrys Inc, San Diego, CA, 2005 Search PubMed.
- J. Choi, S. Yu, S. Yang and M. Cho, The glass transition and thermoelastic behavior of epoxy-based nanocomposites: A molecular dynamics study, Polymer, 2011, 52, 5197–5203 CrossRef CAS.
- H. B. Fan and M. F. Yuen Matthew, Material properties of the cross-linked epoxy resin compound predicted by molecular dynamics simulation, Polymer, 2007, 48, 2174–2178 CrossRef CAS.
- N. C. Karayiannis, V. G. Mavrantzas and D. N. Theodorou, Detailed atomistic simulation of the segmental dynamics and barrier properties of amorphous poly(ethylene terephthalate) and poly(ethylene isophthalate), Macromolecules, 2004, 37, 2978–2995 CrossRef CAS.
- J. M. G. Cowie and I. J. McEwen, Molecular Relaxations in Partially Hydrogenated cis-1,4-Polybutadienes. A Guide to the Glass Transition Temperature of Amorphous Polyethylene, Macromolecules, 1977, 10, 1124–1128 CrossRef CAS.
- A. R. Leach, Molecular modelling principles and applications, Addison Wesley Longman Limited, London, 1996 Search PubMed.
- H. Morita, K. Tanaka, T. Kajiyama, T. Nishi and M. Doi, Study of the glass transition temperature of polymer surface by coarse-grained molecular dynamics simulation, Macromolecules, 2006, 39, 6233–6237 CrossRef CAS.
- Z. Wang and R. G. Larson, Constraint Release in Entangled Binary Blends of Linear Polymers: A Molecular Dynamics Study, Macromolecules, 2008, 41, 4945–4960 CrossRef CAS.
- A. V. Lyulin, N. K. Balabaev and M. A. J. Michels, Correlated Segmental Dynamics in Amorphous Atactic Polystyrene: A Molecular Dynamics Simulation Study, Macromolecules, 2002, 35, 9595–9604 CrossRef CAS.
- A. Soldera, Energetic analysis of the two PMMA chain tacticities and PMA through molecular dynamics simulations, Polymer, 2002, 43, 4269–4275 CrossRef CAS.
- K. Q. Yu, Z. S. Li and J. Sun, Polymer structures and glass transition: A molecular dynamics simulation study, Macromol. Theory Simul., 2001, 10, 624–633 CrossRef CAS.
- H. Yang, Z. S. Li, H. J. Qian, Y. B. Yang, X. B. Zhang and S. C. Sun, Molecular dynamics simulation studies of binary blend miscibility of poly(3-hydroxybutyrate) and poly(ethylene oxide), Polymer, 2004, 45, 453–457 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |