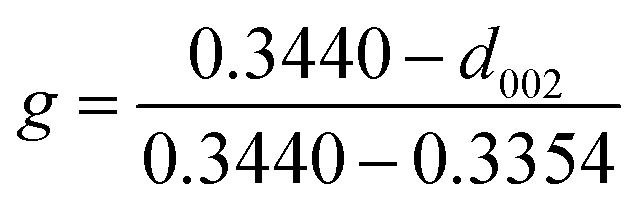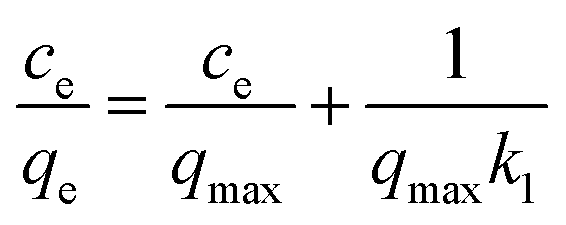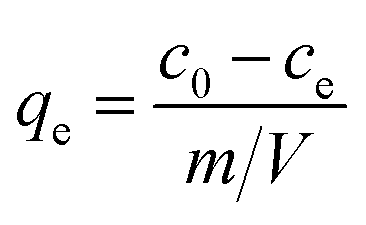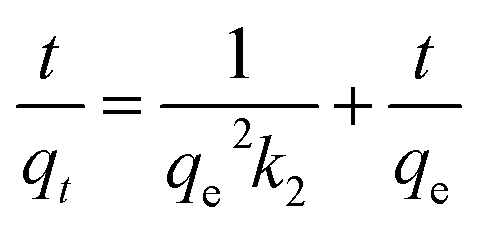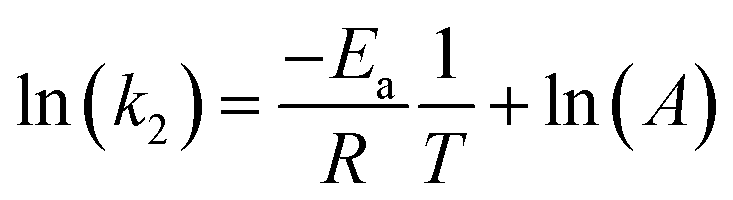DOI:
10.1039/C5RA19616G
(Paper)
RSC Adv., 2016,
6, 2259-2269
Facile synthesis of mesoporous carbon nanocomposites from natural biomass for efficient dye adsorption and selective heavy metal removal†
Received
23rd September 2015
, Accepted 20th December 2015
First published on 22nd December 2015
Abstract
Mesoporous carbon with embedded iron carbide nanoparticles was successfully synthesized via a facile impregnation–carbonization method. A green biomass resource, cotton fabric, was used as a carbon precursor and an iron precursor was implanted to create mesopores through a catalytic graphitization reaction. The pore structure of the nanocomposites can be tuned by adjusting the iron precursor loadings and the embedded iron carbide nanoparticles serve as an active component for magnetic separation after adsorption. The microstructure of the nanocomposites was carefully investigated by various characterization techniques including electron microscopy, X-ray diffraction, surface analyzer, magnetic property analyzer and etc. The newly created mesopores are demonstrated as a critical component to enhance the adsorption capacity of organic dyes and embedded iron carbide nanoparticles are responsible for the selective removal of heavy metal ions (Zn2+, Cu2+, Ni2+, Cr6+ and Pb2+). Isotherm adsorption, kinetic study at three different temperatures (25, 45 and 65 °C) and cycling retention tests were performed to understand the adsorptive behavior of the nanocomposites with organic dyes (methylene blue and methyl orange). Together with the preferable removal of more toxic heavy metal species (Cr6+ and Pb2+), these mesoporous nanocomposites show promising applications in pollutant removal from water. The facile material preparation allows convenient scale-up manufacturing with low cost and minimum environmental impact.
1. Introduction
Sustainable clean water supply has been an increasingly serious issue nowadays since significant amounts of surface and ground water have been severely polluted by industrialization of human society. Water pollution has been recognized as the leading worldwide cause of death and diseases, which accounts for the deaths of more than 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 people daily.1–3 The hazards can be roughly classified into three categories: organic, inorganic and biological pollutants. Targeting different pollutants, miscellaneous water remediation technologies have been developed including adsorption,4–9 coagulation,10 membrane separation,11–13 ion exchange,14,15 electrochemical precipitation16,17etc. Among these technologies, adsorption seems the most promising process due to its low cost, simple operation as well as its capability can produce water of high quality.
000 people daily.1–3 The hazards can be roughly classified into three categories: organic, inorganic and biological pollutants. Targeting different pollutants, miscellaneous water remediation technologies have been developed including adsorption,4–9 coagulation,10 membrane separation,11–13 ion exchange,14,15 electrochemical precipitation16,17etc. Among these technologies, adsorption seems the most promising process due to its low cost, simple operation as well as its capability can produce water of high quality.
Porous carbon, especially activated carbon, is well accepted as effective adsorbent in water purification. However, the relatively high manufacturing cost restricts its wide applications simply because the corrosive chemicals such as ZnCl2 or KOH have to be used during the activation process. These chemicals not only add up the cost but also pose potential secondary environmental pollutions. Over the past decade, biomass derived carbon has aroused great interest in the research field and also find wide application in energy storage such as sodium ion battery,18 supercapacitor19,20 as well as environmental remediation in adsorption based water purification.16,21–23 Researchers have fabricated carbon adsorbents from various biomass resources, such as cotton,21 bamboo,24 vetiver roots,25 oil palm wood,26 rattan sawdust,27 rice husk,28,29 banana stalk,30 peanut shell31 and successfully used them as adsorbents for either heavy metal or organic pollutants removal from polluted water. Adsorbents those are capable of removing both organic and inorganic pollutants are of great interest and practically useful, while very rare work has been reported so far with both capabilities.
In general, carbon materials show superior adsorption capacity after activation, which is attributed to the significantly enlarged surface area and increased surface hydrophilicity that facilitates the diffusion and adhesion of pollutant molecules inside the adsorbent. However, the overall adsorption performance of biomass derived carbon is still not satisfying compared to commercial activated carbon, which demands further structural and functional design to realize practical applicability. In general, macropores and mesopores are considered access points to micropores. It is the micropores in carbon that plays a significant role in adsorption.32 Previous study reveals that mesopores, with pore size in the range of 2–50 nm, could significantly improve the diffusivity of small molecules inside the pore channels.33 To realize efficient diffusion and adsorption of small molecules in biomass derived carbon, the existence of macro-/meso-pore channels and microporous adsorption sites are equally important. Even though mesopore size can be well controlled from bottom-up synthesis via hard-template34–36 and soft-template methods,37–39 it still remains a great challenge to create mesopores in biomass derived carbon from a top-down approach. Conventional physical and chemical activation generally creates micropores those are not ideal structure for internal mass transfer. Our previous work demonstrated that penetrating mesoporous structure can be created via a simple thermal oxidation process in spruce-pine-fir derived carbon, which out-performs commercial activated carbon in organic dye adsorption.40 However, this simple method relies on the thermal degradation of lignin to create mesopores, which does not necessarily works for other biomass derived carbons. One typical example is natural cotton that consists of almost 100% cellulose without lignin. Therefore, an alternative approach needs to be developed to create mesopores in lignin-free biomass.
In this work, mesopores have been successfully created in cotton derived carbon through an in situ catalytic graphitization approach. The microstructure of the synthesized materials was carefully investigated by different characterization techniques. Adsorption isotherms and kinetic studies were conducted to quantify the adsorption capacity and rate, respectively. These materials show outstanding adsorption performance in organic dyes (positively charged methylene blue and negatively charged methyl orange) and more interestingly selective heavy metal adsorption. The pollutants removal mechanisms were also studied in this work.
2. Experimental
2.1 Preparation of carbon and carbon nanocomposites
Cotton fabric was cut from commercially available T-shirt made of 100% cotton. Iron nitrate (>98%) was purchased from Sigma Aldrich. Methylene blue (MB) and methyl orange (MO) were purchased from Fisher Scientific. Nickel(II) chloride hexahydrate (>98%), zinc chloride (>97%), copper(II) chloride dihydrate (98%) and lead chloride (99%) were purchased from Acros Organics. Potassium dichromate (99.8%) was purchased from Fisher Scientific. All chemicals were used as-received without further treatment. Carbon nanocomposites were prepared from a facile two-step procedure, impregnation and carbonization. First, 4.0 g of cotton fabrics were soaked in 40 mL 0.1, 0.3, 0.5 and 1.0 M iron nitrate solutions in a vacuum oven for 24 hours. Then the soaked cotton fabrics were carefully blotted and dried at room temperature overnight. Then the dried cotton fabrics were carbonized at 800 °C for 2 hours in nitrogen atmosphere with a heating rate of 5 °C min−1. As a control, original cotton fabric was carbonized directly without the soaking process and the sample was named C-00M. Carbonized samples pre-soaked with 0.1, 0.3, 0.5 and 1.0 M iron nitrate solutions were denoted as C-01M, C-03M, C-05M and C-10M, respectively. To investigate the pore structure after removing metal nanoparticles, 1.0 g of nanocomposite (C-01M, C-03M, C-05M and C-10M) was mixed with 10.0 mL 1.0 M HCl aqueous solution for 12 hours. Then, the samples were filtered, washed with deionized water until the rinsing water was neutral and dried at 80 °C for 12 hours. Samples after acid washing were named C-01M(w), C-03M(w), C-05M(w) and C-10M(w), respectively.
2.2 Material characterization
The microstructure of C-00M, C-01M, C-03M, C-05M and C-10M was characterized by scanning electron microscopy (SEM, JEOL-7401). Transmission electron microscopy (TEM) images of the prepared materials were obtained by JEOL JEM-1230 microscope. High-resolution TEM (HRTEM) images were taken using a FEI Tecnai G2 F20 microscope operated at 200 kV. The nanoparticle loading in the composites was determined by thermogravimetric analysis (TGA, TA instrument Q500) in air atmosphere from 20 to 800 °C with a ramp rate of 10 °C min−1. The powder X-ray diffraction analysis was carried out with a Bruker AXS D8 Discover diffractometer with GADDS (General Area Detector Diffraction System) operating with a Cu-Kα radiation source filtered with a graphite monochromator. The specific surface area and pore-size distribution were measured on a TriStar II 3020 by nitrogen adsorption at 77.3 K. The pore size distribution was calculated from the adsorption branch of the isotherm. Raman spectrum was obtained using a Horiba LabRam HR Micro Raman Spectrometer, equipped with a CCD camera detector within the range of 400–3000 cm−1. Zeta potential of C-00M and nanocomposites were determined by Malvern Zetasizer Nano-ZS90. To quantify the amount of iron ions leached into the polluted water during adsorption, inductively coupled plasma (ICP) analysis was performed on ICP-OES (Perkin Elmer P-400). The magnetic property was measured in a 9 T physical properties measurement system (PPMS) by Quantum Design.
2.3 Organic dye and heavy metal adsorption
To determine the adsorption capacity of the synthesized materials, 1.0 g L−1 of C-00M, C-01M, C-03M, C-05M and C-10M were used to treat 10 mL MB and MO solutions with different initial concentrations at room temperature. The treatment time was set at 12 hours to ensure equilibrium has reached. After treatment, the mixture was filtered and the solution was characterized by UV-Vis spectrometer to determine the remaining dye concentration. For kinetic studies, 1.0 g L−1 of C-01M, C-03M, C-05M and C-10M were used to treat 40 mL 50 ppm MB or MO solution with sampling time of 1, 3, 5, 7, 10, 15 and 30 min. Due to the low adsorption capacity of C-00M, the initial MB concentration was chosen as 10 ppm with sampling time of 1, 3, 10, 60, 120 and 180 min. Temperate effect on the dye removal kinetics was performed with 1.0 g L−1 C-10M in 50 ppm MB/MO solutions at different temperatures of 25, 45 and 65 °C. Standard curves relating the dye concentration and UV-Vis adsorption peak intensity at 664 nm (MB) and 465 nm (MO) are constructed to determine the remaining dye concentration after removal tests.
Adsorption of heavy metal ions were comparatively investigated with C-00M and C-03M. Specifically, 0.05 g C-00M or C-03M was added to 50 mL aqueous mixture with 25 ppm of each following ions: Cu2+, Zn2+, Cr6+, Pb2+ and Ni2+ under magnetic stirring. 3.0 mL of the mixture was collected, filtered and acidified for ICP analysis at time interval of 1, 2, 4, 6 and 10 hours.
For cycling tests, 50 ppm MB and 25 ppm metal ion mixtures were used separately. Adsorbent after MB adsorption was regenerated in a tube furnace at 500 °C for 1 hour under nitrogen atmosphere. Adsorbent after metal ion adsorption was regenerated by acid washing with 0.1 M HCl. The adsorption capacity from first cycle was recorded as 100%. Adsorption capacity in following cycles versus the first cycle value was calculated as retention.
3. Results and discussion
3.1 Microstructure investigation
Fig. 1(a) showed the microstructure of pure cotton fiber after annealing. The fibrous structure of cotton was well maintained after the annealing process with helical structure and the fiber diameter is about 10 μm. Focusing on the surface, wrinkled texture appeared along the axial direction of fiber, which is consistent with the natural assembly pattern of cellulose fibrils. Compared to the relatively smooth surface of C-00M (Fig. 1(b)), uniformly distributed nanoparticles were observed in the nanocomposites, Fig. 1(c–f). The nanoparticle size largely depends on the concentration of the iron precursor. Specifically, the particle size is only 22.9 ± 5.2 and 49.0 ± 8.1 nm for C-01M and C-03M. With further increasing initial precursor concentration, the particle size increased to 104.8 ± 18.2 and 178.3 ± 36.6 nm for C-05M and C-10M, respectively. The larger particle size at higher precursor concentration is mainly due to the nanoparticle aggregation during annealing. Besides, it is worth mentioning that deeper wrinkled surface texture was developed after introducing nanoparticles. It's well known that volume shrinkage usually occurred during carbonization, which is a homogeneous shrinking process. After introducing nanoparticles, catalytic graphitization accelerates the shrinking process and meanwhile induces heterogeneous shrinkage. Thus, much rougher surface textures were observed in the nanocomposites as compared to C-00M.
 |
| | Fig. 1 SEM microstructures of (a & b) C-00M, (c) C-01M, (d) C-03M, (e) C-05M and (f) C-10M. | |
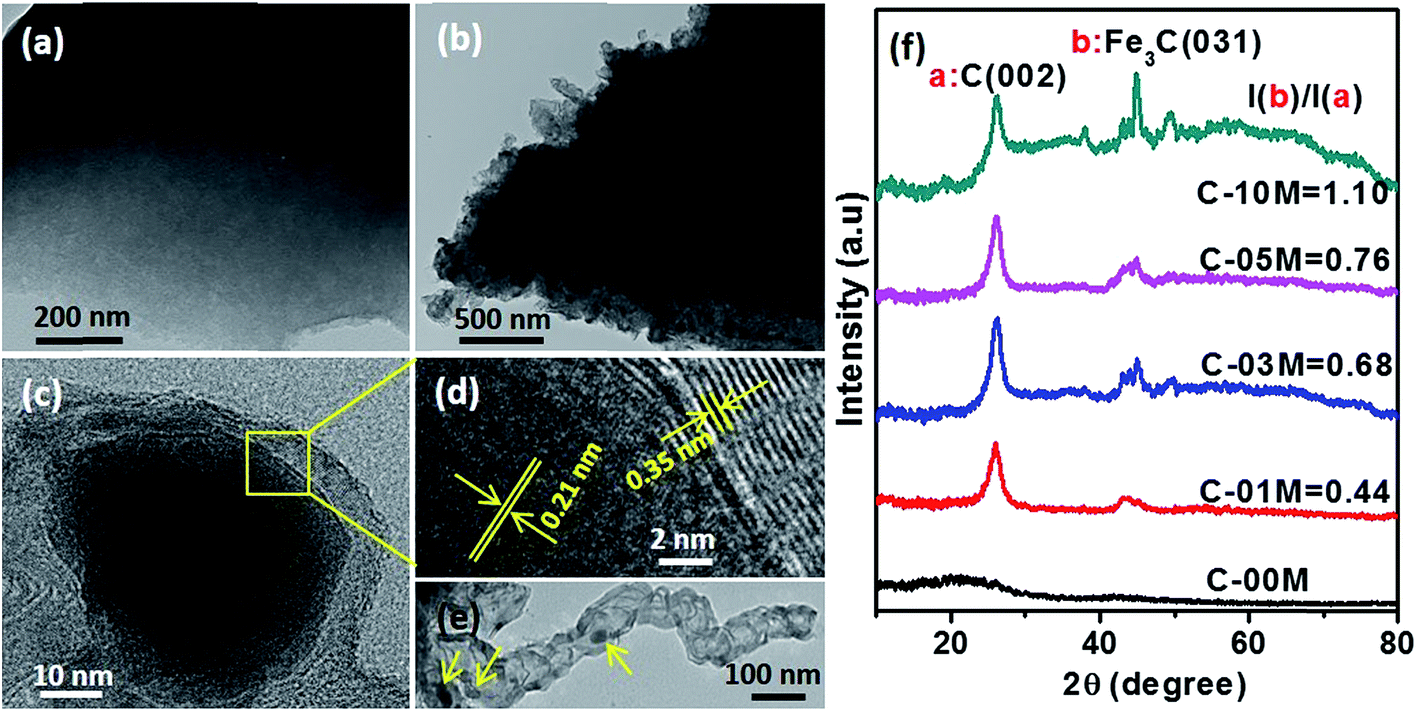
To further examine the effect of nanoparticles on the morphology and crystalline structure of nanocomposites, comparative TEM and HRTEM studies were performed on C-00M and C-03M. C-00M showed amorphous feature, where no lattice fringe could be observed in Fig. 2(a). In C-03M, nanoparticles were uniformly distributed in the carbon matrix, Fig. 2(b). HRTEM images focusing on one nanoparticle show typical core–shell structure, Fig. 2(c). The lattice fringe of the shell was measured as 0.35 nm, which is indexed to the lattice spacing of graphite. The lattice fringe of the encapsulated core area was determined as 0.21 nm that can be assigned to the (031) crystalline plane of Fe3C, Fig. 2(d).18,41 The catalytic graphitization of other biomass like soft wood was conducted by Thompson et al.22 and our group23 recently and both observed graphitized carbon and mesopore formation in the carbonized products. A few key steps for the catalytic graphitization has been proposed: (1) thermal decomposition of iron precursor Fe(NO3)3 to form iron oxide nanoparticles; (2) carbothermal reduction of iron oxide to produce Fe3C nanoparticles. Firstly, iron oxide is reduced by carbon reductant to form elemental iron nanoparticles, and subsequently elemental iron further reacts with carbon to form Fe3C nanoparticles; (3) migration of Fe3C nanoparticles to catalyze the reaction from amorphous carbon to graphitized carbon through the carbon matrix. It is worth mentioning that Fe3C is in liquid state at 800 °C and can migrate freely on carbon matrix. Along the migration path, amorphous carbon will be catalyzed into graphite carbon and leaves behind a hollow tube structure as seen in Fig. 2(e). Accompanies with the catalytic reaction, carbon volume shrink would be expected due to the more orderly-packed crystalline structure and mesopores will be generated between graphitized carbons that will be discussed in later section of this work.
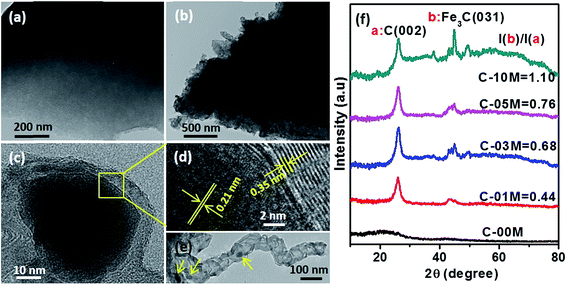 |
| | Fig. 2 TEM images of (a) C-00M, (b) C-03M, (c & d) HRTEM images of C-03M, (e) Fe3C nanoparticles in the center of a graphitized carbon tube with arrows indicated and (f) XRD patterns of C-00M, C-01M, C-03M, C-05M and C-10M. I(b)/I(a) indicates the ratio of peak intensity Fe3C(031)/C(002). | |
To further confirm the crystalline structure of nanoparticles, X-ray diffraction patterns of annealed samples were recorded, Fig. 2(f). Only one broad peak at 2θ ≈ 20° was observed in C-00M, indicating its amorphous nature. This is in good agreement with TEM results. For all the other nanocomposites, the appearance of a sharp graphite peak at around 2θ = 25.9° (PDF # 41-1487) confirmed the formation of graphitic carbon C(002). Also, the peaks appeared at 2θ = 43.7, 45.0 and 49.1° are indexed to the (102), (031) and (221) crystal planes of Fe3C (PDF # 35-0772). Besides, the peak intensity ratio of Fe3C (031)/graphite C(002) increased gradually from 0.44 to 1.10 with increasing nanoparticle loading, Fig. 2(f), indicating the increased portion of Fe3C in the nanocomposites relative to the graphitized carbon. The graphitization degree in nanocomposites can be calculated by Mering–Maire eqn (1):42
| | 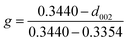 | (1) |
where
g is graphitization degree, 0.3440 indicates the layer distance of non-graphitized carbon, 0.3354 is the layer distance of 100% graphitized carbon and
d002 is the layer distance of sample, which can be calculated from Bragg
eqn (2):
| | | 2dsinθ = nλ | (2) |
where
d is the spacing between the planes in the atomic lattice,
θ is the angle between the incident ray and the scattering planes,
n is an integer, and
λ is the wavelength of incident wave (
λ = 1.54056 Å). The graphitization degree for C-01M, C-03M, C-05M and C-10M was calculated as 12.1, 38.6, 50.8 and 45.6%, respectively. It seems that larger graphitization degree can be achieved by doping more nanoparticles (C-01M → C-05M) due to the provided more active sites for catalytic graphitization. However, further increasing nanoparticle loading (C-10M) leads to severe aggregation and reduced active sites and thus decreased graphitization degree was observed in C-10M than that of C-05M.
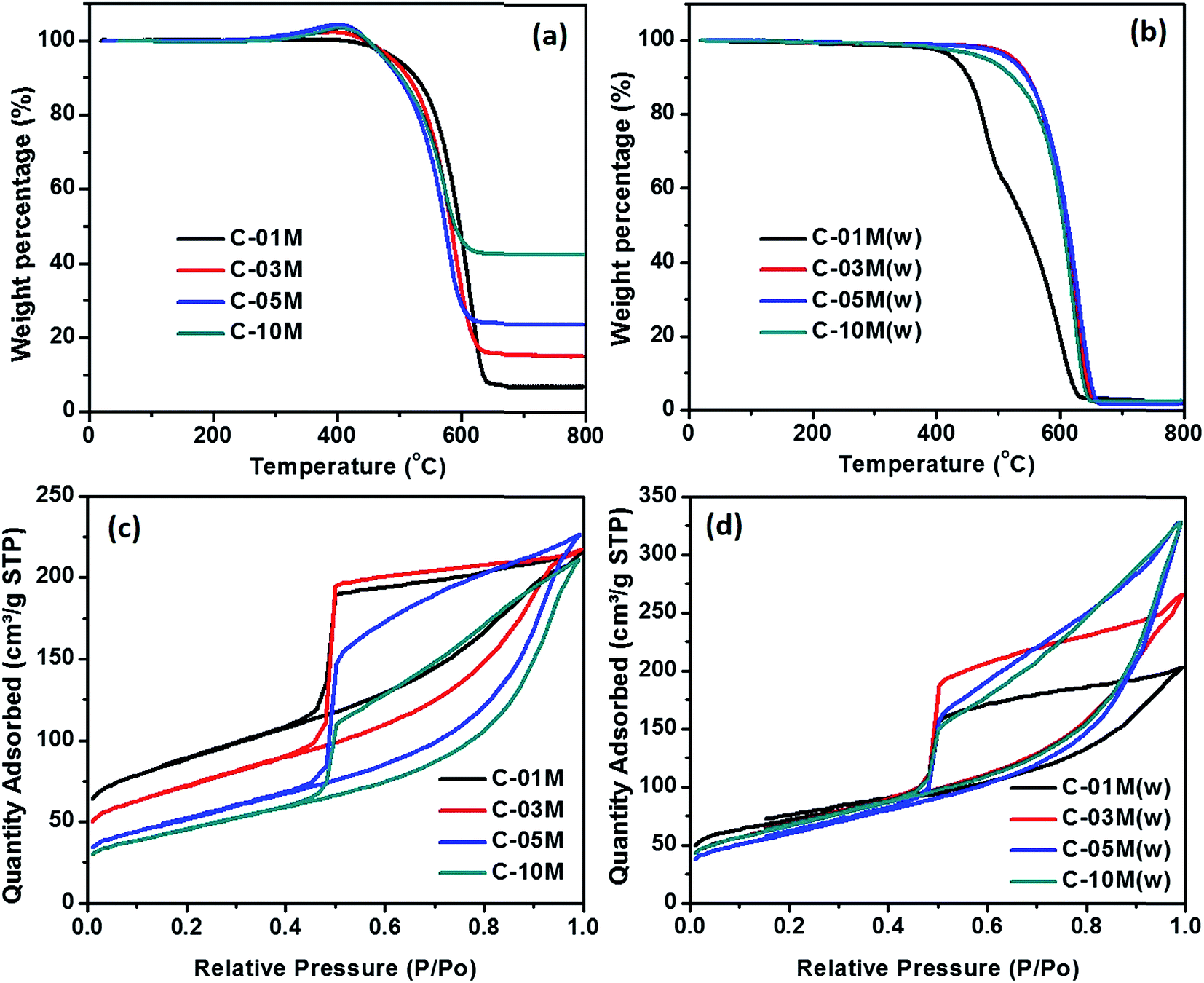
TGA analysis was performed to determine the actual Fe3C loading in the nanocomposites, as shown in Fig. 3. C-00M began to degrade at ∼400 °C and the 1.6% final residue is attributed to the non-degradable inorganics, Fig. S1.† In the nanocomposites, a slight weight increase was observed starting from 250 °C, which is caused by the oxidation of Fe3C in air. With further increasing temperature to ∼400 °C, the Fe3C in nanocomposites was fully oxidized to Fe2O3 and the carbon was completely burned out afterwards, Fig. 3(a). The final residue at 800 °C is composed of Fe2O3 and non-degradable inorganics. To calculate Fe3C loading in the nanocomposites, weight fraction of non-degradable inorganics was subtracted and weight percentage of Fe2O3 was converted to Fe and Fe3C in corresponding nanocomposite as summarized in Table 1.
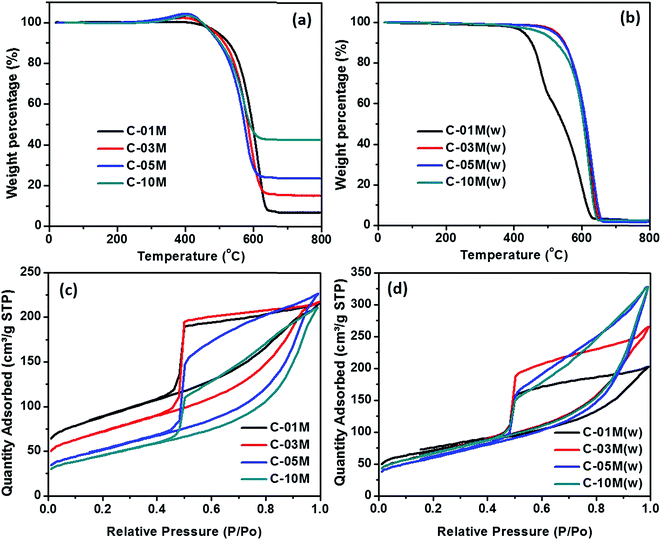 |
| | Fig. 3 TGA curves of C-xxM (xx = 00, 01, 03, 05 and 10) nanocomposites (a) before and (b) after acid washing with 1 M HCl acid. BET isotherms of C-xxM nanocomposites (c) before and (d) after acid washing. The TGA and BET results of C-00M are provided in ESI Fig. S1 and S2.† | |
Table 1 Summary of material properties before and after acid washinga
| Sample |
Residues@800 °C, wt% |
Fe2O3, wt% |
Fe3C, wt% |
S
total, m2 g−1 |
S
Int, m2 g−1 |
S
Ext., m2 g−1 |
S
apparent*, m2 g−1 |
V
pore, cm3 g−1 |
V
apparent*, cm3 g−1 |
D
pore, nm |
|
*Surface area calculated on per gram of pure carbon. **Average pore size not available due to the large portion of micropores. C-xxM(w) indicates the C-xxM after 1.0 M HCl acid washing.
|
| C-00M |
1.6 |
0 |
0 |
396.5 |
351.0 |
45.5 |
396.5 |
0.03 |
0.03 |
—** |
| C-01M |
7.4 |
5.8 |
4.4 |
410.0 |
209.0 |
201.0 |
429.1 |
0.16 |
0.17 |
3.9 |
| C-03M |
15.1 |
13.5 |
10.1 |
251.7 |
46.9 |
204.8 |
278.0 |
0.25 |
0.29 |
5.4 |
| C-05M |
23.7 |
22.1 |
16.6 |
176.2 |
1.0 |
175.2 |
211.3 |
0.24 |
0.29 |
5.9 |
| C-10M |
42.6 |
41 |
30.8 |
154.0 |
0.3 |
153.8 |
222.5 |
0.21 |
0.30 |
5.8 |
| C-01M(w) |
1.9 |
0.3 |
0.2 |
254.9 |
72.1 |
182.8 |
255.4 |
0.20 |
0.20 |
5.0 |
| C-03M(w) |
1.9 |
0.3 |
0.2 |
229.0 |
0 |
229.0 |
229.5 |
0.30 |
0.30 |
5.6 |
| C-05M(w) |
1.7 |
0.1 |
0.1 |
204.4 |
0 |
204.4 |
204.6 |
0.31 |
0.31 |
5.8 |
| C-10M(w) |
2.4 |
0.8 |
0.6 |
228.0 |
0 |
228.0 |
229.4 |
0.40 |
0.40 |
5.8 |
The specific surface area and average pore size of C-01M, C-03M, C-05M and C-10M were determined by nitrogen adsorption–desorption isotherm at 77.3 K, Fig. 3(c). C-00M shows typical microporous structure with a specific surface area of 396.5 m2 g−1, Fig. S2.†Fig. 3(c) reveals that all nanocomposites show typical type-IV curves, which confirmed the existence of mesopores. At relatively low pressure, samples first experience monolayer adsorption, followed by multilayer adsorption, while at higher pressure region, the adsorbed volume increased continuously due to capillary condensation inside mesopores.43,44 All the hysteresis loops closed sharply at P/P0 ≈ 0.44 due to the existence of “ink-bottle” pores, which usually has a narrow entrance but large internal space. The BET surface area, apparent surface area (excluding the mass of Fe3C), average pore diameter, pore volume and apparent pore volume (excluding the mass of Fe3C) obtained from adsorption branches were summarized in Table 1. Among the nanocomposites, the highest surface area of 410.0 m2 g−1 was achieved with lowest nanoparticle loading in C-01M. The specific surface area decreases gradually with increasing nanoparticle loading, e.g. surface area of 251.7, 176.2 and 154.0 m2 g−1 was obtained in C-03M, C-05M and C-10M, respectively. The decreased specific surface area is majorly attributed to the expanded pore size as well as the mass contribution of heavier Fe3C nanoparticles. By excluding the weight fraction of Fe3C from the nanocomposites, apparent surface area was calculated and summarized in Table 1. As discussed before, the catalytic graphitization facilitates the mesopore formation. Therefore, samples with higher Fe3C loading possesses more mesopores instead of micropores and thus smaller specific surface area, which is in good agreement with the apparent surface area in Table 1 except for C-10M where nanoparticle aggregation became dominant. Similarly, by excluding the mass of Fe3C, apparent pore volume was calculated, Table 1. The apparent pore volume increased sharply from 0.03 (C-00M) to 0.17 cm3 g−1 (C-01M) and then stabilized at 0.29–0.30 cm3 g−1 for nanocomposites with higher loadings. The pore size distribution in the micropore region is calculated by density functional theory (DFT) method from CO2 adsorption branch, and the one in the mesopore region is calculated by Barret Joyner and Halenda (BJH) method from N2 adsorption branch. The combined pore size distribution is shown in Fig. S3.† The average pore diameter increased gradually from 3.9 to 5.9 nm with increasing nanoparticle loading majorly because of the expanded pore structure induced by the heterogeneous shrinkage during catalytic graphitization.
To understand the contribution of nanoparticle to the microstructure formation of the nanocomposites, an acid etching step by 1.0 M HCl was applied to remove the metallic component of the nanocomposites. The solid residue after thermal decomposition is about the same for all the etched samples as compared to C-00M, Fig. 3(b), indicating the iron species can be completely removed. The total surface area distributes within the range of 204.4–254.9 m2 g−1 with negligible internal surface area in C-03M(w), C-05M(w) and C-10M(w), Fig. 3(d) and Table 1. Comparing the apparent pore volume results of the nanocomposites before and after acid washing, it is obvious that new pores were generated after removing the iron element. Especially, 33% higher apparent volume is observed in C-10M(w) than that of C-10M, indicating a large portion of nanoparticles were embedded inside the bulk carbon rather than at the surface. These embedded nanoparticles are responsible for the newly generated pores.
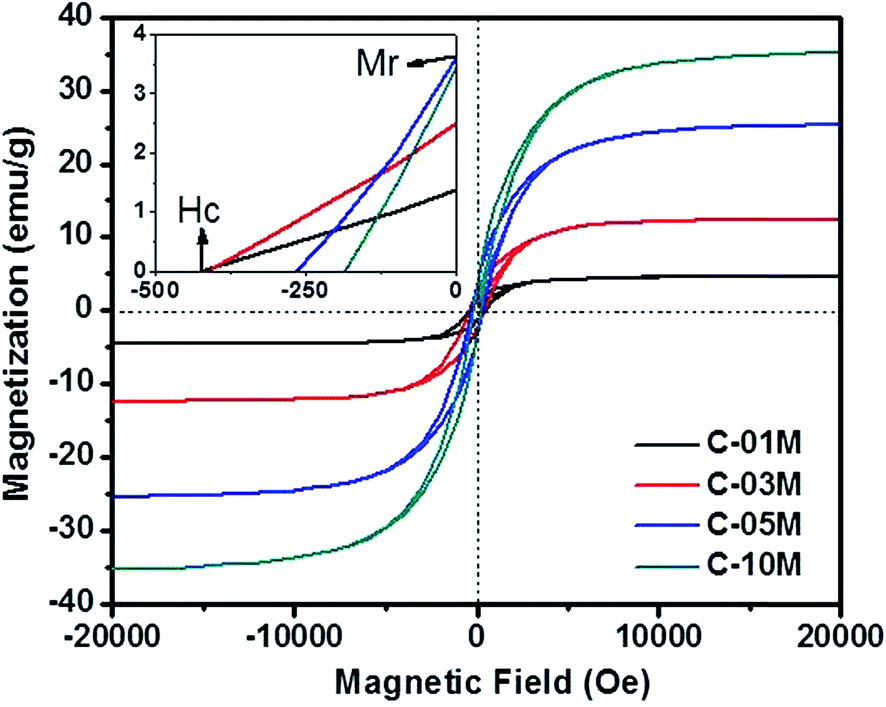
The magnetic property of the nanocomposites is plotted in Fig. 4. Considering the weight fraction of Fe3C in each C-xxM (xx = 01, 03, 05 and 10) nanocomposites and the saturated magnetization of 140 emu g−1 of bulk Fe3C,41 the calculated magnetization value is in good agreement with the measured results of 4.6, 12.4, 25.4 and 35.3 emu g−1 in C-01M, C-03M, C-05M and C-10M, respectively. The coercivity (Hc, Oe) indicates the external applied magnetic field necessary to return the material to a zero magnetization condition, and the remnant magnetization (Mr) is the residue magnetization after the applied field is reduced to zero. Both values can be read from the axes crossing points, inset of Fig. 4. The coercivity decreases continuously from 425 to 184 Oe with increasing nanoparticle loading. The decreased coercivity indicates the Fe3C nanoparticles become magnetically softer due to the increased interparticle dipolar interaction arising from the decreased nanoparticle spacer distance for the single domain nanoparticles.45,46
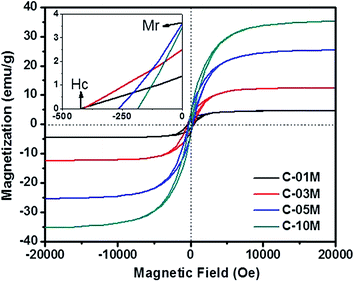 |
| | Fig. 4 Magnetic property of C-01M, C-03M, C-05M and C-10M. | |
3.2 Organic dye adsorption
To determine the dye concentration in polluted water, highly linear standard curves relating dye concentration and UV-Vis peak intensity have been plotted for MB at wavelength of 664 nm and MO at 450 nm, Fig. S4 and S5.† Remaining dye concentration in treated water can be obtained by referencing to the corresponding peak intensity from standard curves. To quantify the adsorption capacity of C-00M, C-01M, C-03M, C-05M and C-10M, isotherm adsorption was performed at room temperature for 12 hours. The isotherm adsorption results were then analyzed by Langmuir model, eqn (3):| | 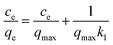 | (3) |
where ce and qe are dye concentration remained in the solution (mg L−1) and the amount of dye adsorbed onto adsorbent (mg g−1) at equilibrium, respectively. qmax is the maximum adsorption capacity (mg g−1) and k1 represents the energy constant related to the energy of adsorption. qe can be calculated by eqn (4):| | 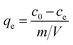 | (4) |
where c0 and ce are initial and equilibrium pollutant concentrations (mg L−1), m is the mass of adsorbent (g) and V is volume of polluted solution (L). qmax and k1 can be determined from the slope and interception of the linearly fitted ce/qe ∼ ce curve.
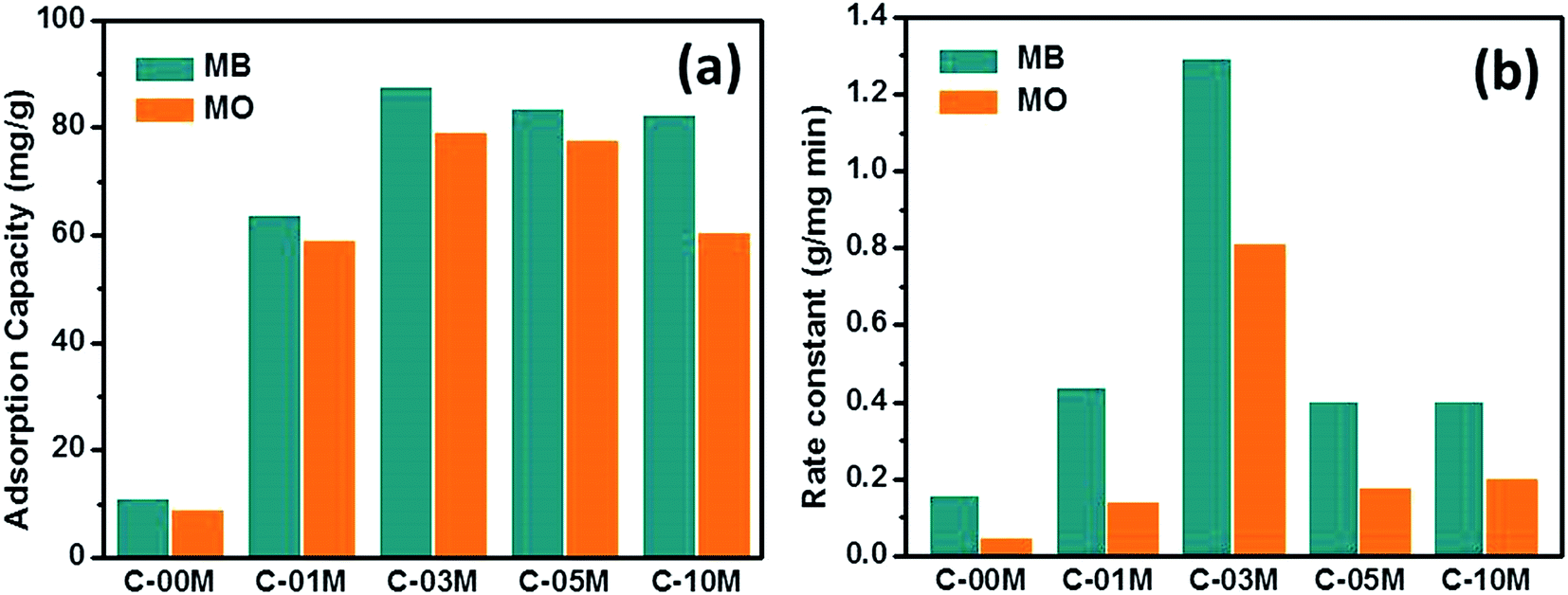
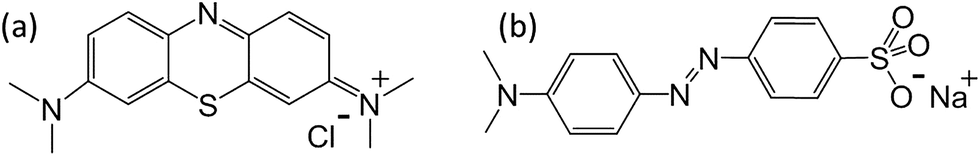
The qmax obtained from Langmuir model and rate constant calculated from pseudo-second order model were shown in Fig. 5(a and b). The details of the non-linear fitting are provided in Fig. S6–S9.† For pure carbon C-00M, the qmax is measured as 10.7 and 8.7 mg g−1 for MB and MO, respectively. The nanocomposites show more than 5 times larger adsorption capacity compared to C-00M. The improved adsorption capacity is mainly attributed to the newly created mesoporous structure, which provides sufficient large pore channels for dye molecule diffusion and extra active sites for adsorption. Although the maximum adsorption capacity is relatively low compared to activated carbon, the adsorption efficiency (defined as: mg dye per m2 adsorbent) is relatively larger compared to activated carbon manufactured from other biomass resources, Table S1.† All the tested samples show higher adsorption capacity for MB rather than MO, even though MB molecule has relatively larger hydrodynamic radius of ∼4.9 Å (ref. 47) compared to ∼4.5 Å (ref. 48) of MO. Organic dye adsorption on carbon materials has been explained by strong π–π stacking in previous literature.49 Considering the molecular structures of MB and MO as seen in Scheme 1, it is obvious that MB shows higher order of conjugation, where the free electrons are more centralized within the domain of benzene and six-membered hetero-ring. Thus, stronger π–π interaction could be expected between MB and adsorbents that leads to higher adsorption capacity.
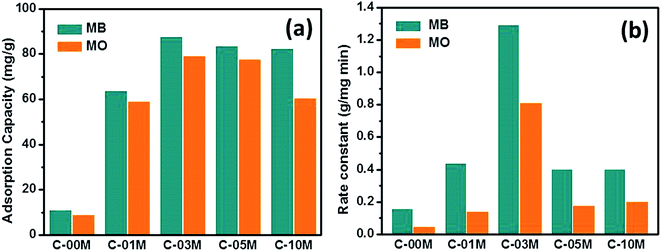 |
| | Fig. 5 (a) Adsorption capacity and (b) adsorption rate constant of C-xxM (xx = 00, 01, 03, 05 and 10) with MB and MO. | |
 |
| | Scheme 1 Molecular structure of (a) MB and (b) MO. | |
Similar to the results from isotherm study, rate constant of the nanocomposites is much higher than pure carbon and highest adsorption rate was achieved in C-03M, Fig. 5(b). Different from isotherm results, all samples show obviously higher adsorption rate for MB, which could be attributed to the different interaction modes between adsorbate and adsorbent. Zeta potential was then measured on each sample and it was found that all the samples exhibit negative surface potential, Fig. S10,† indicating the surfaces are negatively charged. When negative surface encounters cationic MB molecules, charge attraction dominates the interfacial interaction and consequently leads to faster adsorption rate. However, when negative surface encounters anionic MO molecules, charge repulsion force dominates the interfacial behavior and thus results in smaller rate constant.
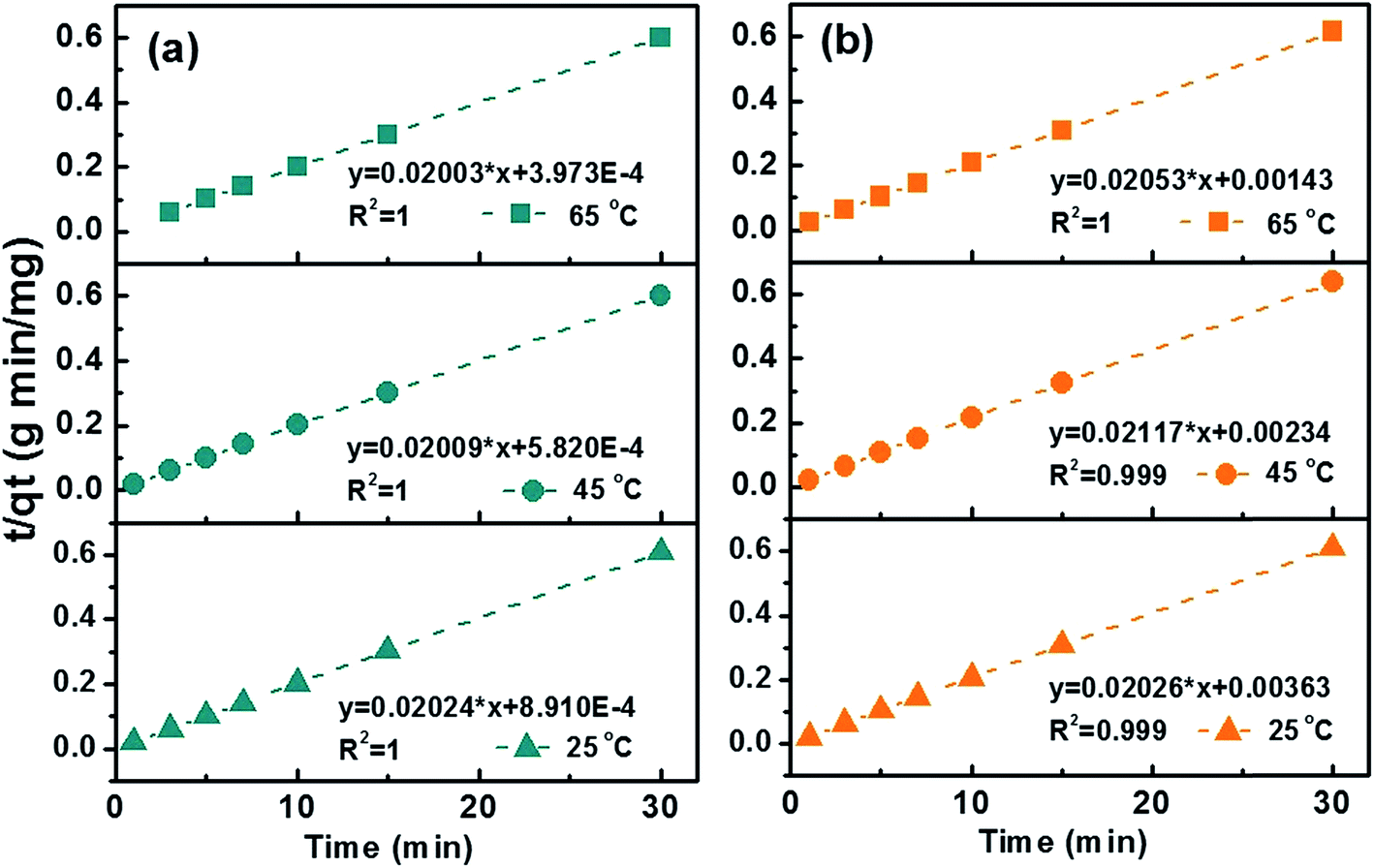
Kinetic studies were then conducted at three different temperatures, 25, 45 and 65 °C to quantify adsorption rate and activation energy, Fig. 6. Pseudo-second-order model demonstrated the highest fitting accuracy (R2 > 0.999) among the four tested models including pseudo-first-order, pseudo-second-order, intraparticle diffusion, and Elovich models, which can be described with eqn (5):
| | 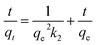 | (5) |
where
t is the sampling time (min),
qt is the dye concentration at time
t,
qe is the amount of dye adsorbed on to adsorbent (mg g
−1) at equilibrium.
k2 is the pseudo-second-order rate constant (g mg
−1 min
−1). The values of
qe and
k2 can be determined from the slope and intercept of the plot of
t/
qt against
t. Generally, the rate constant increases with increasing adsorption temperature for both MB (0.46, 0.69 and 1.01 g mg
−1 min
−1 at 25, 45 and 65 °C) and MO (0.11, 0.19 and 0.29 g mg
−1 min
−1 at 25, 45 and 65 °C). Compared to previous literatures,
50–52 the expedited adsorption rate could be attributed to the hybrid meso-/micro-porous structure which facilitate the fast diffusion of pollutant molecules to active sites. The increased rate constant at higher temperature is attributed to the accelerated diffusion of dye molecules.
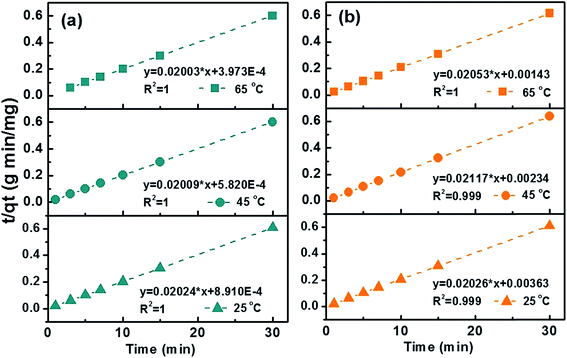 |
| | Fig. 6 Transformed rate plot t/qtvs. t with linear fitting for (a) MB and (b) MO at 25, 45 and 65 °C with C-10M. | |
With known rate constant at different temperatures, activation energy Ea can be determined based on the Arrhenius equation, eqn (6):
| | 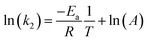 | (6) |
where
T is the absolute temperature (K),
A is the pre-exponential factor,
Ea is the activation energy, and
R is the universal gas constant. By linear fitting the ln(
k) ∼ 1/
T plot,
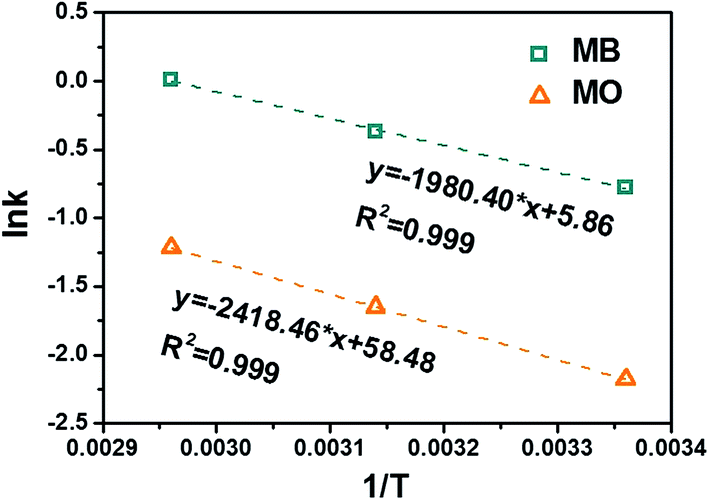
Fig. 7, the
Ea can be determined as 16.5 kJ mol
−1 for MB and 20.1 kJ mol
−1 for MO. The higher
Ea in removing MO is likely caused by the electrostatic repulsion between the adsorbent and MO molecules.
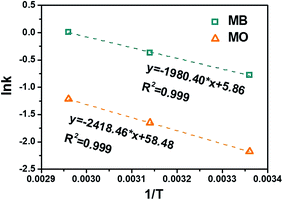 |
| | Fig. 7 Arrhenius plot of MB and MO with C-10M. | |
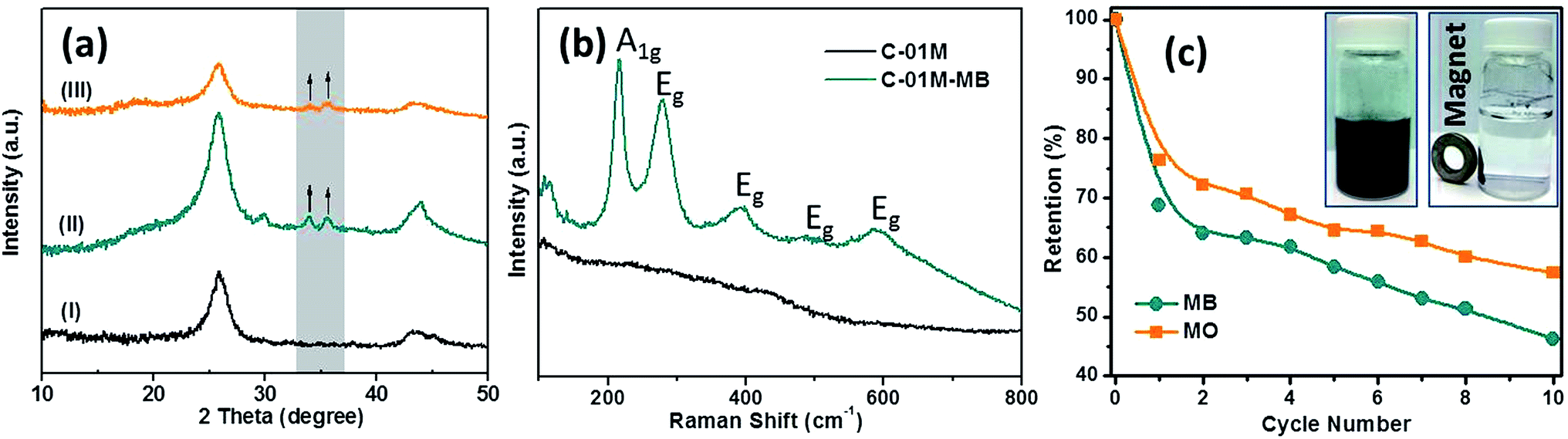
To understand the removal mechanism, XRD patterns of C-01M were collected before and after dye adsorption, Fig. 8. The newly formed peaks at 2θ = 30.5, 33.8 and 35.7° after absorbing MB are indexed to (220), (310) and (311) crystal planes of Fe2O3 (PDF # 02-1047), Fig. 8(a-II), indicating the redox reaction also occurred between Fe3C nanoparticles and MB molecules during adsorption. Similar peaks were also found in C-01M after absorbing MO, Fig. 8(a-III). Raman spectrum further confirms the oxidized form of iron with the newly formed Raman modes of A1g and Eg,53Fig. 8(b). To test recyclability of adsorbent, C-01M was firstly exposed to excess amount of MB (or MO) solutions for 12 hours and then regenerated in N2 atmosphere at 500 °C for 1 hour with a heating rate of 10 °C min−1. The adsorption capacity retention up to 10 cycles was shown in Fig. 8(c). The major adsorption capacity loss is occurred during 1st cycle, which accounts for 31.3% for MB and 23.7% for MO. Afterwards, slower capacity loss was observed in the following 9 cycles with 10th cycle retention of 57% and 46% for MO and MB respectively. To monitor the possible iron leaching during adsorption, the iron concentration was continuously measured with ICP in the testing solution for up to 10 hours. Results reveal that the iron level is <0.1 ppm with organic dye adsorption and <0.4 ppm with metal ion adsorption, Fig. S11.† Therefore, the secondary pollution by possible iron leaching could be neglected.
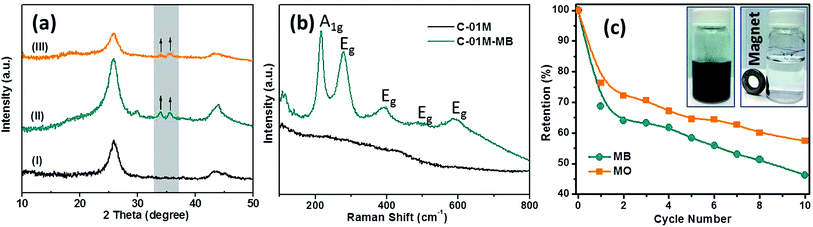 |
| | Fig. 8 XRD patterns of C-01M (a-I) before and after absorbing (a-II) MB and (a-III) MO, (b) Raman spectrum of C-01M before and after absorbing MB, and (c) cycling retention tests of C-01M, the two insets showed C-01M in water before (left) and after (right) attraction by a permanent magnet. | |
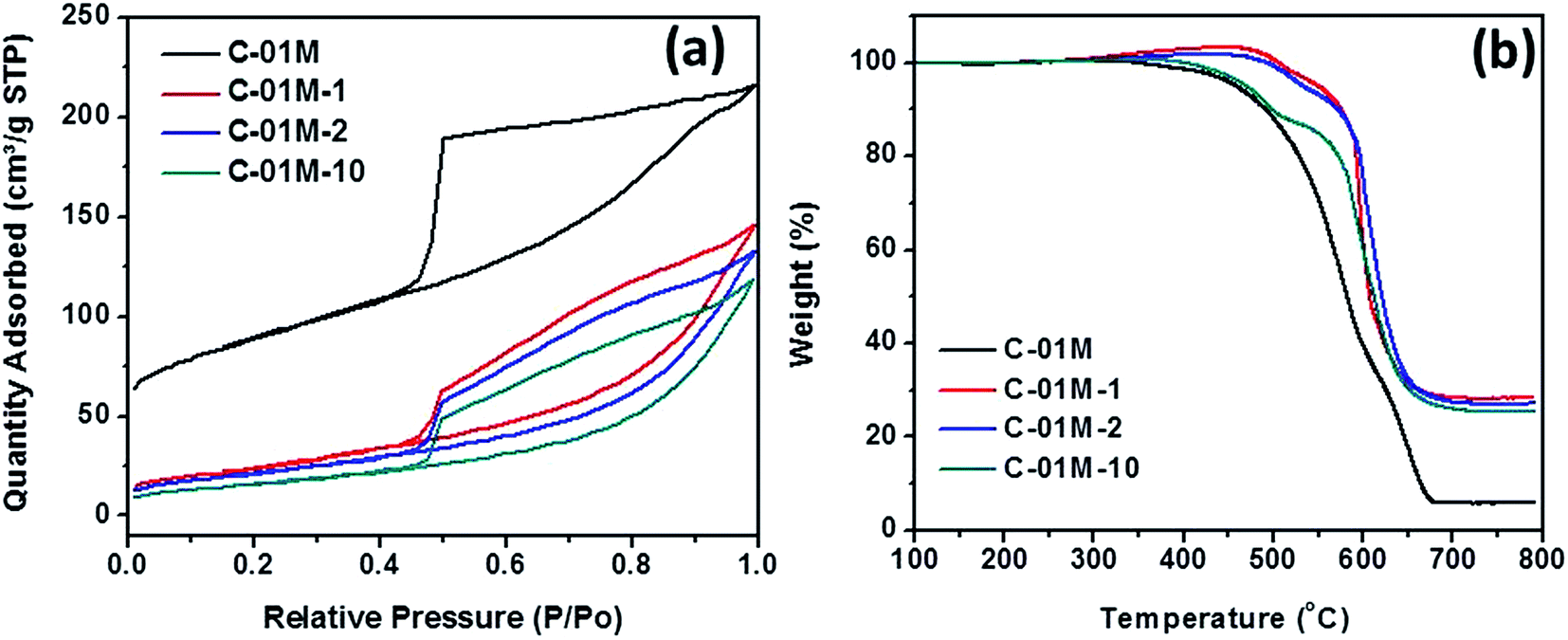
The adsorbent C-01M is further characterized by BET and TGA to analyze the structure and component change after regeneration. The as-prepared C-01M and C-01M after 1st, 2nd and 10th regeneration (named C-01M, C-01M-1, C-01M-2 and C-01M-10) were selected for analysis. It is obvious that the major pore structural change occurred after 1st regeneration with significant decrease of surface area from 410 to 79 m2 g−1 and increase of average pore size from 3.9 to 6.1 nm, Fig. 9(a). After 1st regeneration, the weight percentage of carbon in C-01M dropped from 94.6 to 80.0 wt%, Fig. 9(b). The structural and composition of C-01M do not change significantly from 2nd to 10th regeneration (Fig. 9), which well explains the relatively slow decay of adsorption capacity after 1st regeneration.
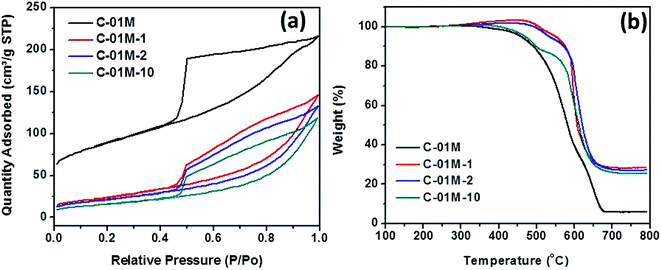 |
| | Fig. 9 (a) BET isotherms and (b) TGA of C-01M after different regeneration cycles. | |
3.3 Selective heavy metal removal
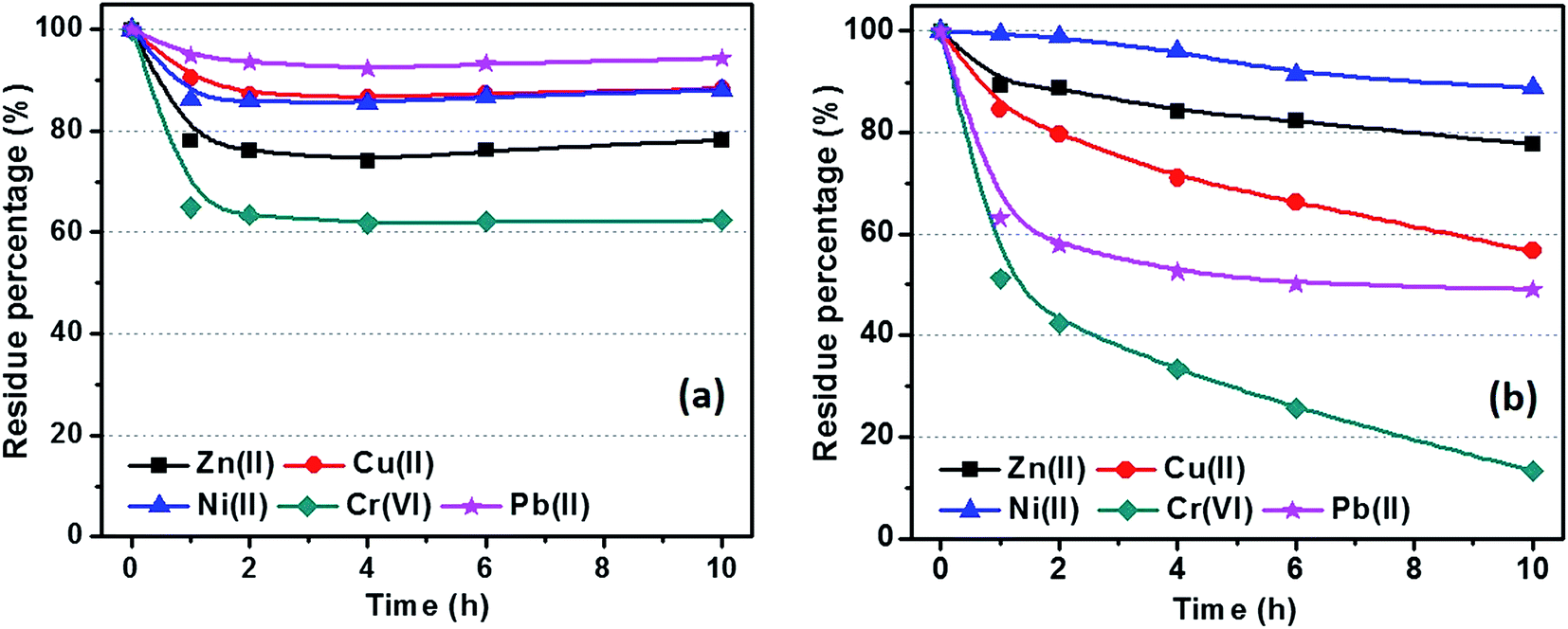
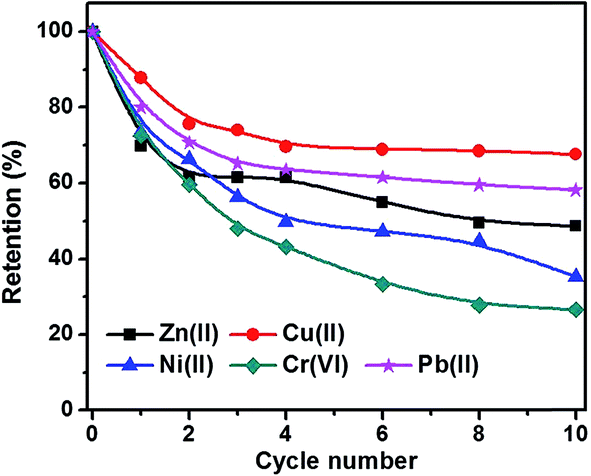
Fig. 10 shows the metal ion residue percentage in solution as a function of adsorption time. The adsorption equilibrium can be reached within 2 hours by using C-00M adsorbent and it shows selective adsorption for different metal ions Cr(VI) > Zn(II) > Ni(II) > Cu(II) > Pb(II), Fig. 10(a). After 10 hours adsorption, over 60% of all the metal ions are still remained in the water, especially for Pb(II) with over 90% remained. For C-03M, the adsorption continuously proceeds within the first 10 hours probably due to the gradual diffusion of pollutants from external surface (mesopores) to inner surface (micropores). More interestingly, it shows higher selectivity towards more toxic species such as Cr(VI) and Pb(II) rather than less toxic Cu(II), Zn(II) and Ni(II) ions. Also, it's worth mentioning that no Cr(VI) can be detected in the remaining solution after 24 hours adsorption. Two possible mechanisms are proposed to explain the higher adsorption capacity of C-03M compared to C-00M. Firstly, the larger pore size of C-03M provides easy-accessible adsorption sites for heavy metal ion adsorption. Secondly, the more negative zeta potential of C-03M (−25.64 mV) than C-00M (−11.28 mV), Fig. S10,† leads to stronger electrostatic interaction between adsorbent and cationic heavy metals ions and thus larger adsorption capacity. The adsorption capacity of metal ions with C-03M follows the order of Cr(VI) > Pb(II) > Cu(II) > Ni(II) > Zn(II). Isotherm was then performed with different initial metal ion concentrations of 25, 50, 75 and 100 ppm and the fitting results with Freundlich isotherm model were summarized in, Table S2.† The adsorption capacity parameter Kf in Table S2† shows high consistency with the adsorption results in Fig. 10(b). Generally, selective adsorption of heavy metals can be achieved by grafting functional groups54–57 and imprint specific ion species,58 which require extra step for surface functionalization not even mention the chemicals involved during material preparation. Since the mesopore size in the nanocomposites (in nm order) is much larger than the hydrodynamic radius of the metal ions (in Å order), it is not supposedly to restrict ion movement through gating-effect. Therefore, the selectivity is attributed to the interfacial interaction, e.g. electrostatic attraction and ion exchange,59 between embedded iron carbide nanoparticles and adsorbates. The adsorption capacity for each metal ion decreases with increasing regeneration cycles with observed major decrease during the first three cycles, Fig. 11. The adsorption of Cr(VI) degrades the fastest among the five metal ions (∼30% remaining after 10 cycles), followed by Ni(II), Zn(II), Pb(II) and Cu(II). One major advantage of the nanocomposites prepared in this work is the achieved superior selectivity towards more toxic metal ion species without additional surface modification procedure.
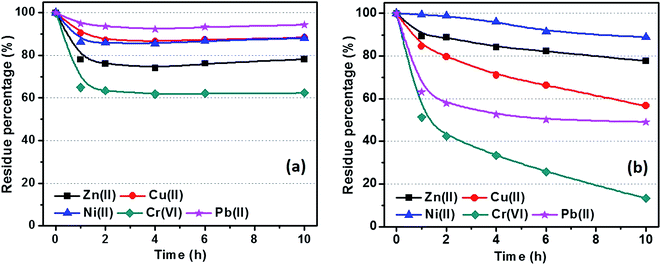 |
| | Fig. 10 Isothermal adsorption of metal ions with (a) C-00M and (b) C-03M. [Adsorbent] = 1.0 g L−1, [metal ion] = 25.0 ppm and volume of tested solution: 40.0 mL. | |
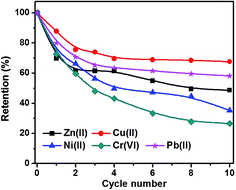 |
| | Fig. 11 Cycling retention tests for heavy metal ion adsorption. | |
4. Conclusion
In sum, mesoporous carbon/iron carbide nanocomposites have been synthesized via a facile impregnation–carbonization process by using sustainable biomass-cotton as source material. The pore structure of the nanocomposites can be easily regulated by adjusting iron catalyst loading during carbonization (catalytic graphitization) and large surface area of >300 m2 g−1 can be obtained. The graphitization induced mesopores are demonstrated essential structural component for the significantly enhanced adsorption capacity for organic dyes. The embedded iron carbide are responsible for the highly selectivity towards toxic heavy metal ions. Kinetic study reveals the pseudo-second-order adsorption behavior with organic dyes. Adsorption capacitance retention can be stabilized after regeneration. Besides, the magnetic property of these nanocomposites facilitates their easy separation from pollutant solution by a permanent magnet. The unique dual function towards both organic dye and heavy metal adsorption allows this mesoporous carbon/iron carbide nanocomposites promising applications in practical polluted water treatment not only because the effective adsorption property but also the widely accessible raw material, easy fabrication, low cost and minimum environmental impacts.
Acknowledgements
Acknowledgement is made to the Donors of the American Chemical Society Petroleum Research Fund for support of this research (#55570-DNI10). Partial support from start-up fund, Faculty Research Committee, Biomimicry Research Incentive Center and Firestone Faculty Research Fellowship from The University of Akron are also acknowledged. DPY acknowledges support from the NSF under grant no. DMR1306392. HRTEM was performed at the Liquid Crystal Institute, Kent State University supported by the Ohio Research Scholars Program Research Cluster on Surfaces in Advanced Materials. The authors appreciate the technical support from Dr Min Gao with HRTEM.
References
- A. J. Whelton, L. McMillan, M. Connell, K. M. Kelley, J. P. Gill, K. D. White, R. Gupta, R. Dey and C. Novy, Environ. Sci. Technol., 2015, 49, 813–823 CrossRef CAS PubMed.
- N. S. Galbraith, N. J. Barrett and R. Stanwell-Smith, Water Environ. J., 1987, 1, 7–21 CrossRef.
-
L. West, http://www.environment.about.com/od/environmentalevents/a/waterdayqa.htm, 2006.
- J. Li, C. Chen, S. Zhang and X. Wang, Environ. Sci.: Nano, 2014, 1, 488–495 RSC.
- S. Zhu, S. Jiao, Z. Liu, G. Pang and S. Feng, Environ. Sci.: Nano, 2014, 1, 172–180 RSC.
- J. Zhu, S. Wei, H. Gu, S. B. Rapole, Q. Wang, Z. Luo, N. Haldolaarachchige, D. P. Young and Z. Guo, Environ. Sci. Technol., 2011, 46, 977–985 CrossRef PubMed.
- J. Zhu, H. Gu, S. B. Rapole, Z. Luo, S. Pallavkar, N. Haldolaarachchige, T. J. Benson, T. C. Ho, J. Hopper, D. P. Young, S. Wei and Z. Guo, RSC Adv., 2012, 2, 4844–4856 RSC.
- M. Li, C. Wang, M. J. O'Connell and C. K. Chan, Environ. Sci.: Nano, 2015, 2, 245–250 RSC.
- S. S. Lee, W. Li, C. Kim, M. Cho, J. G. Catalano, B. J. Lafferty, P. Decuzzi and J. D. Fortner, Environ. Sci.: Nano, 2015, 2, 500–508 RSC.
- A. G. El Samrani, B. S. Lartiges and F. Villiéras, Water Res., 2008, 42, 951–960 CrossRef CAS PubMed.
- W.-P. Zhu, S.-P. Sun, J. Gao, F.-J. Fu and T.-S. Chung, J. Membr. Sci., 2014, 456, 117–127 CrossRef CAS.
- Z. Thong, G. Han, Y. Cui, J. Gao, T.-S. Chung, S. Y. Chan and S. Wei, Environ. Sci. Technol., 2014, 48, 13880–13887 CrossRef CAS PubMed.
- W.-P. Zhu, J. Gao, S.-P. Sun, S. Zhang and T.-S. Chung, J. Membr. Sci., 2015, 487, 117–126 CrossRef CAS.
- H. Li and L. Zou, Desalination, 2011, 275, 62–66 CrossRef CAS.
- X. Meng, D. A. Vaccari, J. Zhang, A. Fiume and X. Meng, Environ. Sci. Technol., 2014, 48, 1541–1548 CrossRef CAS PubMed.
- C. E. Barrera-Díaz, V. Lugo-Lugo and B. Bilyeu, J. Hazard. Mater., 2012, 223–224, 1–12 CrossRef PubMed.
- N. Kongsricharoern and C. Polprasert, Water Sci. Technol., 1995, 31, 109–117 CrossRef CAS.
- X. Wang, P. Zhang, W. Wang, X. Lei and H. Yang, RSC Adv., 2015, 5, 57828–57832 RSC.
- J. Zhu, M. Chen, H. Wei, N. Yerra, N. Haldolaarachchige, Z. Luo, D. P. Young, T. C. Ho, S. Wei and Z. Guo, Nano Energy, 2014, 6, 180–192 CrossRef CAS.
- J. Zhu, M. Chen, N. Yerra, N. Haldolaarachchige, S. Pallavkar, Z. Luo, T. C. Ho, J. Hopper, D. P. Young, S. Wei and Z. Guo, Nanoscale, 2013, 5, 1825–1830 RSC.
- J. Zhu, H. Gu, J. Guo, M. Chen, H. Wei, Z. Luo, H. A. Colorado, N. Yerra, D. Ding, T. C. Ho, N. Haldolaarachchige, J. Hopper, D. P. Young, Z. Guo and S. Wei, J. Mater. Chem. A, 2014, 2, 2256–2265 CAS.
- E. Thompson, A. E. Danks, L. Bourgeois and Z. Schnepp, Green Chem., 2015, 17, 551–556 RSC.
- L. Chen, H. Wang, H. Wei, Z. Guo, M. A. Khan, D. P. Young and J. Zhu, RSC Adv., 2015, 5, 42540–42547 RSC.
- B. H. Hameed, A. T. M. Din and A. L. Ahmad, J. Hazard. Mater., 2007, 141, 819–825 CrossRef CAS PubMed.
- S. Altenor, B. Carene, E. Emmanuel, J. Lambert, J.-J. Ehrhardt and S. Gaspard, J. Hazard. Mater., 2009, 165, 1029–1039 CrossRef CAS PubMed.
- A. L. Ahmad, M. M. Loh and J. A. Aziz, Dyes Pigm., 2007, 75, 263–272 CrossRef CAS.
- B. H. Hameed, A. L. Ahmad and K. N. A. Latiff, Dyes Pigm., 2007, 75, 143–149 CrossRef CAS.
- N. R. Bishnoi, M. Bajaj, N. Sharma and A. Gupta, Bioresour. Technol., 2004, 91, 305–307 CrossRef CAS PubMed.
- M. Ahmaruzzaman and V. K. Gupta, Ind. Eng. Chem. Res., 2011, 50, 13589–13613 CrossRef CAS.
- J. M. Salman and B. H. Hameed, J. Hazard. Mater., 2010, 176, 814–819 CrossRef CAS PubMed.
- Z. A. Al-Othman, R. Ali and M. Naushad, Chem. Eng. J., 2012, 184, 238–247 CrossRef CAS.
-
P. Serp and B. Machado, in Nanostructured Carbon Materials for Catalysis, The Royal Society of Chemistry, 2015, ch. 1, pp. 1–45 Search PubMed.
- L. Chuenchom, R. Kraehnert and B. M. Smarsly, Soft Matter, 2012, 8, 10801–10812 RSC.
- S. A. Johnson, E. S. Brigham, P. J. Ollivier and T. E. Mallouk, Chem. Mater., 1997, 9, 2448–2458 CrossRef CAS.
- J. Rodriguez-Mirasol, T. Cordero, L. R. Radovic and J. J. Rodriguez, Chem. Mater., 1998, 10, 550–558 CrossRef CAS.
- Y. Zhai, Y. Dou, D. Zhao, P. F. Fulvio, R. T. Mayes and S. Dai, Adv. Mater., 2011, 23, 4828–4850 CrossRef CAS PubMed.
- J. S. Lee, A. Hirao and S. Nakahama, Macromolecules, 1988, 21, 274–276 CrossRef CAS.
- M. Templin, A. Franck, A. Du Chesne, H. Leist, Y. Zhang, R. Ulrich, V. Schädler and U. Wiesner, Science, 1997, 278, 1795–1798 CrossRef CAS PubMed.
- Y. Wan, X. Cui and Z. Wen, J. Hazard. Mater., 2011, 198, 216–223 CrossRef CAS PubMed.
- L. Chen, T. Ji, L. Brisbin and J. Zhu, ACS Appl. Mater. Interfaces, 2015, 7, 12230–12237 CAS.
- A. Wu, D. Liu, L. Tong, L. Yu and H. Yang, CrystEngComm, 2011, 13, 876–882 RSC.
-
J. Maire and J. Mering, in Chemistry and Physics of Carbon, ed. P. L. Walker and J. A. Thrower, Dekker, New York, 1971, vol. 6, pp. 125–190 Search PubMed.
- K. Morishige and N. Tarui, J. Phys. Chem. C, 2007, 111, 280–285 CAS.
- D. Wallacher, N. Künzner, D. Kovalev, N. Knorr and K. Knorr, Phys. Rev. Lett., 2004, 92, 195704 CrossRef CAS PubMed.
- Z. Guo, K. Lei, Y. Li, H. W. Ng, S. Prikhodko and H. T. Hahn, Compos. Sci. Technol., 2008, 68, 1513–1520 CrossRef CAS.
- J. Zhu, S. Wei, X. Chen, A. B. Karki, D. Rutman, D. P. Young and Z. Guo, J. Phys. Chem. C, 2010, 114, 8844–8850 CAS.
- M. Majumder, P. Sheath, J. I. Mardel, T. G. Harvey, A. W. Thornton, A. Gonzago, D. F. Kennedy, I. Madsen, J. W. Taylor, D. R. Turner and M. R. Hill, Chem. Mater., 2012, 24, 4647–4652 CrossRef CAS.
-
J. F. Urban, Nonlinear, Spatiotemporal Hydrogel Dynamics in Oscillatory Drug Release, University of Minnesota, doctoral dissertation, 2008.
- F. Liu, S. Chung, G. Oh and T. S. Seo, ACS Appl. Mater. Interfaces, 2011, 4, 922–927 Search PubMed.
- H. Deng, J. Lu, G. Li, G. Zhang and X. Wang, Chem. Eng. J., 2011, 172, 326–334 CrossRef CAS.
- L. Liu, Y. Lin, Y. Liu, H. Zhu and Q. He, J. Chem. Eng. Data, 2013, 58, 2248–2253 CrossRef CAS.
- S. Karagöz, T. Tay, S. Ucar and M. Erdem, Bioresour. Technol., 2008, 99, 6214–6222 CrossRef PubMed.
- S.-H. Shim and T. S. Duffy, Am. Mineral., 2002, 87, 318–326 CrossRef CAS.
- A. M. Liu, K. Hidajat, S. Kawi and D. Y. Zhao, Chem. Commun., 2000, 1145–1146 RSC.
- J. Brown, L. Mercier and T. J. Pinnavaia, Chem. Commun., 1999, 69–70 RSC.
- C. Liu, R. Bai and Q. San Ly, Water Res., 2008, 42, 1511–1522 CrossRef CAS PubMed.
- T. Kang, Y. Park and J. Yi, Ind. Eng. Chem. Res., 2004, 43, 1478–1484 CrossRef CAS.
- G.-Z. Fang, J. Tan and X.-P. Yan, Anal. Chem., 2005, 77, 1734–1739 CrossRef CAS PubMed.
- J. Hu, G. Chen and I. Lo, J. Environ. Eng., 2006, 132, 709–715 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra19616g |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence