
Efficient construction of the A/C/D tricyclic skeleton of palhinine A†
Shuangshuang
Duan
a,
Dan
Long
a,
Changgui
Zhao
a,
Gaoyuan
Zhao
a,
Ziyun
Yuan
a,
Xingang
Xie
a,
Jianguo
Fang
a and
Xuegong
She
*ab
aState Key Laboratory of Applied Organic Chemistry, Department of Chemistry, Lanzhou University, Lanzhou, 730000, China. E-mail: shexg@lzu.edu.cn
bCollaborative Innovation Center of Chemical Science and Engineering (Tianjin), Tianjin, 30071, China
First published on 18th July 2016
Abstract
An efficient approach for the synthesis of the 9/6/6 tricyclic structure of Lycopodium alkaloid palhinine A has been accomplished. The developed synthetic route features oxidative dearomatization and tandem hydroxyl oxidation/intramolecular Diels–Alder (IMDA) reactions to assemble the A/C/D tricyclic ring system. Most importantly, the protocol can undergo ring constriction to rapidly construct the highly strained nine-membered azonane ring of palhinine A.
Introduction
The Lycopodium alkaloid1 palhinine A (1) along with palhinine B (2), C (3) and isopalhinine A (4) was isolated from the nodding clubmoss Palhinhaea cernua by Long et al. and Zhao et al., respectively.2 As a novel C16N-type Lycopodium alkaloid, palhinine A (1) was found to have a unique 9/5/6/6 tetracyclic ring system (Fig. 1). However, little is known about the biological activities of palhinines, with the exception of their ineffectiveness against acetylcholinesterase, butyryl-cholinesterase and human chronic myelogenous leukemia K562 cells.2a As a consequence, there is an urgent need to identify and produce the promising palhinine family and its analogues in a large quantity.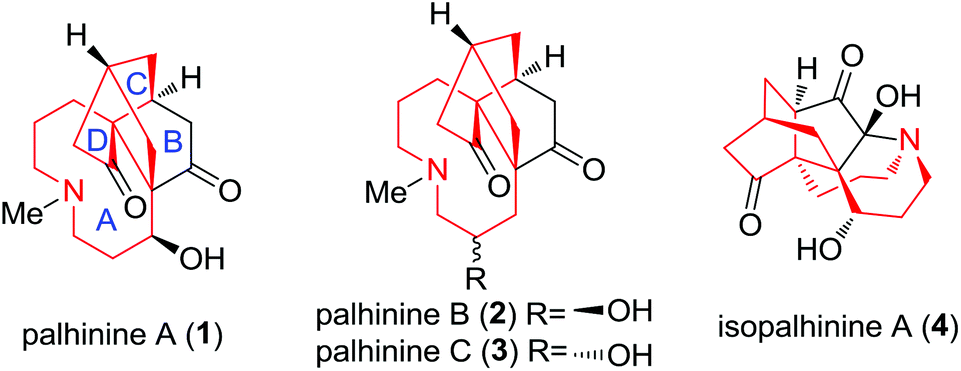 | ||
| Fig. 1 Chemical structures of palhinines. | ||
Given their biological activity and intriguing molecular architecture, the synthesis of palhinines has been widely explored.3 Our group accomplished the 6/6/5 tricyclic ring system by a tandem oxidative dearomatization/intramolecular Diels–Alder reaction4,5 and an intramolecular radical cyclization. At the same time, Fan and co-workers developed a concise strategy to build the isotwistane core3b,6 using the Nozaki–Hiyama–Kishi/IMDA reaction. The Maier group described their approach for constructing the isotwistane core by employing a domino Michael/Arndt–Eistert homologation/intramolecular aldol sequence. Rychnovsky and co-workers utilized a Morita–Baylis–Hillman/IMDA strategy to complete the isotwistane core as well.
Unfortunately, all the elegant previous studies were incomplete with respect to the total syntheses of palhinines with the deficiency of the nine-membered azonane ring. After the construction of the isotwistane core, we have made many attempts to achieve the A ring, but none of them gave the desired result, which made the nine-membered azonane ring the most challenging problem. Although a series of studies on the establishment of the medium-sized ring have been reported,7–11 the much more strained nine-membered azonane ring fused with the caged structure of bicycle[2.2.2]octanone has not been accomplished. As a continuation of our previous work,3a we wish to describe the synthesis of the A/C/D tricyclic core of palhinine A (1) through ring constriction from a macro-sized ring in order to overcome the strain of the nine-membered azonane ring.12
Results and discussion
Our retrosynthetic analysis focusing on completion of the most troublesome A ring is illustrated in Scheme 1. We proposed that 1 would be conceived by constructing the cyclopentanone through homologation and cyclization from intermediate 5, while 5 could be generated from dienophile 6via oxidative dearomatization and intramolecular Diels–Alder reactions. This strategy offered an efficient approach to set the required quaternary stereocenters simultaneously and build a [2.2.2] bridged-ring system along with the nine-membered azonane ring in one step. It should be noted that the intramolecular Diels–Alder strategy might provide an alternative route for the establishment of the framework of 1 as well as its structurally related molecules. Installation of the dienophile fragment of 6 might be realized through the Morita–Baylis–Hillman reaction from aldehyde 7, which might be accessible from ester 8via N-alkylation and several standard manipulations.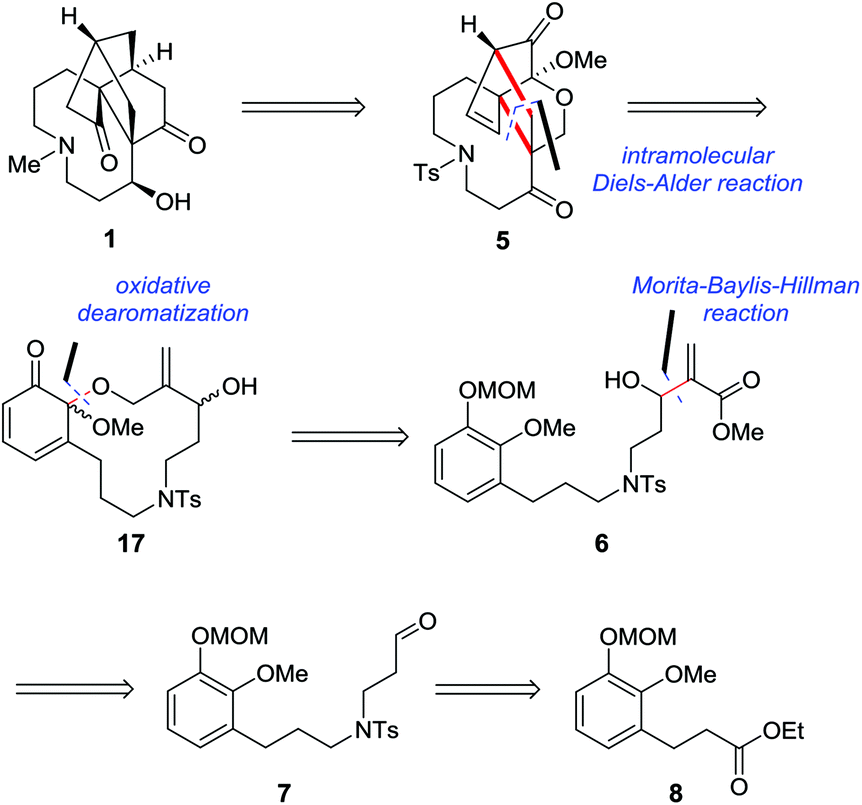 | ||
| Scheme 1 Retrosynthetic analysis of palhinine A (1). | ||
On the basis of the above analysis, our synthesis commenced with the known ester 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 3a (Scheme 2). Reduction of ester 8 with LiAlH4 followed by treatment of the resulting alcohol with mesyl chloride resulted in the formation of the corresponding mesylate 9 in 90% yield over two steps.
3a (Scheme 2). Reduction of ester 8 with LiAlH4 followed by treatment of the resulting alcohol with mesyl chloride resulted in the formation of the corresponding mesylate 9 in 90% yield over two steps.
 | ||
| Scheme 2 Synthesis of dienophile 6. | ||
Conversion of mesylate 9 to sulfonamide 10 was achieved through a three-step sequence involving azide generation, amine formation and sulfonylation in 78% yield. Then sulfonamide 10 was reacted with propylene glycol derivative 1113 to afford 12,14 which was subjected to deprotection to provide alcohol 13 in 90% yield. The Swern oxidation of 13 proceeded smoothly to afford aldehyde 7 in excellent yield. During the period of synthesis of the dienophile fragment of 6, various reaction conditions were screened.3d,15 Among them, the optimized condition was that the reaction should be performed under an atmosphere of Ar with methyl acrylate being used as a solvent and DABCO as a base to provide 6 in 52% yield.
Next, as shown in Scheme 3, we investigated the synthesis of key intermediate 17. Protection of the secondary hydroxyl group of dienophile 6 followed by reduction of the ester group gave the corresponding primary alcohol 15.16 Exposure of 15 to 2 M HCl resulted in diol 16 in 86% yield. When diol 16 was treated with PhI(OCOCF3)2 under open air at 0 °C, an unexpected ortho-quinone was delivered as a major product with a small amount of 17. After many trials, the reaction was found to proceed smoothly to afford 17 as a mixture of diastereoisomers with a solution of PhI(OCOCF3)2 in CH2Cl2 being added dropwise at 0 °C under an inert atmosphere, and the yield could be increased to 60%.
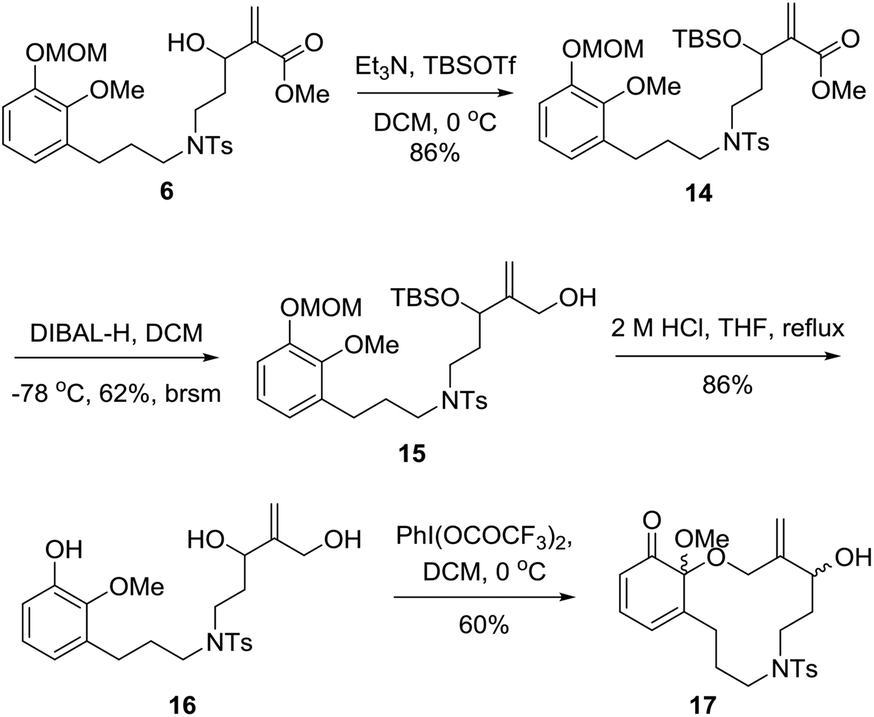 | ||
| Scheme 3 Synthesis of the IMDA precursor 17. | ||
As illustrated in Scheme 4, with 17 in hand, the stage was set for the development of the A/C/D tricyclic skeleton of palhinine A (1). Our initial attempt to apply 17 in refluxed toluene to promote the cyclization failed to give any desired products. In addition, Et2AlCl was also ineffective under typical reaction conditions. We postulated that the failure was attributed to the low reactivity of the substrate. To overcome this obstacle, the hydroxyl group was oxidized to convert the dienophile into the corresponding unsaturated ketone. Fortunately, when the hydroxyl group was oxidized with DMP (Dess–Martin periodinane) at 0 °C, the resulting enone was converted to 5 spontaneously in 42% yield, the structure of which was further confirmed by single-crystal X-ray diffraction analysis.17
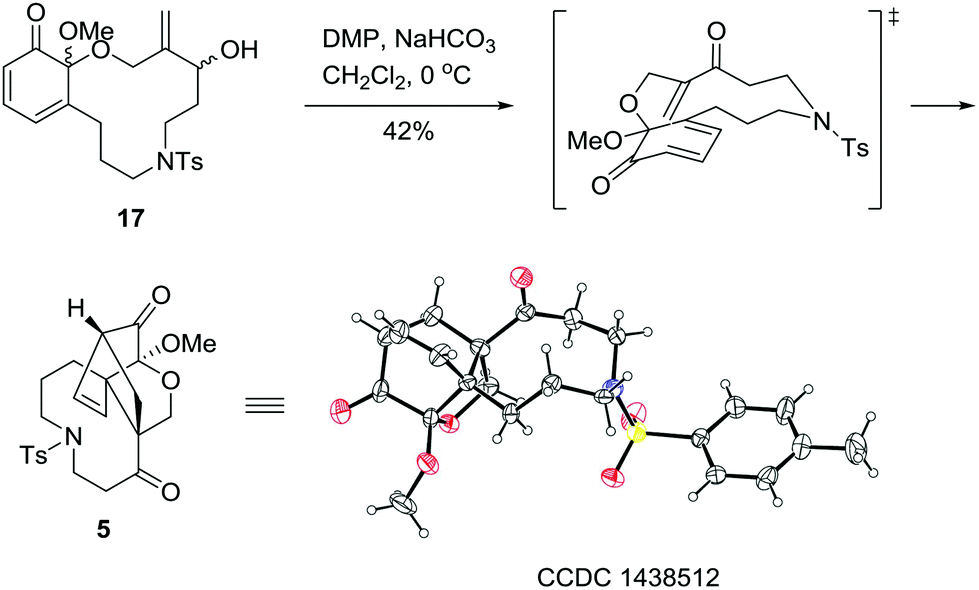 | ||
| Scheme 4 Synthesis of the A/C/D tricyclic core 5. | ||
Conclusion
In summary, an efficient approach based on oxidative dearomatization and tandem hydroxyl oxidation/intramolecular Diels–Alder reactions to construct the A/C/D tricyclic core of palhinine A (1) has been accomplished, while the most challenging nine-membered azonane ring fused with the caged structure of bicycle[2.2.2]octanone was accomplished in one step through ring constriction. Application of this strategy to the synthesis of palhinines is ongoing in our laboratory.Experimental section
General methods
Synthesis of 3-(2-methoxy-3-(methoxymethoxy)phenyl)propan-1-ol (9a). To a stirred solution of ester 8 (3.90 g, 14.5 mmol) in Et2O (150 mL) was added lithium aluminum hydride (660 mg, 17.4 mmol) in portions at 0 °C. After stirring at 25 °C for 3 h, the reaction mixture was quenched by adding H2O (660 mg), aqueous NaOH (w% = 10%, 1.32 g), and H2O (1.98 g) in turn at 0 °C. The solid formed was filtered and washed with EtOAc (5 × 20 mL). The organic layers were combined and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure and the residue was purified through silica gel column chromatography (n-hexane/EtOAc, 1
:1) to give the alcohol 9a (3.21 g, 14.2 mmol, 98% yield) as a colorless oil. IR νmax (neat)/cm−1: 3403, 2935, 2372, 1585, 1477, 1431, 1265, 1221, 1204, 1154, 1120; 1H NMR (400 MHz, CDCl3): δ 6.98 (m, 2H), 6.84 (dd, J = 7.2, 1.6 Hz, 1H), 5.22 (s, 2H), 3.86 (s, 3H), 3.59 (t, J = 5.6 Hz, 2H), 3.52 (s, 3H), 2.74 (t, J = 7.2 Hz, 2H), 2.22 (br, 1H), 1.84 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 150.08, 147.83, 135.55, 124.13, 123.38, 114.44, 95.04, 61.51, 60.84, 56.15, 33.41, 25.63; HRMS (ESIMS) calcd for C12H18O8Na1 [M + Na]+ 249.1097, found 249.1093.
Synthesis of 3-(2-methoxy-3-(methoxymethoxy)phenyl)propylmethanesulfonate (9). 9a, MsCl (1.6 mL, 21 mmol) and Et3N (6.1 mL, 43 mmol) were dissolved in CH2Cl2 (150 mL) at 0 °C and stirred at 25 °C for 5 h. The reaction mixture was quenched by adding H2O (50 mL) and extracted with CH2Cl2 (3 × 100 mL). The combined organic layers were washed with H2O (20 mL) and brine (20 mL), and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure and the residue was purified through silica gel column chromatography (n-hexane/EtOAc, 4
:1) to give 9 (3.98 g, 13.1 mmol, 92% yield) as a colourless oil. IR νmax (neat)/cm−1: 3399, 2938, 1601, 1585, 1478, 1354, 1267, 1222, 1175, 1087, 1006, 923; 1H NMR (400 MHz, CDCl3): δ 7.03 (dd, J = 8.0, 1.2 Hz, 1H), 6.97 (t, J = 8.0 Hz, 1H), 6.83 (d, J = 8.0 Hz, 1H), 5.22 (s, 2H), 4.24 (t, J = 6.4 Hz, 2H), 3.85 (s, 3H), 3.52 (s, 3H), 3.00 (s, 3H), 2.76 (t, J = 7.2 Hz, 2H), 2.06 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 150.31, 147.91, 134.37, 124.00, 123.27, 114.87, 95.02, 69.57, 60.70, 56.19, 37.27, 29.82, 26.03; HRMS (ESIMS) calcd for C13H20O6S1Na1 [M + Na]+ 327.0873, found 327.0865.
Synthesis of 1-(3-azidopropyl)-2-methoxy-3-(methoxymethoxy)benzene (10a). To a solution of 9 (3.98 g, 13.1 mmol) in anhydrous DMF (100 mL) was added NaN3 (1.02 g, 15.7 mmol) and a catalytic amount of NaI in one portion, and then the solution was heated at 70 °C for 2 h. After cooling to 25 °C, H2O (70 mL) was added to quench the reaction. The organic layer was separated and the aqueous layer was extracted with Et2O (3 × 100 mL). The combined organic layers were washed with H2O (20 mL) and brine (20 mL), dried over anhydrous Na2SO4, filtered and concentrated, and the residue was purified through silica gel column chromatography (n-hexane/EtOAc, 20
:1) to give the azide 10a (2.76 g, 11.0 mmol, 84% yield) as a colourless oil. IR νmax (neat)/cm−1: 3341, 2935, 2097, 1585, 1478, 1267, 1154, 1088, 1015, 925; 1H NMR (400 MHz, CDCl3): δ 7.02 (dd, J = 8.0, 1.6 Hz, 1H), 6.96 (t, J = 8.0 Hz, 1H), 6.82 (dd, J = 7.4, 1.6 Hz, 1H), 5.21 (s, 2H), 3.86 (s, 3H), 3.51 (s, 3H), 3.30 (t, J = 6.8 Hz, 2H), 2.71 (t, J = 7.2 Hz, 2H), 1.89 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 150.27, 147.89, 134.92, 123.87, 123.23, 114.69, 95.00, 60.63, 56.11, 50.81, 29.59, 27.11; HRMS (ESIMS) calcd for C12H17N3O3Na1 [M + Na]+ 274.1162, found 274.1158.
Synthesis of N-(3-(2-methoxy-3-(methoxymethoxy)phenyl)propyl)-4-methyl-benzenesulfonamide (10). 10a (2.76 g, 11.0 mmol) was dissolved in THF (100 mL). After the solution was cooled to 0 °C, lithium aluminum hydride (502 mg, 13.2 mmol) was added in portions and the reaction mixture was stirred for 3 h at 25 °C. The reaction mixture was quenched by adding H2O (502 mg), aqueous NaOH (w% = 10%, 1.00 g), and H2O (1.51 g) in turn at 0 °C. The solid formed was filtered and washed with EtOAc (10 × 10 mL). The organic layers were combined and dried over anhydrous Na2SO4. The solvent was removed to give the amine (2.23 g, 9.91 mmol, 90% yield) as a colourless oil without further purification.
The amine obtained, p-TsCl (2.26 g, 11.9 mmol) and Et3N (4.3 mL, 30 mmol) were dissolved in CH2Cl2 (100 mL) and stirred at 25 °C for 8 h. The reaction mixture was taken up in CH2Cl2 and washed successively with 1 M NaOH (2 × 20 mL), H2O (30 mL) and brine (30 mL). The organic layer was dried over anhydrous Na2SO4, filtered and concentrated. The residue was purified through silica gel column chromatography (n-hexane/EtOAc, 4:1) to give 10 (3.49 g, 9.21 mmol, 93% yield) as a clear crystal. M.p. 94–96 °C; IR νmax (neat)/cm−1: 3282, 2933, 1599, 1585, 1477, 1326, 1266, 1158, 1093, 1021; 1H NMR (400 MHz, CDCl3): δ 7.74 (d, J = 8.0 Hz, 2H), 7.27 (d, J = 8.0 Hz, 2H), 6.98 (dd, J = 8.4, 1.6 Hz, 1H), 6.91 (t, J = 7.6 Hz, 1H), 5.19 (s, 2H), 5.11 (t, J = 6.0 Hz, 1H), 3.82 (s, 3H), 3.50 (s, 3H), 2.89 (q, J = 6.4 Hz, 2H), 2.62 (t, J = 4.8 Hz, 2H), 2.40 (s, 3H), 1.75 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 150.09, 147.67, 143.08, 137.04, 134.73, 129.54, 126.98, 124.06, 123.21, 114.57, 94.98, 60.78, 56.13, 42.19, 30.33, 26.36, 21.40; HRMS (ESIMS) calcd for C19H25N1O5S1Na1 [M + Na]+ 402.1346, found 402.1339.
Synthesis of N-(3-((tert-butyldimethylsilyl)oxy)propyl)-N-(3-(2-methoxy-3-(methoxymethoxy)phenyl)propyl)-4-methyl benzenesulfonamide (12). 10 (2.46 g, 6.49 mmol) was dissolved in anhydrous DMF (4 L mol−1 amine) and the mixture was cooled to 0 °C. NaH (338 mg, 8.50 mmol, w% = 60%) was added in portions and the mixture was stirred at 0 °C until gas evolution ceased. A solution of 11 (2.09 g, 7.80 mmol) in DMF (2 L mol−1 amine) was added dropwise. The solution was heated at 70 °C overnight. After cooling to 25 °C, the reaction mixture was quenched with saturated aqueous NH4Cl (15 mL). Et2O (100 mL) was added and the organic layer was separated. The aqueous layer was extracted with Et2O (2 × 100 mL) and the combined organic layer was washed with H2O (5 × 30 mL). Then the organic phase was washed with brine (30 mL) and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure and the residue was purified through silica gel column chromatography (n-hexane/EtOAc, 10
:1) to give 12 (2.85 g, 5.17 mmol, 80% yield) as a colourless oil. IR νmax (neat)/cm−1: 3368, 2954, 2929, 2857, 1599, 1586, 1475, 1342, 1260, 1158, 1090, 1011; 1H NMR (400 MHz, CDCl3): δ 7.81 (d, J = 8.4 Hz, 2H), 7.41 (d, J = 8.0 Hz, 2H), 7.15–7.06 (m, 2H), 6.93 (d, J = 7.6 Hz, 1H), 5.34 (s, 2H), 3.95 (s, 3H), 3.72 (t, J = 6.0 Hz, 2H), 3.65 (s, 3H), 3.33 (q, J = 7.2 Hz, 4H), 2.74 (t, J = 8.0 Hz, 2H), 2.54 (s, 3H), 2.00–1.92 (m, 2H), 1.90–1.83 (m, 2H), 1.00 (s, 9H), 0.15 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 150.25, 147.86, 142.92, 136.85, 135.45, 129.55, 127.16, 123.90, 123.14, 114.58, 95.08, 60.70, 60.37, 56.18, 48.35, 45.36, 31.95, 29.39, 27.22, 25.85, 21.45, 18.19, −5.43, −5.47; HRMS (ESIMS) calcd for C28H45N1O6S1Si1Na1 [M + Na]+ 574.2629, found 574.2634.
Synthesis of N-(3-hydroxypropyl)-N-(3-(2-methoxy-3-(methoxymethoxy)phenyl)-propyl)-4-methylbenzene sulfonamide (13). 12 (2.63 g, 4.77 mmol) was dissolved in THF (50 mL). After the solution was cooled to 0 °C, TBAF·3H2O (1.80 g, 5.71 mmol) in THF (5.7 mL) was added dropwise. Then the reaction mixture was stirred at 25 °C for 3 h. The reaction mixture was quenched with saturated aqueous NH4Cl (10 mL) when it was completed (monitored by TLC), and the aqueous layer was extracted with EtOAc (3 × 70 mL). The combined organic layers were washed with H2O (20 mL) and brine (20 mL), dried over anhydrous Na2SO4. The solvent was removed under reduced pressure and the residue was purified through silica gel column chromatography (n-hexane/EtOAc, 1
:1) to give the alcohol 13 (1.86 g, 4.26 mmol, 90% yield) as a colourless oil. IR νmax (neat)/cm−1: 3531, 2936, 1736, 1599, 1585, 1477, 1336, 1268, 1157, 1018; 1H NMR (400 MHz, CDCl3): δ 7.67 (d, J = 8.0 Hz, 2H), 7.29 (d, J = 8.0 Hz, 2H), 7.00 (dd, J = 8.0, 1.6 Hz, 1H), 6.95 (t, J = 8.0 Hz, 1H), 6.78 (dd, J = 7.6, 1.6 Hz, 1H), 5.21 (s, 2H), 3.81 (s, 3H), 3.74 (q, J = 6.0 Hz, 2H), 3.51 (s, 3H), 3.23 (t, J = 6.4 Hz, 2H), 3.18 (t, J = 7.6 Hz, 2H), 2.61–2.55 (m, 3H), 2.41 (s, 3H), 1.85–1.80 (m, 2H), 1.78–1.71 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 150.18, 147.73, 143.19, 136.24, 135.18, 129.63, 127.01, 123.92, 123.09, 114.61, 94.99, 60.65, 58.73, 56.14, 48.90, 44.97, 31.39, 29.58, 27.30, 21.40; HRMS (ESIMS) calcd for C22H31N1O6S1Na1 [M + Na]+ 460.1764, found 460.1761.
Synthesis of N-(3-(2-methoxy-3-(methoxymethoxy)phenyl)propyl)-4-methyl-N-(3-oxopropyl)benzenesulfon amide (7). To a solution of oxalyl chloride (0.73 mL, 8.5 mmol) in CH2Cl2 (30 mL) was added dropwise DMSO (1.33 g, 17.1 mmol) in CH2Cl2 (10 mL) at −78 °C over 10 min. After 30 min, a solution of 13 (1.86 g, 4.26 mmol) in CH2Cl2 (10 mL) was added to the resulting mixture at −78 °C. After 30 min, Et3N (3.7 mL, 26 mmol) was added to the reaction mixture. The resulting mixture was then warmed to 25 °C for 30 min. After addition of H2O (10 mL), the organic layer was separated, and the aqueous layer was extracted with CH2Cl2 (2 × 60 mL). The combined organic layers were washed with saturated aqueous NaHCO3 (10 mL) and brine (10 mL), and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure and the residue was purified through silica gel column chromatography (n-hexane/EtOAc, 5
:1) to give 7 (1.81 g, 4.16 mmol, 98% yield) as a colourless oil. IR νmax (neat)/cm−1: 3425, 2934, 1723, 1598, 1585, 1478, 1339, 1268, 1158, 1090, 1012, 920; 1H NMR (400 MHz, CDCl3): δ 9.74 (s, 1H), 7.67 (d, J = 8.0 Hz, 2H), 7.30 (d, J = 8.0 Hz, 2H), 7.01 (dd, J = 8.0, 1.6 Hz, 1H), 6.95 (t, J = 8.0 Hz, 1H), 6.79 (dd, J = 7.2, 1.6 Hz, 1H), 5.21 (s, 2H), 3.83 (s, 3H), 3.51 (s, 3H), 3.41 (t, J = 7.2 Hz, 2H), 3.17 (t, J = 7.2 Hz, 2H), 2.82 (t, J = 7.2 Hz, 2H), 2.61 (t, J = 7.6 Hz, 2H), 2.42 (s, 3H), 1.83–1.75 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 200.16, 150.20, 147.77, 143.35, 136.01, 135.10, 129.68, 127.10, 123.90, 123.10, 114.65, 95.01, 60.63, 56.13, 49.02, 43.94, 41.73, 29.38, 27.21, 21.40; HRMS (ESIMS) calcd for C22H29N1O6S1Na1 [M + Na]+ 458.1608, found 458.1614.
Synthesis of methyl-3-hydroxy-5-(N-(3-(2 methoxy-3-(methoxymethoxy)phenyl)-propyl)-4-methylphenyl sulfonamido)-2-methylenepentanoate (6). Under an atmosphere of Ar, a 100 mL flask was charged with 7 (1.81 g, 4.16 mmol), DABCO (470 mg, 4.20 mmol) and methyl acrylate (19.0 mL, 210 mmol, 50 equiv.). The resulting solution was stirred at 25 °C for 2 weeks. After completion of the reaction (monitored by TLC), excess methyl acrylate was removed under reduced pressure and the residue was purified through silica gel column chromatography (n-hexane/EtOAc, 4
:1) to give 6 (1.14 g, 2.19 mmol, 52% yield) as a colourless oil. IR νmax (neat)/cm−1: 3509, 3297, 2952, 2936, 1721, 1478, 1440, 1336, 1267, 1157, 1090, 1018, 926; 1H NMR (400 MHz, CDCl3): δ 7.67 (d, J = 8.0 Hz, 2H), 7.28 (d, J = 8.0 Hz, 2H), 7.00 (dd, J = 8.0, 1.6 Hz, 1H), 6.94 (t, J = 8.0 Hz, 1H), 6.80 (dd, J = 7.6, 1.6 Hz, 1H), 6.26 (s, 1H), 5.90 (s, 1H), 5.20 (s, 2H), 4.58 (t, J = 4 Hz, 1H), 3.81 (s, 3H), 3.74 (s, 3H), 3.51 (s, 3H), 3.48–3.41 (m, 1H), 3.30–3.20 (m, 1H), 3.19–3.07 (m, 3H), 2.60 (t, J = 8.0 Hz, 2H), 2.41 (s, 3H), 2.05–1.97 (m, 1H), 1.89–1.79 (m, 2H), 1.72–1.63 (m, 1H); 13C NMR (100 MHz, CDCl3): δ 166.64, 150.22, 147.85, 143.17, 142.01, 136.42, 135.28, 129.64, 127.12, 125.16, 123.90, 123.16, 114.68, 95.09, 67.76, 60.68, 56.17, 51.80, 48.67, 45.10, 35.35, 29.36, 27.24, 21.44; HRMS (ESIMS) calcd for C26H36N1O8S1 [M + H]+ 522.2156, found 522.2166.
Synthesis of methyl-3-((tert-butyldimethylsilyl)oxy)-5-(N-(3-(2-methoxy-3-(methoxymethoxy)phenyl)propyl)-4-methylphenylsulfonamido)-2-methylenepentanoate (14). 6 (2.89 g, 5.55 mmol) was dissolved in CH2Cl2 (60 mL) and Et3N (2.4 mL, 17 mmol) was added to the solution. The mixture was cooled to 0 °C. TBSOTf was added dropwise and the solution was stirred at 0 °C for 0.5 h. The reaction mixture was quenched with H2O (10 mL), and extracted with CH2Cl2 (3 × 100 mL). The combined organic layers were washed with saturated aqueous NaHCO3 (10 mL) and brine (10 mL), and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure and the residue was purified through silica gel column chromatography (n-hexane/EtOAc, 5
:1) to give 14 (2.97 g, 4.68 mmol, 86% yield) as a colourless oil. IR νmax (neat)/cm−1: 3345, 2953, 2930, 2896, 2857, 1718, 1630, 1599, 1585, 1476, 1439, 1343, 1265, 1158, 1091, 1010; 1H NMR (400 MHz, CDCl3): δ 7.65 (d, J = 8.4 Hz, 2H), 7.27 (d, J = 8.0 Hz, 2H), 7.01–6.92 (m, 2H), 6.80 (dd, J = 7.6, 1.6 Hz, 1H), 6.25 (s, 1H), 5.89 (s, 1H), 5.21 (s, 2H), 4.58 (t, J = 4.8, 1H), 3.82 (s, 3H), 3.73 (s, 3H), 3.52 (s, 3H), 3.20–3.14 (m, 4H), 2.59 (t, J = 8.0 Hz, 2H), 2.41 (s, 3H), 1.93–1.86 (m, 1H), 1.85–1.68 (m, 3H), 0.90 (s, 9H), 0.04 (s, 3H), −0.03 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 166.17, 150.21, 147.88, 142.84, 142.69, 136.84, 135.40, 129.49, 127.12, 125.09, 123.82, 123.09, 114.60, 95.08, 68.40, 60.63, 56.13, 51.72, 48.07, 44.29, 35.68, 29.32, 27.09, 25.71, 21.40, 18.00, −4.87, −5.20; HRMS (ESIMS) calcd for C32H50N1O8S1Si1 [M + H]+ 636.3021, found 636.3038.
Synthesis of N-(3-((tert-butyldimethylsilyl)oxy)-4-(hydroxymethyl)pent-4-en-1-yl)-N-(3-(2-methoxy-3-(methoxymethoxy)phenyl)propyl)-4-methyl-benzenesulfonamide (15). Under an atmosphere of Ar, DIBAL-H (7.8 mL, 11.7 mmol, 1.5 M in toluene) was added dropwise to a solution of 14 (2.97 g, 4.68 mmol) in CH2Cl2 (50 mL) at −78 °C. The mixture was stirred for 1 h at −78 °C. The reaction mixture was quenched by adding a few drops of MeOH, then potassium tartrate tetrahydrate (20 mL) was added to the mixture and the mixture was stirred for another 1 h at 25 °C. The organic layer was extracted with CH2Cl2 (3 × 50 mL), the organic layers were combined, washed with H2O (30 mL) and brine (30 mL), and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure and the residue was purified through silica gel column chromatography (n-hexane/EtOAc, 4
:1) to give 15 (1.74 g, 2.87 mmol, 62% yield, brsm) as a colourless oil. IR νmax (neat)/cm−1: 3522, 2954, 2930, 2857, 2371, 1599, 1585, 1475, 1340, 1265, 1221, 1157, 1089, 1030, 925; 1H NMR (400 MHz, CDCl3): δ 7.65 (d, J = 8.0 Hz, 2H), 7.27 (d, J = 8.0 Hz, 2H), 7.01–6.94 (m, 2H), 6.80 (dd, J = 7.6, 1.6 Hz, 1H), 6.25 (s, 1H), 5.89 (s, 1H), 5.21 (s, 2H), 4.58 (t, J = 5.2 Hz, 1H), 3.82 (s, 3H), 3.73 (s, 3H), 3.52 (s, 3H), 3.17 (q, J = 4.0 Hz, 4H), 2.61 (t, J = 8.0 Hz, 2H), 2.41 (s, 3H), 1.93–1.86 (m, 1H), 1.85–1.68 (m, 3H), 0.90 (s, 9H), 0.04 (s, 3H), −0.03 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 150.21, 149.52, 147.78, 142.97, 136.78, 135.36, 129.55, 127.07, 123.94, 123.19, 114.64, 111.64, 95.05, 72.72, 62.93, 60.67, 56.16, 48.08, 44.50, 35.22, 29.36, 27.22, 25.69, 21.40, 18.00, −4.86, −5.17; HRMS (ESIMS) calcd for C31H50N1O7S1Si1 [M + H]+ 608.3072, found 608.3082.
Synthesis of N-(3-(3-hydroxy-2-methoxyphenyl)propyl)-N-(3-hydroxy-4-(hydroxymethyl)pent-4-en-1-yl)-4-methyl benzenesulfonamide (16). To a solution of 15 (641 mg, 1.06 mmol) in THF (10 mL) was added a few drops of 2 M HCl, and the mixture was heated under reflux for 2 h. After the reaction mixture was cooled to 25 °C, saturated aqueous NaHCO3 (3 mL) was added to quench the reaction. The aqueous layer was extracted by EtOAc (3 × 20 mL). The combined organic layers were washed with saturated aqueous NaHCO3 (10 mL), H2O (10 mL) and brine (15 mL), dried over anhydrous Na2SO4. The solvent was removed under reduced pressure and the residue was purified through silica gel column chromatography (n-hexane/EtOAc, 1
:1) to give 16 (425 mg, 0.947 mmol, 86% yield) as a colourless oil. IR νmax (neat)/cm−1: 3429, 2935, 2874, 1720, 1592, 1473, 1331, 1290, 1155, 1088, 1004; 1H NMR (400 MHz, CDCl3): δ 7.66 (d, J = 8.0 Hz, 2H), 7.29 (d, J = 8.0 Hz, 2H), 6.92 (t, J = 8.0 Hz, 1H), 6.82–6.80 (m, 1H), 6.65 (d, J = 8.0 Hz, 1H), 6.07–5.99 (m, 1H), 5.12 (s, 1H), 5.09 (s, 1H), 4.41–4.40 (m, 1H), 4.21 (dd, J = 32.0, 13.2 Hz, 2H), 3.76 (s, 3H), 3.44–3.36 (m, 1H), 3.24–3.17 (m, 1H), 3.13–3.10 (m, 1H), 3.08–3.00 (m, 1H), 2.62 (t, J = 7.6 Hz, 2H), 2.42 (s, 3H), 1.93–1.72 (m, 4H); 13C NMR (100 MHz, CDCl3): δ 149.28, 149.07, 145.23, 143.40, 136.00, 134.23, 129.73, 127.10, 124.90, 121.25, 114.03, 112.37, 70.44, 64.20, 61.13, 48.92, 45.45, 34.91, 29.39, 26.99, 21.48; HRMS (ESIMS) calcd for C23H31N1O6S1Na1 [M + Na]+ 472.1764, found 472.1768.
Synthesis of (7aR*,9aS*,11R*)-9a-methoxy-4-tosyl-3,4,5,6,9a,11-hexahydro-1H-7a,11-methanobenzofuro[3,3a–e]azonine-7,10(2H,8H)-dione (5). Under an atmosphere of Ar, a 50 mL flask was charged with 16 (124 mg, 0.276 mmol) and CH2Cl2 (28 mL). After the solution was cooled to 0 °C, PIFA (129 mg, 0.290 mmol) in CH2Cl2 (1 mL) was added dropwise, and then the reaction temperature was slowly raised to 25 °C over an hour. After reaching 25 °C, H2O (5 mL) was added to quench the reaction. The mixture was extracted with CH2Cl2 (3 × 30 mL), washed with water (3 mL) and brine (3 mL), and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure to give the crude product 17 (75 mg, 0.17 mmol, 60% yield) as a yellow oil without further purification.
17 (75 mg, 0.17 mmol) was dissolved in CH2Cl2 (10 mL), and NaHCO3 (71 mg, 0.85 mmol) was added. After the solution was cooled to 0 °C, DMP (0.36 g, 0.85 mmol) was added in portions, and then the reaction temperature was raised to 25 °C over an hour. The reaction mixture was quenched with sodium thiosulphate solution (10 mL) after 2 h (monitored by TLC), and the mixture was stirred at 25 °C for another 0.5 h, and extracted with CH2Cl2 (3 × 15 mL). The organic layers were combined and washed with water (3 mL) and brine (3 mL), and dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure and the residue was purified through silica chromatography (n-hexane/EtOAc, 1:1) to give 5 (32 mg, 0.072 mmol, 42% yield) as a clear crystal. M.p. 208–210 °C. IR νmax (neat)/cm−1: 3458, 3383, 2948, 2373, 1740, 1703, 1597, 1452, 1338, 1269, 1160, 1090, 1043, 1002; 1H NMR (400 MHz, CDCl3): δ 7.67 (d, J = 8.0 Hz, 2H), 7.33 (d, J = 8.0 Hz, 2H), 6.45 (t, J = 8.0 Hz, 1H), 6.36 (br, 1H), 4.81 (br, 1H), 4.12 (d, J = 7.2 Hz, 1H), 3.61 (s, 3H), 3.44–3.43 (m, 1H), 3.30–3.28 (m, 1H), 3.13 (m, 3H), 2.44 (s, 3H), 2.27–2.18 (m, 2H), 2.04–1.97 (m, 2H), 1.96–1.88 (m, 1H), 1.63 (m, 1H); 13C NMR (100 MHz, CDCl3): δ 201.95, 143.70, 134.58, 133.63, 129.75, 128.42, 127.16, 100.83, 73.68, 60.59, 54.48, 48.86, 47.12, 45.69, 23.35, 22.25, 21.42, 14.10; HRMS (ESIMS) calcd for C23H27N1O6S1H1 [M + H]+ 446.1632, found 446.1629.
Acknowledgements
This work was supported by the National Science Foundation of China (21125207, 21372103, 21472079, and 21572088), SRFDP (20130211110018), IRT_15R28, and lzujbky-2015-50.Notes and references
- Reviews on Lycopodium alkaloids: (a) M. Kitajima and H. Takayama, in Topics in Current Chemistry, 2012, vol. 309, pp. 1–32 Search PubMed; (b) Y. Hirasawa, J. Kobayashi and H. Morita, Heterocycles, 2009, 77, 679–729 CrossRef CAS; (c) W. A. Ayer, Nat. Prod. Rep., 1991, 8, 455–463 RSC; (d) W. A. Ayer and L. S. Trifonov, in Lycopodium Alkaloids, In The Alkaloids: Chemistry and Pharmacology, ed. G. A. Cordell, A. Brossi, Academic Press, San Diego, CA, 1994, ch. 3, vol. 45, pp. 233–266 Search PubMed; (e) J. Kobayashi and H. Morita, in The Lycopodium Alkaoids, In The Alkaloids: Chemistry and Biology, ed. G. A. Cordell, Elsevier, San Diego, CA, 2005, ch. 1, vol. 61, pp. 1–57 Search PubMed; (f) C.-H. Tan and D.-Y. Zhu, Chin. J. Nat. Med., 2003, 1, 1 CAS; (g) X. Ma and D. R. Gang, Nat. Prod. Rep., 2004, 21, 752–772 RSC; (h) D. B. MacLean, Alkaloids, 1985, 26, 241–298 CAS.
- (a) F.-W. Zhao, Q.-Y. Sun, F.-M. Yang, G.-W. Hu, J.-F. Luo, G.-H. Tang, Y.-H. Wang and C.-L. Long, Org. Lett., 2010, 12, 3922–3925 CrossRef CAS PubMed; (b) L.-B. Dong, X. Gao, F. Liu, J. He, X.-D. Wu, Y. Li and Q.-S. Zhao, Org. Lett., 2013, 15, 3570–3573 CrossRef CAS PubMed; (c) X.-J. Wang, L. Li, S.-S. Yu, S.-G. Ma, J. Qu, Y.-B. Liu, Y. Li, Y. Wang and W. Tang, Fitoterapia, 2013, 91, 74–81 CrossRef CAS PubMed.
- (a) C. Zhao, H. Zheng, P. Jing, B. Fang, X. Xie and X. She, Org.Lett., 2012, 14, 2293–2295 CrossRef CAS PubMed; (b) G.-B. Zhang, F.-X. Wang, J.-Y. Du, H. Qu, X.-Y. Ma, M.-X. Wei, C.-T. Wang, Q. Li and C.-A. Fan, Org. Lett., 2012, 14, 3696–3699 CrossRef CAS PubMed; (c) D. Gaugele and M. E. Maier, Synlett, 2013, 24, 955–958 CrossRef CAS; (d) N. Sizemore and S. D. Rychnovsky, Org. Lett., 2014, 16, 688–691 CrossRef CAS PubMed.
- For selected reviews on the oxidative dearomatization/intramolecular Diels–Alder reaction, see: (a) C.-C. Liao and R. K. Peddinti, Acc. Chem. Res., 2002, 35, 856–866 CrossRef CAS PubMed; (b) C.-C. Liao, Pure Appl. Chem., 2005, 77, 1221–1234 CAS and references therein.
- For selected examples of the oxidative dearomatization/intramolecular Diels–Alder reaction strategy in total synthesis, see: (a) C.-C. Wang, Y.-C. Ku and G. J. Chuang, J. Org. Chem., 2015, 80, 10979–10991 CrossRef CAS PubMed; (b) E. Georgopanou, K.-I. Martini, P. Pantazis, P. Pelagias, P. Voulgari and L. P. Hadjiarapoglou, J. Org. Chem., 2015, 80, 9682–9690 CrossRef CAS PubMed; (c) Y.-Y. Chou and C.-C. Liao, Org. Lett., 2013, 15, 1584–1587 CrossRef CAS PubMed; (d) D.-S. Hsu, Y.-Y. Chou, Y.-S. Tung and C.-C. Liao, Chem. – Eur. J., 2010, 16, 3121–3131 CrossRef CAS PubMed; (e) S. A. Snyder and F. Kontes, J. Am. Chem. Soc., 2009, 131, 1745–1752 CrossRef CAS PubMed; (f) S. P. Cook, A. Polara and S. J. Danishefsky, J. Am. Chem. Soc., 2006, 128, 16440–16441 CrossRef CAS PubMed; (g) C.-F. Yen and C.-C. Liao, Angew. Chem., Int. Ed., 2002, 41, 4090–4093 CrossRef CAS.
- For selected examples for the construction of the isotwistane core, see: (a) H. Niwa, K. Wakamatsu, T. Hida, K. Niiyama, H. Kigoshi, M. Yamada, H. Nagase, M. Suzuki and K. Yamada, J. Am. Chem. Soc., 1984, 106, 4547–4552 CrossRef CAS; (b) S.-L. Hsieh, C.-T. Chiu and N.-C. Chang, J. Org. Chem., 1989, 54, 3820–3823 CrossRef CAS; (c) T. Yoshimitsu, S. Sasaki, Y. Arano and H. Nagaoka, J. Org. Chem., 2004, 69, 9262–9268 CrossRef CAS PubMed; (d) V. Singh, S. Pal and S. M. Mobin, J. Org. Chem., 2006, 71, 3014–3025 CrossRef CAS PubMed; (e) B. Mitasev and J. A. Porco Jr., Org. Lett., 2009, 11, 2285–2288 CrossRef CAS PubMed.
- For selected reviews on the synthesis of the medium-sized ring, see: (a) Y. Wang and Z.-X. Yu, Acc. Chem. Res., 2015, 48, 2288–2296 CrossRef CAS PubMed; (b) A. Archambeau, F. Miege, C. Meyer and J. Cossy, Acc. Chem. Res., 2015, 48, 1021–1031 CrossRef CAS PubMed; (c) A. Hussain, S. K. Yousuf and D. Mukhherjee, RSC Adv., 2014, 4, 43241–43257 RSC.
- For selected methods for the synthesis of the medium-sized ring via rhodium-catalysis, see: F. Medina, C. Besnard and J. Lacour, Org. Lett., 2014, 16, 3232–3235 CrossRef CAS PubMed.
- For selected methods for the synthesis of the medium-sized ring via gold-catalysis, see: (a) Y.-W. Sun, X.-Y. Tang and M. Shi, Chem. Commun., 2015, 51, 13937–13940 RSC; (b) C. Zhao, X. Xie, S. Duan, H. Li, R. Fang and X. She, Angew. Chem., Int. Ed., 2014, 53, 10789–10793 CrossRef CAS PubMed.
- For selected methods for the synthesis of a medium-sized ring via RCM reaction, see: B. Laroche, M. Detraz, A. Blond, L. Dubost, P. Mailliet and B. Nay, J. Org. Chem., 2015, 80, 5359–5363 CrossRef CAS PubMed.
- For selected methods for the synthesis of a medium-sized ring via ring expansion, see: (a) B. A. Bhat, L. Samantha, St. E. J. Germain, P. Maity and S. D. Lepore, J. Org. Chem., 2014, 79, 9402–9407 CrossRef CAS PubMed; (b) Z. A. Kasun, L. M. Geary and M. J. Krische, Chem. Commun., 2014, 50, 7545–7547 RSC; (c) D. M. Zubrytski, D. G. Kananovich and O. G. Kulinkovich, Tetrahedron, 2014, 70, 2944–2950 CrossRef CAS; (d) N. P. Tsvetkov, A. Bayir, S. Schneider and M. Brewer, Org. Lett., 2012, 14, 264–267 CrossRef CAS PubMed.
- For selected examples of intramolecular Diels–Alder reactions with ring constriction, see: (a) A. Toró, P. Nowak and P. Deslongchamps, J. Am. Chem. Soc., 2000, 122, 4526–4527 CrossRef; (b) R. Munakata, H. Katakai, T. Ueki, J. Kurosaka, K. Takao and K. Tadano, J. Am. Chem. Soc., 2003, 125, 14722–14723 CrossRef CAS PubMed; (c) D. A. Evans and J. T. Starr, Angew. Chem., Int. Ed., 2002, 41, 1787–1790 CrossRef CAS; (d) D. J. Mergott, S. A. Frank and W. R. Roush, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 11955–11959 CrossRef CAS PubMed; (e) M. E. Layton, C. A. Morales and M. D. Shair, J. Am. Chem. Soc., 2002, 124, 773–775 CrossRef CAS PubMed.
- S. A. Wacowich-Sgabi and D. R. Bundle, J. Org. Chem., 1999, 64, 9080–9089 CrossRef.
- J. J. Verendel, T. Zhou, J.-Q. Li, A. Paptchikhine, O. Lebedev and P. G. Andersson, J. Am. Chem. Soc., 2010, 132, 8880–8881 CrossRef CAS PubMed.
- (a) B. Das, J. Banerjee, N. Chowdhury, A. Majhi and G. Mahender, Helv. Chim. Acta, 2006, 89, 876–883 CrossRef CAS; (b) A. Bouzide, Org. Lett., 2002, 4, 1347–1350 CrossRef CAS PubMed; (c) R. Jogireddy and M. E. Maier, J. Org. Chem., 2006, 71, 6999–7006 CrossRef CAS PubMed.
- If the secondary hydroxyl group was not protected, only a trace of 15 could be obtained due to the 1,4-addition of the hydride to the α, β unsaturated ester.
- CCDC 1438512 (5) was contained in the supplementary crystallographic data for this paper.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1438512. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qo00222f |
| This journal is © the Partner Organisations 2016 |