
Potassium-mediated stereochemical assistance to form one indenonaphthacene isomer from rubrene with complementary diastereoselectivity to the acid based protocol†
J.
Zhang
,
K. V.
Luzyanin
and
S.
Jansat
*
Department of Chemistry, University of Liverpool, Crown Street, Liverpool, L697ZD, UK. E-mail: sjansats@gmail.com
First published on 18th May 2016
Abstract
Diastereoselective C–H and C–C bond formation has been assessed from rubrene to form selectively one indenonaphthacene derivative R*, in the presence of metallic potassium stabilised by THF at room temperature. The potassium salt [K(rac-R*)(THF)x] contains R* as anion with a (1S,2S) and (1R,2R) configuration, on the two new chiral carbon centers, which has been unambiguously confirmed by selective 1D NOESY, 1H, 13C-NMR, 1H, 13C-HMBC and polarimetry. The stated chirality could only originate from a face selective intramolecular reaction, which takes place on one side of the original tetracene core. The diastereomeric excess for the potassium salt has been quantified by 1H NMR to be at least 97%. Potassium removal affords the neutral ((S,S)+(R,R))-R*. In contrast, the acid-based protocol furnishes two structural isomers, ((S,R)+(R,S))-R* and rac-R′. The former compound has been isolated pure with a yield of 56% by precipitation. The complementary chirality in R* is a direct consequence of the inter- and intramolecular character for both face-selective processes, along with potassium assistance in the latter case.
Undecorated C–H activation (taking place in a selective way with no external assistance) to selectively create new bonds is a field that has continuously provided challenges in synthetic chemistry. Up to now C–X bond activations have been performed efficiently using catalytic routes of general applicability under “traditional conditions”.1 Although bare C–H activation is appealing as it does not require any additional substrate functionalization,2 regioselectivity and activity rely mainly on directing group assistance.3 Straightforward C–H activation becomes extremely challenging when Polycyclic Aromatic Hydrocarbons (PAH) are used as substrates because all of the atoms present almost similar reactivity.4 This situation can be amended with the use of photocatalytic reactions in the presence of lanthanides,5 or the use of established alkaline metal procedures.6 In line with the latter example, solvated radical anions could be oxidized and/or protonated in the presence of a protic media in the well-known Birch reduction with alkaline metals in NH3/alcohol mixtures. This is a reliable way to obtain trans alkenes from alkynes, 1,4-cyclohexadienes from benzenoid rings, and to achieve a complete chiral induction for 1,2-syn hydrogenated products with particular substrates. Underlining its importance different groups have developed both established variants such as the Benkeser reaction in the presence of amines7 (PAH hydrogenation) and air stable solids with adsorbed alkali metals to perform the reaction.8
In the present communication, we describe the diastereoselective synthesis of one structural indenonaphthacene isomer (rac-R*), bearing two chiral centers, from rubrene (R). Application of metallic potassium at room temperature allowed the selective formation of one C–H and one C–C bond, without the use of the typical experimental Birch conditions requiring ammonia, amines or, alcohols as liquid media. This synthetic approach could be complemented by an acid-catalysed procedure leading to the other pair of diastereomers possible for the same compound R*.9
The expected reactivity for a THF solution of R in the presence of alkali metals is the transformation to the corresponding salt containing the chemically unchanged organic anion (R1−).10 The most common stabilization for IA and IIA cations is covalent/electrostatic binding with the anionic organic counterpart, although sometimes cations can be encapsulated by neutral organic ligands.11
Precedents to form related compounds to R* bearing a comparable skeleton with the concomitant formation of aliphatic protons and/or, quaternary carbons are scarce despite their great potential applications in materials. Published examples involve reduction using NaBH3CN and ZnI2,12 allene dimerization in combination with rearrangement followed by oxidation,13 and “in situ oxidation” during rubrene crystal growth.14 In none of these cases has there been an attempt to control the stereochemical course of the reaction with an external chiral inductor, and no information about the chirality of the product has been specified when asymmetric centres were created.
In a special case, such as our current study, the prediction of the stereochemical course of a reaction with PAH compounds could be rationalized. Factors to take into account are the mechanism, nature of the chemical process and substrate structure. This information might be particularly important for PAH molecules, to increase the number of established protocols.
To the best of our knowledge this is the first report regarding the regio- and diastereoselective synthesis of ((S,S)+(R,R))-R* where R* = 4b,9,10-triphenyl-4b,9-dihydroindeno[1,2,3-fg]tetracene. The only synthetic precedent found in the literature is the acid-catalysed version which furnishes the other pair of diastereomers ((S,R)+(R,S))-R* in the presence of another structural isomer (R′). The present communication contains full spectroscopic characterization for both R* diastereomer pairs.‡
Rubrene (R) has been successfully and quantitatively converted into the corresponding potassium salt derivative stabilized by the organic anion R* using 1.2 equivalents of metallic potassium in THF. As a main feature R* comprises two new chiral centres, C1 and C2, the latter being a strained quaternary carbon. Its core includes 7 fused rings, 4 of these being rings with five and six members (structure shown in Fig. 1 and Scheme 1). Three 1D selective NOESY contacts of the new moiety H1, two strong with ortho3 and ortho1 at 4 and 7 bonds of connectivity (pink arrows in Fig. 1), and one less intense for H4 at 7 bonds, could be uniquely explained by a molecule with an (S,S) configuration (dashed pink arrow in Fig. 1).
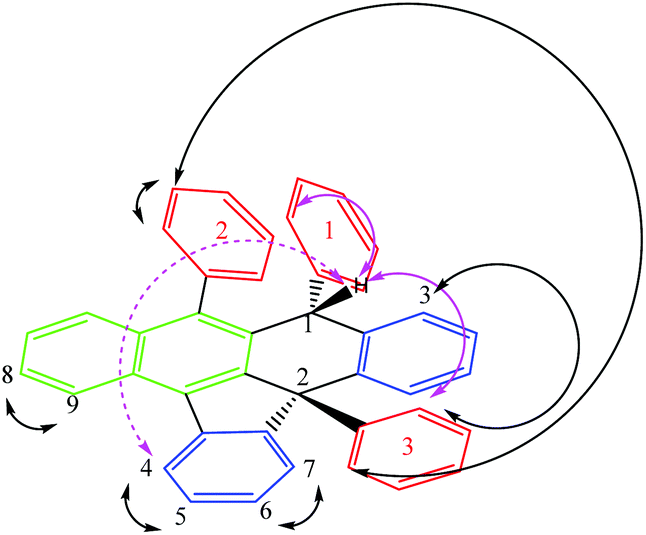 | ||
| Fig. 1 Selective 1D NOESY contacts for the structural isomer isolated by the K procedure. Pink arrows for NOE contacts of the H1 nucleus. Only the organic anion is shown. Numbering for aromatic lateral rings in red. Type of aromatic rings highlighted in different colours. | ||
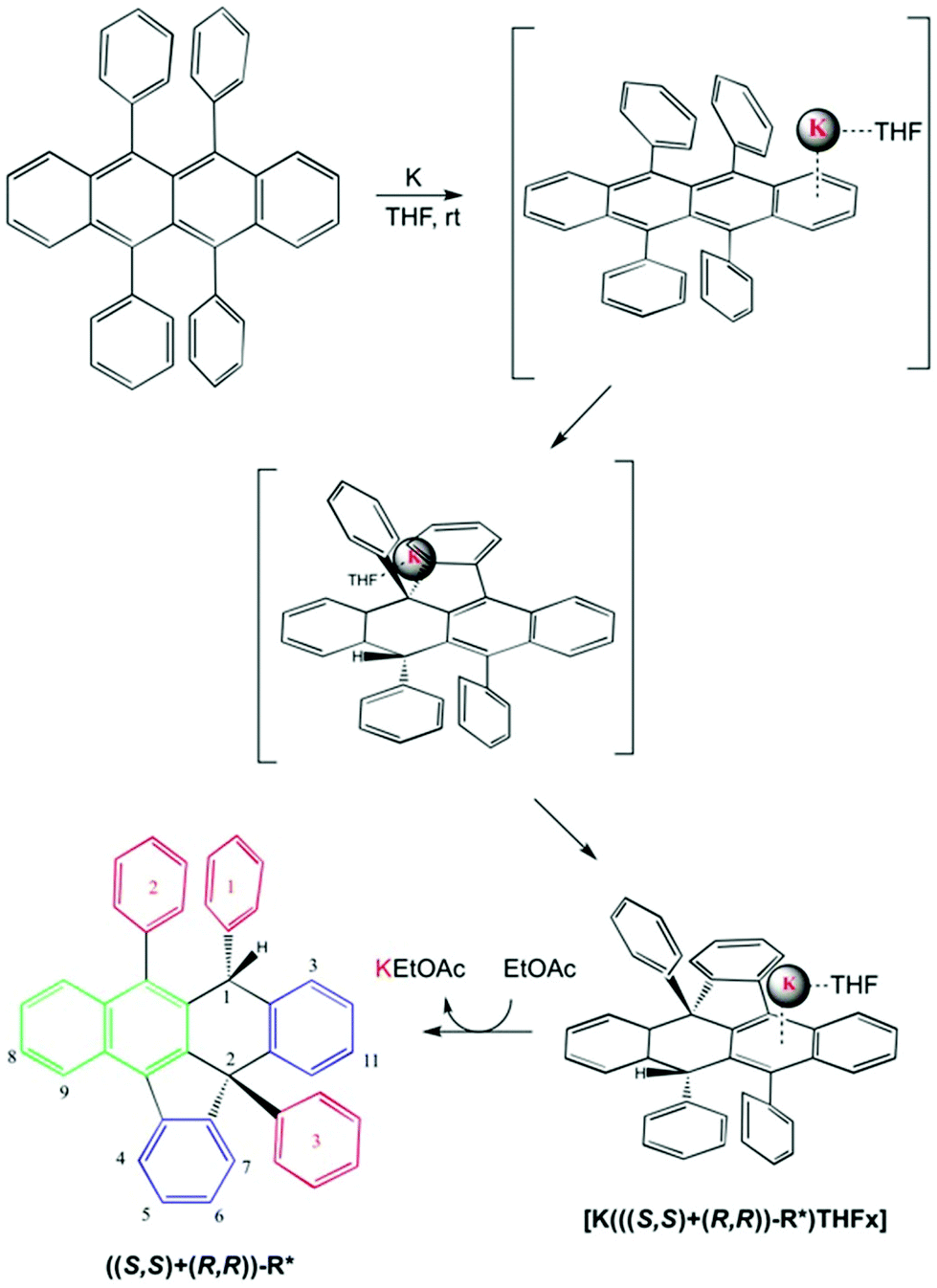 | ||
| Scheme 1 Proposed mechanism for [K(((S,S)+(R,R))-R*)(THF-d8)x] formation and rac-(S,S)-R* isolation. The nature of the aromatic rings highlighted in different colours as in Fig. 1. The numbering system used in the text for ((S,S)+(R,R))-R* molecule is shown. | ||
The last observation evidences the important skeleton distortion and corroborates definitely the proposed configuration in combination with the additional NOE contacts meta2–ortho2, meta2–ortho3 and H3-ortho3. The para-protons do not show an NOE effect in agreement with their spatial disposition (Fig. 1).
As R* contains two chiral centres it could be obtained as a mixture of 4 enantiomers or, two pairs of diastereomers, but spectroscopic characterization, and a nil value in optical activity, confirm that the isolated potassium salt contains the racemic mixture ((S,S)+(R,R))-R* with no presence of any other diastereomer such as ((S,R)+(R,S))-R*. Skeletal core distortion is disclosed by the chirality of C1 and C2, and for each diastereomer pair is governed mainly by the geometry of the strained chiral quaternary centre C2. This encumbered carbon is bonded to four aromatic rings and shares simultaneously the edge of a cyclopentane and cyclohexane ring. Bending is higher in ((S,R)+(R,S))-R* where the two aromatic rings, Ar1 and Ar3, are pointing in the identical sense. MM2 modelling (ESI 4†) and comparison with experimental values measured in the published crystal structure of (S,R)-R* (121.35(6)°) are consistent with this afirmation.9 The diastereomeric excess for the crude potassium salt has been quantified as a minimum up to 97% by 1H NMR in the reactive THF solution.
1H and 13C NMR spectra of the salt support that the organic anion has three kinds of aromatic rings proven by the three ipso carbons at low field (associated with typical ortho, meta and para nucleus), the expected signals for a naphthalene moiety and the pattern for ortho-disubstituted aromatic rings (Fig. 2 in red, green and blue colours, respectively). The structure of ((S,S)+(R,R))-R* presents various perfect sites to stabilize a cation simultaneously by encapsulation and/or, π–cation interactions. When the [K(((S,S)+(R,R))-R*)(THF-d8)x] species was freshly formed a dynamic process in solution was evidenced by two experimental facts, the change of solution colour from red to green dark,15 and the NMR quaternary carbon signals shift from 77 to 97 ppm. This has been linked with K passing from a metastable accommodation, close to a reactive site, to the final stabilization as a naphthalenide compound. First K settlement might be visualised as encapsulation between the chiral quaternary carbon and the adjacent ortho-disubstituted ring. This configuration will allow likely additional interactions with ipso and α-carbons found typically in potassium aromatic salts.16 In fact, the ionic radii for K(+) (0.138–0.151 nm for coordination number 6–7 respectively) might fit on the space created by C2, C10 and ipso-3 on the ((S,S)+(R,R))-R* anion structure. The final naphthalenide dark-green compound stays in solution for more than four months in anhydrous conditions (Fig. 2). Its NMR spectra shows one species with 22 protons magnetically non equivalent in agreement with the proposed structure (Scheme 1). 39K NMR gave no signal as a consequence of the broadening associated with the K quadrupolar nature, and the extreme asymmetric environment with nearly all nuclei magnetically different (ESI 2†).
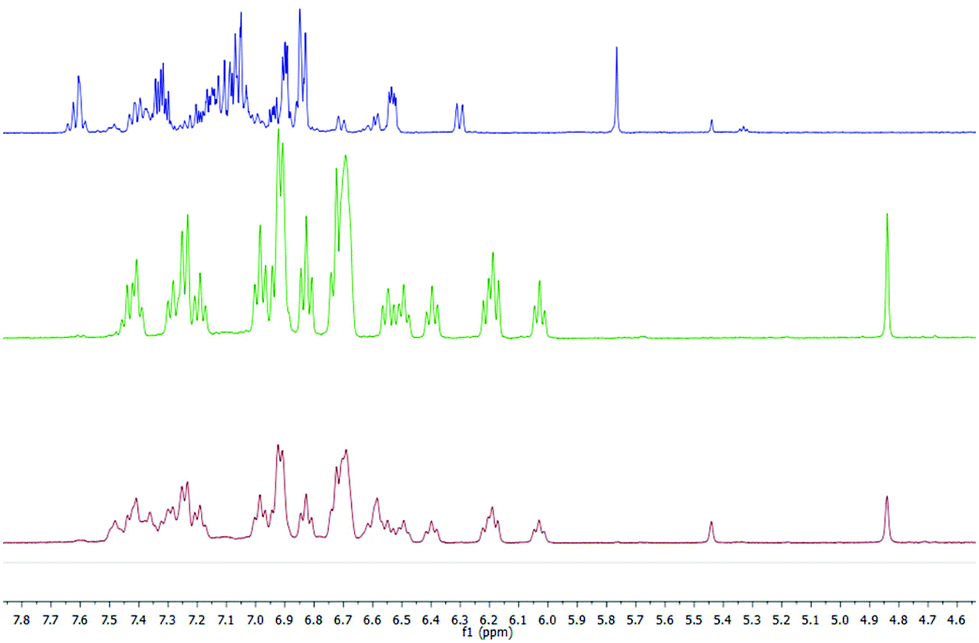 | ||
| Fig. 2 1H NMR spectra measured at different times of reaction, red during and green four months after [K(((S,S)+(R,R))-R*)(THF)x] formation. Blue profile corresponding to neutral ((S,S)+(R,R))-R* and minor hydrogenated compound. | ||
The published calorimetric measurements for some naphthalenide potassium salt derivatives shown that they are about 9.6 kcal mol−1 more stable than their free neutral counterparts. This difference won't allow the in situ reduction of the cation with no help from an external agent and can explain the stability of our compound in these conditions (Fig. 2).17 Removal of K(+) was done by metallation of ethyl acetate via[K(((S,S)+(R,R))-R*)(THF-d8)x] under mild conditions and maintains an unchanged chemical structure of R*. However, a simultaneous process of hydrogenation takes place, as demonstrated by MS-TOF(+) mass measurement for the minor species present after work up at 10% (ESI 2.2†). Ethyl acetate salt was eliminated by column chromatography eluting with hexanes. 1H, 13C-NMR, 1D selective NOESY for H1, and IR are in agreement with a recovered neutral ((S,S)+(R,R))-R* molecule which conserves the original stereochemistry, as shown by the presence of the H1–H4 NOESY contact (Scheme 1).
A well-defined intermediate compound was visualized prior to the formation of [K(((S,S)+(R,R))-R*)(THF-d8)x] by NMR via a sole singlet resonance at 5.8 ppm which was measurable during five days (red profile in Fig. 2). Its disappearance was directly related to the increase of H1 in (((S,S)+(R,R))-R*)−1. Such chemical shift is in the range area for some published alkaline rubrene salts10b and, ruthenium dihydrogen germylene complexes.18 And therefore it might be tentatively assigned to an ionic species containing R and K in a 1/1 stoichiometry (Fig. 2). The analogous Cs derivative has been reported as a stable organometallic compound in solid and liquid state.10b The smaller size of potassium cation (0.138 nm for K versus 0.167 for Cs in a coordination number 6), might allow its approach towards the π-cloud enabling a proper and kinetically accessible agostic interaction which will further develop until ((S,S)+(R,R))-R* formation.19 THF molecules are expected to attenuate the rate of the C–H activation, allowing intermediate visualization by NMR in the liquid state.
The ((S,S)+(R,R))-R* isolated structure implies a face selective process where the organic molecule needs to twist and bend surrounding the cation in a dynamic progression governed mainly by ionic attraction. The latter configuration permits reaction uniquely on the side where K is laying presumably in an intramolecular and concerted mechanism. Once that one ortho hydrogen of one lateral ring has been activated, there is no possibility of potassium isomerization to the other face. Overall the reaction mechanism might be seen as an intramolecular Birch modification where the organic substrate acts as a proton donor. Due to the lack of a second proton and electron donors the reaction passes through a C–C coupling.
From the above statements, the most likely activation pathway which might potentially take place is one stable and accessible anagostic interaction in one ortho position and concomitant stabilization of a positive encapsulated potassium during formation of the five membered ring. Simultaneously the cation would interact with a partially developed hydride ligand and, a neutral THF molecule. This proposition may explain the face selective nature of the process, initiation site on one ortho aromatic position, reactive sites happening in para disposition, and axial attack of the hydride located onto C2 aided by cation interaction. The latter attack will form the new C–H bond with identical sense orientation as Ar3. At the last stage, chirality induction is exerted by the first asymmetric centre formed and explains the isolation of the product as racemic mixtures (and not diasteromeric mixtures).
In line with the anticipated mechanism, the use of external proton sources as KH gave complex mixtures of diverse products indicating that a different mechanism is acting and, regio- and stereo-selectivity is controlled to a much more minor extend.
In order to verify the exact nature of ((S,S)+(R,R))-R* a comparison with isolated products from the acid catalysed synthetic protocol has been carried out. Reaction at different times using diverse stoichiometric ratios for R/CF3COOH furnishes a mixture of two products in a similar percentage. Both products are structural isomers of R and R*, as evidenced by MS-TOF(+) measurements.
The major product has been isolated as a pure compound by precipitation with a yield of 56%. Assignation as ((S,R)+(R,S))-R* is based on its characterization and its published crystal structure. ((S,R)+(R,S))-R* shows the same experimental MS-TOF(+) as ((S,S)+(R,R))-R*, an analogous nonsymmetric molecule proved by its 1H and 13C NMR, but with a clear different spatial disposition for Ar1 and Ar3, and finally, an identical IR pattern for C–H stretching and C–C–C bending. All these facts indicate in a strong enough way that use of potassium allows for the isolation of ((S,S)+(R,R))-R*, meanwhile the acid catalysed protocol furnishes ((S,R)+(R,S))-R* as the major isomer in agreement with the crystal structure published.9 The best separation protocol to get pure ((S,R)+(R,S))-R* is precipitation directly from the concentrated crude reaction mixture using THF.
1H NMR of the reaction mixture shows a minor structural isomer. The proposed isomer structure rac-R′ is displayed in Fig. 3, and has been assigned to (4R,8S,12R,16S)-R′ as the main species (R′ = ((8bR,12bS)-4b,12b-diphenyl-4b,8b,12b,16b-tetrahydrodiindeno[1,2,3-fg:1′,2′,3′-op]tetracene)) based on: a distinctive 2H loss for the molecular peak in its MS-TOF(+) analysis, a meso-type geometry shown in the NMR analysis and a singular intensity pattern mode in the IR for the C–H stretching area (ESI 2†). Its formation can be described by two consecutive SNu attacks to the external H+ of the acid present in excess on the reaction (ESI 3†). After the first C–H bond formation, the positive charge might rearrange to the para position, and final SNu attack from one lateral ring to the carbocation position furnishes R′. The proposed arrangement contains 4 stereocenters embedded in a core of 8 fused rings (Fig. 3 as R′). An interesting structural feature of this compound is that the absolute chirality of the carbons might change the core profile in a singular manner. As a result, the skeleton shape can form a concave surface, or several near planar configurations bearing steps along different directions.
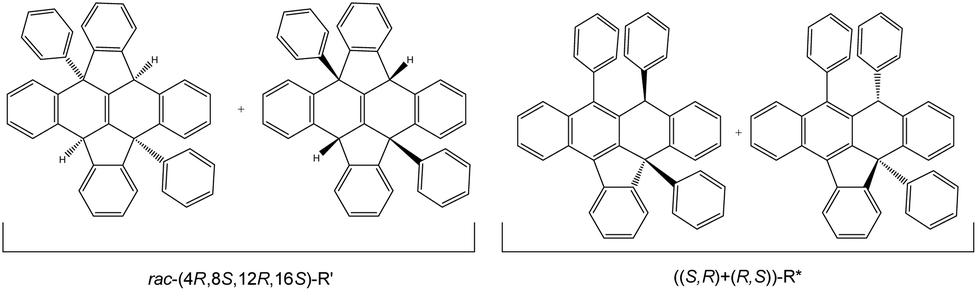 | ||
| Fig. 3 Proposed isolated isomers with the acid catalysed protocol for rac-(4R,8S,12R,16S)-R′ and ((S,R)+(R,S))-R*. | ||
From the perspective of the optical purity, two consecutive reactive cycles starting in orthogonal positions can form selectively nine types of diastereomer pairs, 5 of them being meso. The characterization performed seems to indicate that R′ is formed as a racemic mixture of one compound but its characterization has not been optimized in this respect. Although the chemical shifts in 1H and 13C NMR for positions 1 and 2 in all products are sensitive to the solvation media, simultaneous proton and carbon comparison for all isolated compounds on the same solvent (when possible), allowed the identification of rac-R′, ((S,R)+(R,S))-R* and ((S,S)+(R,R))-R* as different compounds as shown by the rest of the characterization data. Further contrast with the unique structural isomer found in the literature agrees with this. The stated compound has two C–H functions in para disposition with a core that includes four lateral aromatic rings.8 Interestingly, the chemical shift for quaternary carbon C2 and, ipso1 and ipso3 carbons in R* and R′, are dependent on compound structure reflecting both C2 strain and Ar1 and Ar3 spatial disposition dictated by the 6-membered ring (Fig. 4 and experimental). Accordingly, C2 signals appear at 97.0, 84.7, 60.8 and 60.9 ppm for the salts, both R* and rac-R′ respectively. In contrast, C1 signals for the same compounds emerge in a small scale range from 48.4 to 51.3 ppm. Concerning the ipso carbons ((S,S)+(R,R))-R* presents a unique signal at higher ppm meanwhile, the other diastereomer shows two in the same area indicating similar geometric neighbouring (Ar1 and Ar3).
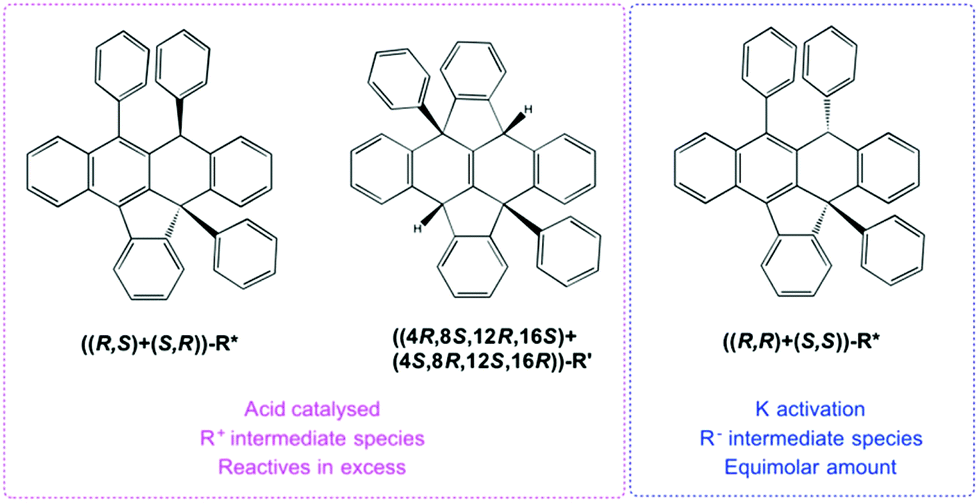 | ||
| Fig. 4 Chemical structure of products R′ and R* which may be isolated with acid or K protocols using the same substrate R. | ||
Nil values for polarimetry measurements corroborate that both R* isomers are obtained as racemic mixtures. This fact is explained by the lack of absolute asymmetric control in the formation of the first bond in the two different synthetic methods.
As a summary, two distinctive protocols, acid catalysed and intramolecular reaction assisted by potassium, have been defined to form, in a controlled stereochemical manner, the two diastereomeric couples possible for one indenonaphthacene isomer R*. The potassium based process might be regarded as a Birch variation that combines a C–C and a C–H bond formation, in an intramolecular process where the organic substrate is the origin of the proton. K permits a face-selective mechanism with no presence of asymmetric external inductors, allowing the isolation of only two enantiomers of the four initially possible as ((S,S)+(R,R))-R* (with at least 97% de). The organic product is isolated as the anion of the corresponding potassium salt. Further ethyl acetate treatment releases the neutral organic moiety as ((S,S)+(R,R))-R*. In contrast the acid catalysed protocol yields, in a racemic way, two couples of diastereomers of two different structural isomers, ((S,R)+(R,S))-R* and rac-R′. In both mechanisms, the formation of racemic mixtures is explained by the chiral induction exerted from the first asymmetric centre created on the second bond formation and lack of induction on absolute chirality in the first reactive site. Complementary diastereoselectivity for R* is based on the origin source of the proton (external or internal), and assistance of the cation to allow uniquely to form racemic ((S,S)+(R,R))-R* in the intramolecular process (Fig. 4). For the intermolecular reaction, products and lack of regioselectivity, are independent of the chemical nature of the external hydrogen: proton and/or hydride. Other substrates have not been attempted. Further work will be devoted to scaling up the present procedures and exploit the potential with other organic substrates.
Experimental section
Synthesis of [K(THF-d8)x(((S,S)+(R,R))-R*)]
THF-d8 freshly distilled from metallic sodium (0.5 mL) was added into a Schlenk flask containing potassium metal (1.0 mg, 0.025 mmol, 1.25 equivalents) and rubrene (10.6 mg 0.02 mmol). The resulting mixture orange in colour, similar to that of rubrene, was stirred for 12 hours. It produced a dark red solution that developed to dark green after one day. The solution maintained this colour for four months until the removal of K and was stored in a fire sealed glass NMR tube. Quantitative yield as seen by 1H-NMR. Product was not weighed. 1H NMR (500 MHz; THF-d8; Me4Si) δH 7.44 (1H, pd, J = 6.0), 7.40 (1H, pt, J = 8.0), 7.28 (1H, t, J = 7.3), 7.24 (2H, d, J = 7.2), 7.19 (s, 1H), 7.19 (1H, J = 7.2), 6.98 (2H, t, J = 7.7 Hz), 6.91 (5H, m), 6.83 (2H, t, J = 7.0), 6.73 (2H, t, J = 7.0), 6.70 (3H, m), 6.54 (1H, pt, J = 7.5), 6.49 (1H, t, J = 7.2), 6.40 (1H, s, J = 7.5), 6.21 (2H, m), 6.07 (1H, t, J = 8.5), 4.84 (1H, s). 13C NMR (126 MHz, THF-d8; Me4Si) δCq, 149.4, 148.6, 147.0, 143.6, 141.8, 138.6, 136.1, 136.1, 129.4, 128.9, 128.2, 116.50, 97.0; δCH, 134.9, 133.9, 132.7, 131.4, 128.7, 128.3, 128.2, 128.2, 127.9, 127.8, 127.6, 127.0, 126.7, 126.2, 125.6, 124.4, 123.8, 121.8, 121.4, 116.0, 114.9, 112.3, 51.3.Isolation of ((S,S)+(R,R))-R* from [K(THF-d8)x(((S,S)+(R,R))-R*)]
A THF solution was placed in a bath at −45 °C. Then 0.5 mL of ethyl acetate were added. After 5 minutes, two drops of IPA and two of water were further added and the bath removed to allow the solution to reach room temperature. The sample was filtered through a pad of silica with hexanes as eluent, dried with anhydrous Na2SO4 and concentrated for characterization. The solid was isolated as a white slightly yellow solid. MS-TOF(+) m/z 533.2285 (MH+, 100%), 455.1808 (MH+ − Ph, 44.45%). 1H NMR (400 MHz; CDCl3; Me4Si) δH 7.58 (1H, ptd, J = 4), 7.52 (1H, pd, J = 4), 7.42–7.34 (7H, m), 7.20–6.84 (14H, m), 6.72 (1H, t, 4), 6.49 (2H, m), 6.31 (1H, d, 4), 5.76 (s, 1H). 13C NMR (126 MHz; CDCl3; Me4Si) δC 149.0, 140.4, 139.6, 139.3, 138.5, 136.2, 133.4, 133.3, 132.3, 132.0, 133.0, 131.2, 130.0, 129.4, 128.5, 128.5, 128.1, 127.8, 127.5, 127.2, 127.0, 126.9, 126.6, 126.5, 126.1, 126.1, 125.2, 125.1, 124.7, 84.7, 48.4. vmax/cm−1 (IR): 3056, 2926, 2850, 1595, 1456, 1446, 1261, 1026, 766, 724, 697. [α]D 0 (1 in CHCl3).Synthesis of ((S,R)+(S,R))-R*
In short, a solution of rubrene (53.2 mg, 0.1 mmol) and anhydrous dichloromethane (0.7 mL) was stirred in the dark in the presence of CF3COOH (0.3 mL, mmol) for 12 hours (or up to 48 hours). The reaction can be followed by TCL (hexanes/acetone, 0.1%). Concentration under vacuum and precipitation after THF addition affords pure ((S,R)+(S,R))-R* (30 mg, 56% yield). EA calc. for C42H28(C4H8O)0.6(H2O)0.7: C, 90.61; H, 5.86. Found: C, 90.58; H, 5.84%. MS-TOF(+) m/z 533.2266 (MH+, 100%), 455.1813 (MH+ − Ph, 86.56%). 1H NMR (400 MHz; DMF-d7; Me4Si) δH 9.08 (1H, d, J = 8.0), 8.68 (1H, d, J = 8.0), 8.18 (1H, m), 7.84 (1H, t, J = 8.3), 7.87–7.38 (13H, m), 7.25 (1H, d, J = 4), 6.75–6.62 (7H, m), 6.39 (2H, d, J = 8), 5.32 (1H, s). 13C NMR (101 MHz; DMF-d7; Me4Si) δC 152.9, 149.2, 143.1, 141.0, 140.0, 138.8, 138.4, 138.2, 134.1, 134.0, 131.0, 130.6, 130.4, 128.8, 128.7, 128.4, 128.0, 128.0, 127.9, 127.9, 127.7, 127.3, 127.2, 127.1, 127.0, 126.8, 126.6, 126.1, 125.8, 125.1, 124.4, 123.9, 60.8, 50.2. vmax/cm−1 (IR): 3060, 3008, 2972, 2849, 1598, 1492, 1449, 1058, 775, 764, 738, 694, 669, 599. [α]D 0 (1 in DMF).Acknowledgements
We thank the EPSRC for financial support under EP/K027212/1. Comments from Dr Baltasar Bonillo are acknowledged. The authors express their gratitude to Prof. Matthew Rosseinsky for his kind support.Notes and references
- (a) E.-i. Negishi, Angew. Chem., Int. Ed., 2011, 50, 6738 CrossRef CAS PubMed; (b) A. Suzuki, Angew. Chem., Int. Ed., 2011, 50, 6723 Search PubMed; (c) V. Snieckus, Nature, 2015, 527, 306 CrossRef CAS PubMed.
- I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890 CrossRef CAS PubMed.
- I. Funes-Ardoiz and F. Maseras, Angew. Chem., Int. Ed., 2016, 55, 2764 CrossRef CAS PubMed.
- Y. Segawa, T. Maekawa and K. Itami, Angew. Chem., Int. Ed., 2015, 54, 66 CrossRef CAS PubMed.
- P. L. Arnold, M. W. McMullon, J. Rieb and F. E. Kühn, Angew. Chem., Int. Ed., 2015, 54, 82 CrossRef CAS PubMed.
- T. J. Donohoe, R. Garg and C. A. Stevenson, Tetrahedron: Asymmetry, 1996, 7, 317 CrossRef CAS.
- E. M. Kaiser, Synthesis, 1972, 391 CrossRef CAS.
- (a) M. Lefenfeld and J. L. Dye, Titanium Oxide and alumina alkali metal compositions, US7560606B2, 2009 Search PubMed; (b) J. L. Dye, K. D. Cram, S. A. Urbin, M. Y. Redko, J. E. Jackson and M. Lefenfeld, J. Am. Chem. Soc., 2005, 127, 9338 CrossRef CAS PubMed.
- T. Hosokawa, H. Nakano, K. Takami, K. Kobiroa and A. Shiga, Tetrahedron Lett., 2003, 44, 1175 CrossRef CAS.
- (a) H. Bock, K. Gharagozloo-Hubmann, C. Näther, N. Nagel and Z. Havlas, Angew. Chem., Int. Ed. Engl., 1996, 35, 631 CrossRef CAS; (b) A. V. Zabula, N. J. Sumner, A. S. Filatov, S. N. Spisak, V. M. Grigoryants and M. A. Petrukhina, Eur. J. Inorg. Chem., 2012, 4675 CrossRef CAS.
- R. Bertermann, H. Braunschweig, P. Constantinidis, T. Dellermann, R. D. Dewhurst, W. C. Ewing, I. Fischer, T. Kramer, J. Mies, A. K. Phukan and A. Vargas, Angew. Chem., Int. Ed., 2015, 54, 13090 CrossRef CAS PubMed.
- Y. Hou, X. Chi, X. Wana and Y. Chen, J. Mol. Struct., 2008, 889, 265 CrossRef CAS.
- P. Capdevielle and J. Rigaudy, Tetrahedron, 1979, 35, 2093 CrossRef CAS.
- D. Braga, A. Jaafari, L. Miozzo, M. Moret, S. Rizzato, A. Papagni and A. Yassar, Eur. J. Org. Chem., 2011, 4160 CrossRef CAS.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877 CrossRef CAS PubMed.
- F. Feil and S. Harde, Organometallics, 2000, 19, 5010 CrossRef CAS.
- G. R. Stevenson, C. R. Wiedrlch, S. S. Zlgler, L. Echegoyen and R. Maldonado, J. Phys. Chem., 1983, 87, 4995 CrossRef CAS.
- K. A. Smart, E. Mothes-Martin, L. Vendier, R. N. Perutz, M. Grellier and S. Sabo-Etienne, Organometallics, 2015, 34, 4158 CrossRef CAS.
- C. Lepetit, J. Poater, M. E. Alikhani, B. Silvi, Y. Canac, J. Contreras-García, M. Sola and R. Chauvin, Inorg. Chem., 2015, 54, 2960 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: HMBC, 1H, 39K, 13C NMR. See DOI: 10.1039/c6qo00130k |
| ‡ See ESI† for additional and raw spectroscopic data for ((S,S)+(R,R))-R*, ((S,R)+(S,R))-R*, [K(THF)x(((S,S)+(R,R))-R*)] and rac-R′. |
| This journal is © the Partner Organisations 2016 |