
Pd(II)-catalyzed direct functionalization of C–H bonds of benzamides for synthesis of 1,1-difluoro-1-alkenes†
Chunpu
Li‡
,
Dengyou
Zhang‡
,
Wei
Zhu
,
Penghui
Wan
and
Hong
Liu
*
CAS Key Laboratory of Receptor Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, 555 Zuchongzhi Road, Shanghai 201203, China. E-mail: hliu@simm.ac.cn
First published on 20th June 2016
Abstract
Pd-catalyzed direct difluoroallylation of C–H bonds of benzamides with readily available Rf-Br reagents is reported. The reaction proceeds via Pd-catalyzed C–H activation, followed by reaction of the resulting intermediate with 3-bromo-3,3-difluoropropene (BDFP) in a γ-selective fashion. The C–H functionalization process was characterized by a broad substrate scope as well as an excellent functional-group tolerance.
Transition-metal-catalyzed direct functionalization of substrates by activation of C–H bonds would eliminate the need for prefunctionalization of coupling partners. Direct functionalization involving C–H bond activation would provide simple and atom-economical pathways to produce functionalized molecules.1
The advantages of allyl moieties for various useful synthetic modifications in functionalization have led to broad interest in developing an efficient strategy to introduce these attractive structural motifs. Consequently, a series of synthetic methods for allyl arenes have been developed, such as classical Friedel–Crafts reaction2 or cross-coupling reactions.3 However, these methods can suffer from competing formation of conjugated double-bond isomers, harsh reaction conditions, and limited reaction scope.
Chelation-assisted transformation has become a common and powerful method for the regioselective functionalization of ortho C–H bonds.4 Bidentate directing groups have recently attracted the attention of researchers because they offer the possibility of developing new types of C–H bond transformations.5 Significant advances in transition-metal-catalyzed C–H allylation using bidentate directing groups have been made, among which the recent Ni- and Fe-catalyzed allylation reactions reported by Chatani,6 Nakamura,7 Zeng8 and Sundararaju9 are particularly attractive (Scheme 1).
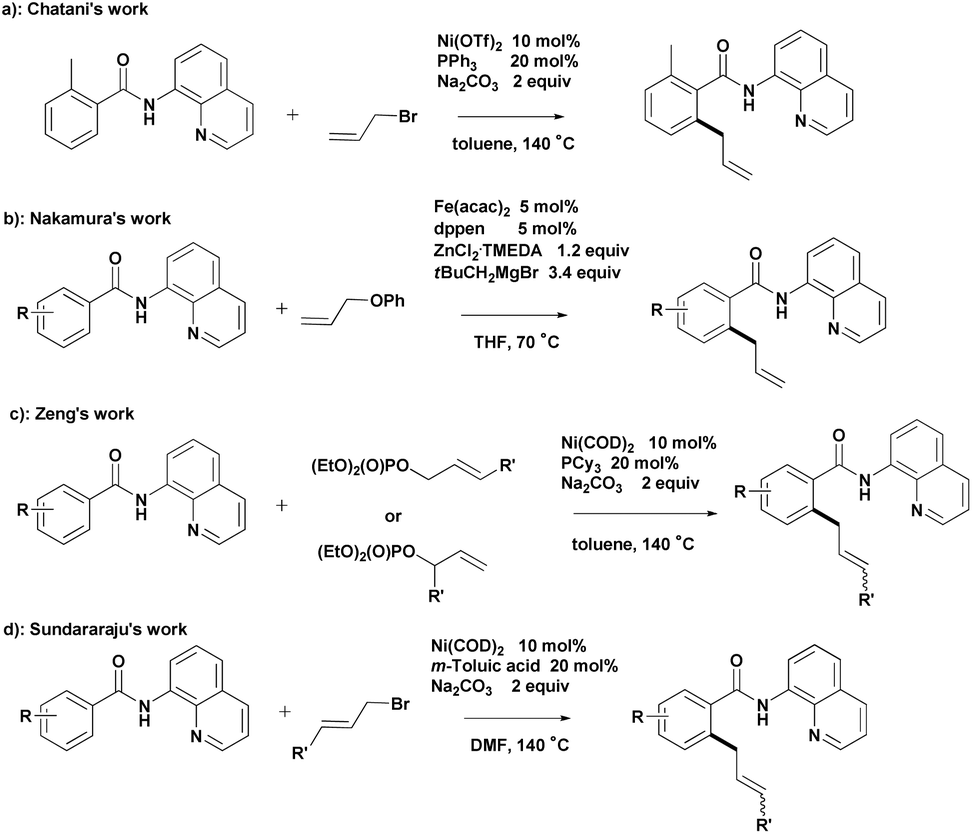 | ||
| Scheme 1 Transition-metal-catalyzed C–H allylation using bidentate directing groups. | ||
Organofluorine chemistry is a rapidly expanding field. The ubiquity of fluorinated organic molecules has been highlighted in the fields of pharmaceutical research and agrochemistry.10 Recently, Zhang developed Pd-catalyzed highly selective gem-difluoroallylation of aryl boronic acids using bromodifluoromethylated alkenes (Scheme 2).11 This efficient and practical protocol provides an easy route for the synthesis of fluorinated bioactive compounds in drug development. However, the direct difluoroallylation of C–H bonds to introduce difluoroallyl groups to organic molecules remains relatively unexplored. Inspired by the recent studies on the use of the 8-aminoquinoline directing group to aid catalytic functionalizations of C–H bonds,12 we have developed a palladium-catalyzed direct functionalization of C–H bonds of benzamides with 3-bromo-3,3-difluoroprop-1-ene 2a (Scheme 2). It provides an alternative approach for the synthesis of 1,1-difluoro-1-alkenes compounds from simple precursors. Herein, we report the first example of Pd-catalyzed direct functionalization of C–H bonds with 3-bromo-3,3-difluoroprop-1-ene 2a using a readily removable 8-aminoquinoline directing group.
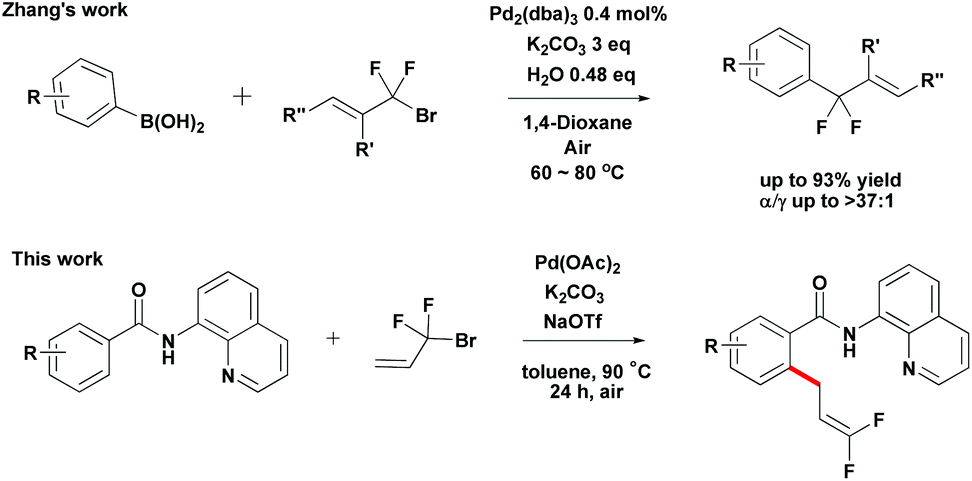 | ||
| Scheme 2 Theme of this work. | ||
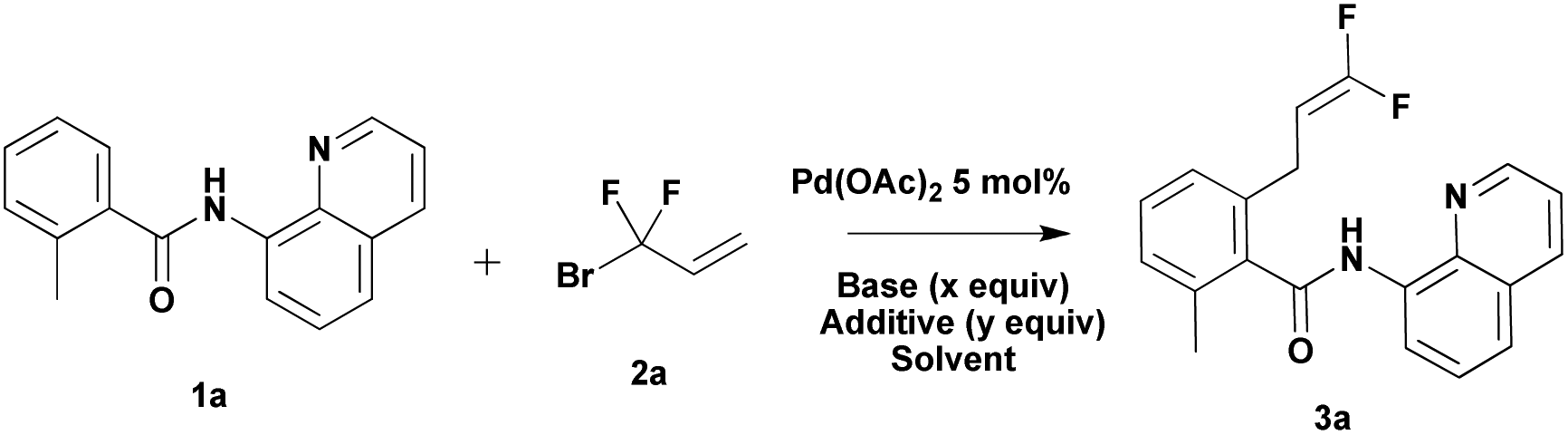
Our preliminary studies focused on the palladium-catalyzed cross-coupling of 8-aminoquinoline-containing benzamide 1a with 3-bromo-3,3-difluoroprop-1-ene 2a (Table 1). We obtained 2-(3,3-difluoroallyl)-6-methyl-N-(quinolin-8-yl)benzamide 3a in 52% yield when Ag2CO3 (1 equiv.) was employed as an additive in the presence of 5 mol% of Pd(OAc)2 (Table 1, entry 1). The use of larger amounts of 2a (3 equiv.) resulted in an increase in yield (65%, entry 2 vs. 52%, entry 1). The yield decreased to 59% when the reaction was conducted in toluene (Table 1, entry 3). Interestingly, application of 1 equiv. of Ag2CO3 and 0.2 equiv. of (BnO)2PO2H showed different effects in different solvents.13 The use of 20 mol% (BnO)2PO2H in t-amylOH lowered the yield by 19% (Table 1, entry 4); however, the use of 20 mol% (BnO)2PO2H in toluene improved the yield by 18% (Table 1, entry 5). Replacing 1 equiv. of Ag2CO3 with 2 equiv. of K2CO3 additive, 3a was obtained in 64% (t-amylOH as the solvent) and 68% (toluene as the solvent) yield, respectively (Table 1, entries 6 and 7). Encouraged by these results, we attempted to carry out this reaction without expensive silver salts and ligands. To further improve the yield, we turned our attention to additives. The addition of NaOTf or NaI improved the yield to 84% and 81%, respectively, whereas the addition of NaOAc decreased the yield by 6% (Table 1, entries 8–10). Increasing the temperature to 120 °C resulted in a lower yield of 3a (Table 1, entry 11 vs. entry 10).
|
|
||||
|---|---|---|---|---|
| Entry | Base (equiv.) | Additive (equiv.) | Solvent | Yieldb (%) |
| a Reaction conditions: 1a (0.2 mmol), 2a (0.6 mmol), Pd(OAc)2 (0.01 mmol), solvent (2 mL), 24 h, 90 °C, air. b Isolated yield. c 2a (0.3 mmol). d 120 °C. e Without Pd(OAc)2. | ||||
| 1c | Ag2CO3 (1) | — | t-AmylOH | 52 |
| 2 | Ag2CO3 (1) | — | t-AmylOH | 65 |
| 3 | Ag2CO3 (1) | — | Toluene | 59 |
| 4 | Ag2CO3 (1) | (BnO)2PO2H (0.2) | t-AmylOH | 46 |
| 5 | Ag2CO3 (1) | (BnO)2PO2H (0.2) | Toluene | 77 |
| 6 | K2CO3 (2) | — | t-AmylOH | 64 |
| 7 | K2CO3 (2) | — | Toluene | 68 |
| 8 | K2CO3 (2) | NaOAc (3) | Toluene | 62 |
| 9 | K2CO3 (2) | NaOTf (3) | Toluene | 84 |
| 10 | K2CO3 (2) | NaI (2) | Toluene | 81 |
| 11d | K2CO3 (2) | NaOTf (3) | Toluene | 61 |
| 12e | K2CO3 (2) | — | Toluene | NR |
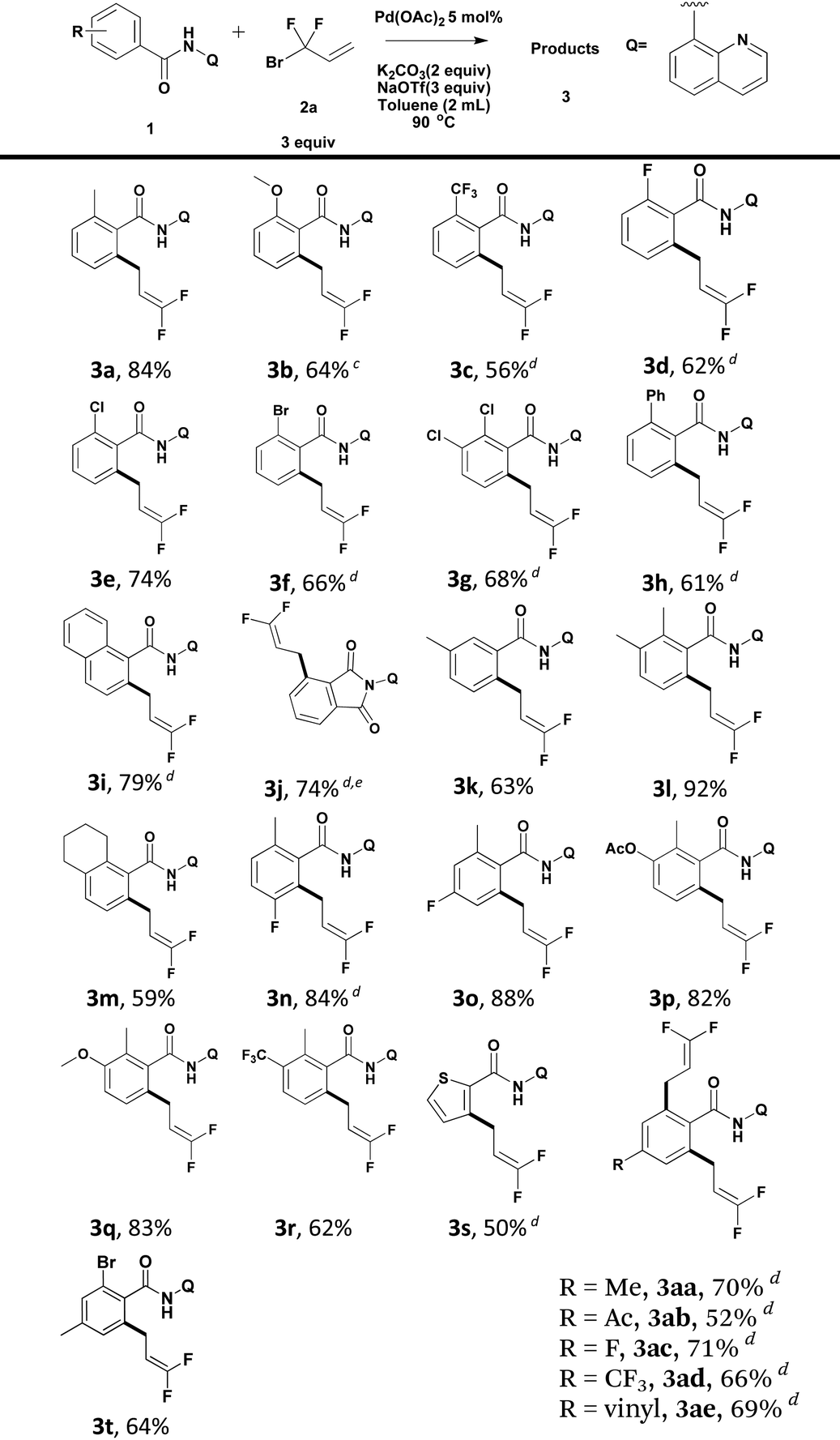
Using the optimized reaction conditions, we explored the scope of various benzamides (Table 2). As shown in Table 2, both electron-rich and -deficient benzamides produced the desired products in moderate to excellent yields. The introduction of stronger electron-donating groups (–OMe) at the ortho-position of the benzamide resulted in a reduced yield of the target compound (3b). ortho-Trifluoromethyl benzamide underwent direct functionalization to furnish the corresponding cross-coupling product in 56% yield (3c). It is worth noting that halides such as fluoride (3d), chloride (3e, 3g) and bromide (3f), remained intact under the standard reaction conditions, producing the desired products in moderate to good yields. The tolerance of the reaction to these active groups in the substrates meant that they could be further transformed into other different functional groups. 1-Naphthalene carboxamide produced the desired product regioselectively at the 2-position in 79% yield (3i). In the case of ortho-ester benzamide 1j, in situ intramolecular nucleophilic attack of the nitrogen atom in the amide moiety to the ester group produced phthalimide 3j in 74% yield. When meta-methyl benzamide 1k was used, the reaction occurred predominantly at the less hindered position (3k). A series of polysubstituted benzamides were also compatible, and gave good to excellent yields (3l–3r). Moreover, heteroarenes, such as thiophene (3s), could serve as suitable substrates, albeit in moderate yields. In the case of para-substituted benzamides, the formation of di-difluoroallylated benzamides (3aa–3ae) was preferred. 4-Acetyl and vinyl-bearing benzamides proved to be tolerant, leading to difluoroallylated products in good yields (3ab and 3ae). Mono-difluoroallylated para-substituted benzamides could be produced conveniently by debromination of product 3t.
| a Reaction conditions (unless otherwise specified): 1 (0.2 mmol), K2CO3 (0.4 mmol), NaOTf (0.6 mmol), Pd(OAc)2 (0.01 mmol), toluene (2 mL), 24 h, 90 °C, air. b Isolated yield. c Reaction conditions: 1 (0.2 mmol), Pd(OAc)2 (0.01 mmol), Ag2CO3 (0.2 mmol), (BnO)2PO2H (0.04 mmol), toluene (2 mL), 24 h, 120 °C, air. d T = 120 °C. e 1j = methyl 2-(quinolin-8-ylcarbamoyl)benzoate. |
|---|
|
To improve the utility of this protocol, the 8-aminoquinoline directing group needs to be removed for further elaboration. A sequential two-step deprotection procedure (Boc activation of the amide, then LiOH/H2O2 hydrolysis conditions) could readily cleave the 8-aminoquinoline directing group from 3k to obtain the corresponding free acid 5 (Scheme 3).14
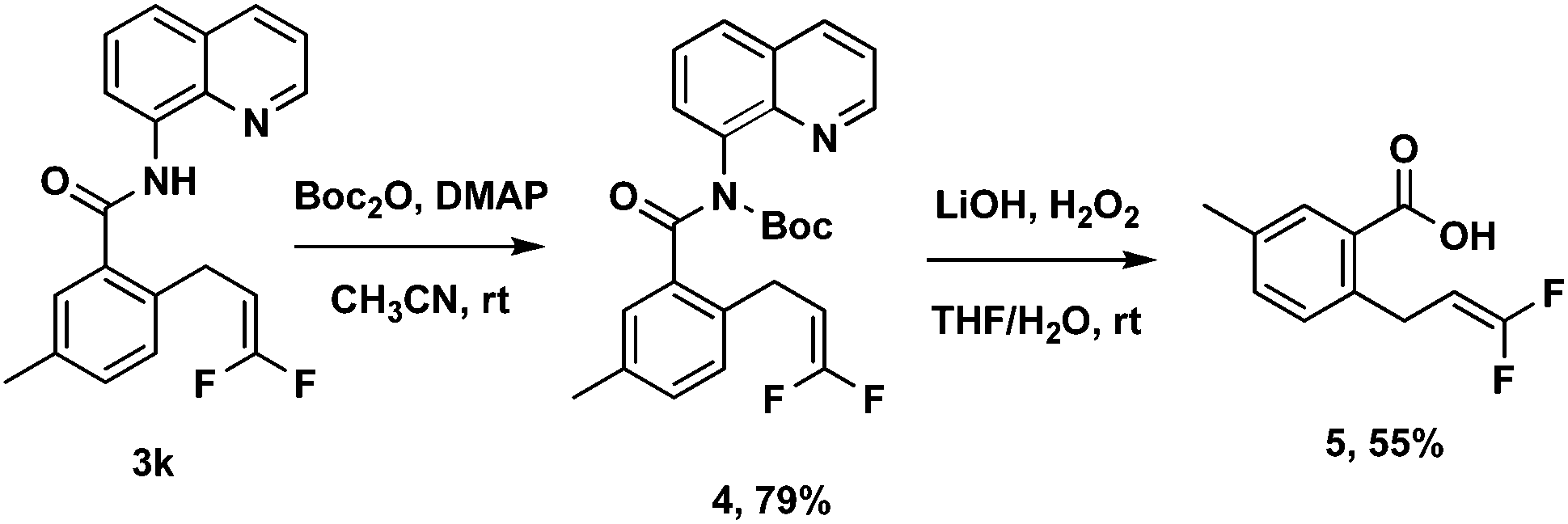 | ||
| Scheme 3 Removal of the directing group. | ||
Meanwhile, difluoroalkene 3a was subjected to further reduction under simple Pd-C/H2 conditions to produce difluoroalkane 6 (Scheme 4a). This provides an efficient route to the synthesis of terminally difluoroalkane derivatives without using toxic reagents such as diethylaminosulfur trifluoride (DAST). Compound 3l could be converted into monofluoroalkene 7 easily through selective hydrodefluorination (Scheme 4b).15 Nickel-catalyzed Suzuki cross-coupling of 3m with phenylboronic acid produced 8 in moderate yield with good regioselectivity (Scheme 4c).16
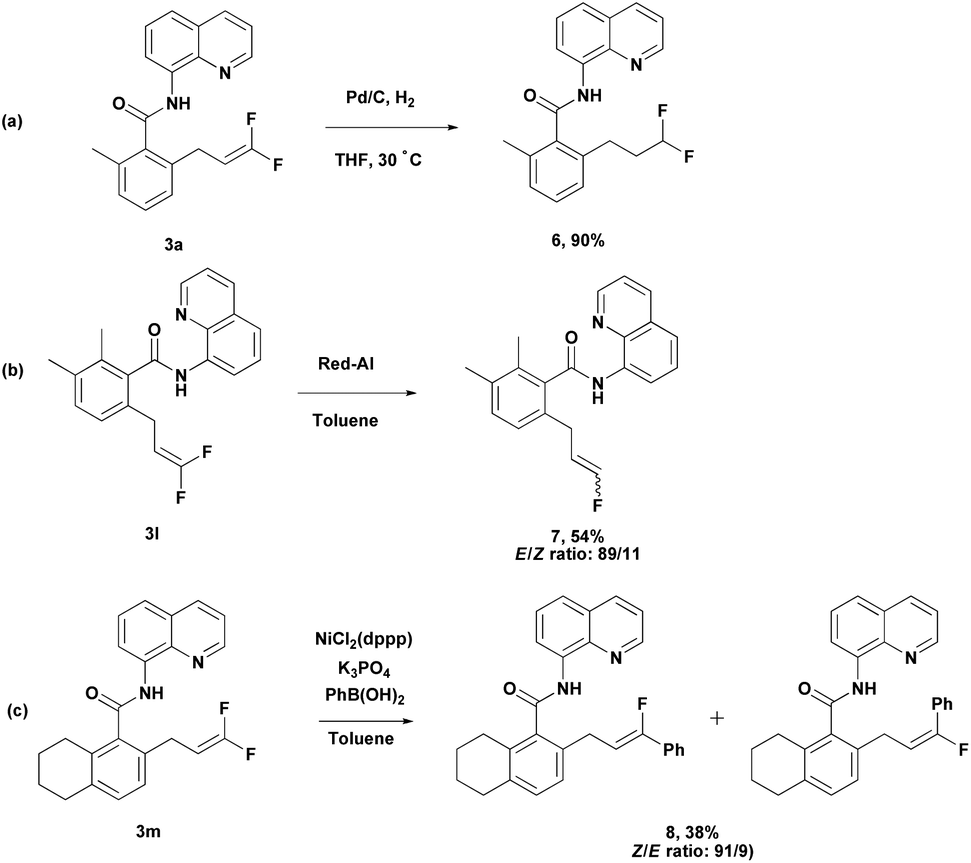 | ||
| Scheme 4 Synthetic applications. | ||
Although the mechanism details remain to be ascertained, a possible mechanism for this reaction can be depicted (Scheme 5).17 First, coordination of Pd(II) with the bidentate DG provided intermediate A. The palladacycle intermediate B, presumably generated from Avia a concerted metallation–deprotonation (CMD) mechanism, undergoes oxidative addition with 2a to provide intermediate Cvia an SN2′ mechanism. Reductive elimination of this Pd(IV)-intermediate C would then provide intermediate D, which provides product 3a and the Pd(II) catalyst subsequently.
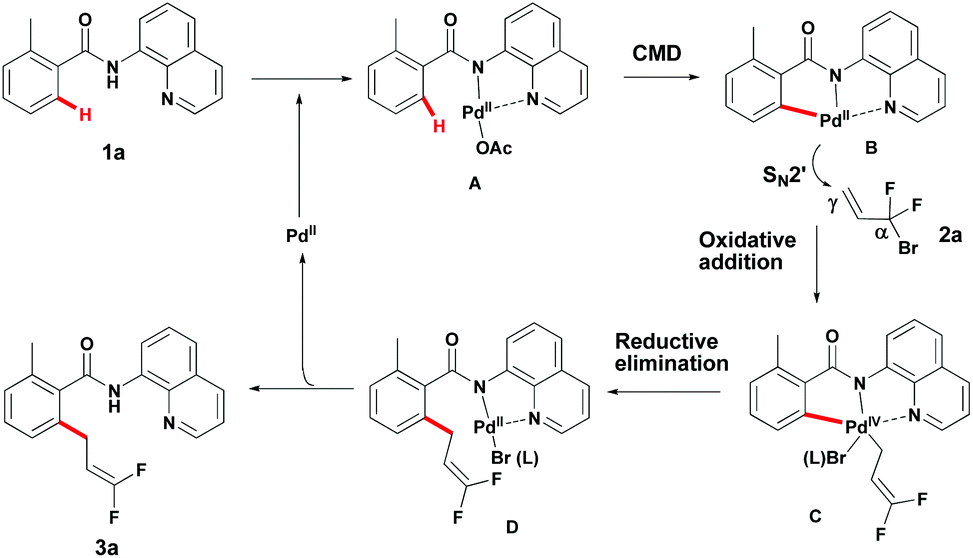 | ||
| Scheme 5 Plausible mechanism. | ||
In summary, we have developed a Pd-catalyzed direct functionalization of C–H bonds with 3-bromo-3,3-difluoropropene 2a using a readily removable 8-aminoquinoline directing group under mild and operationally simple conditions. The benzamide substrates tolerate a wide range of functional groups. This method provides an alternative approach for the synthesis of 1,1-difluoro-1-alkenes compounds, which are valuable synthetic intermediates in a variety of organic reactions and have various potential uses in the pharmaceutical research and agrochemistry. This finding paves the way for developing Pd-catalyzed direct functionalization of C–H bonds with fluorinated building blocks using directing groups.
Notes and references
- (a) X. Chen, K. M. Engle, D. H. Wang and J. Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094 CrossRef CAS PubMed; (b) Y. Segawa, T. Maekawa and K. Itami, Angew. Chem., Int. Ed., 2015, 54, 66 CrossRef CAS PubMed; (c) C. Liu, H. Zhang, W. Shi and A. Lei, Chem. Rev., 2011, 111, 1780 CrossRef CAS PubMed; (d) J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369 CrossRef CAS PubMed.
- T. B. Poulsen and K. A. Jorgensen, Chem. Rev., 2008, 108, 2903 CrossRef CAS PubMed.
- (a) F. C. Pigge, Synthesis, 2010, 1745 CrossRef CAS; (b) J. L. Farmer, H. N. Hunter and M. G. Organ, J. Am. Chem. Soc., 2012, 134, 17470 CrossRef CAS PubMed.
- L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS PubMed.
- (a) O. Daugulis, J. Roane and L. D. Tran, Acc. Chem. Res., 2015, 48, 1053 CrossRef CAS PubMed; (b) O. Daugulis, H. Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074 CrossRef CAS PubMed.
- (a) Y. Aihara, J. Wuelbern and N. Chatani, Bull. Chem. Soc. Jpn., 2015, 88, 438 CrossRef CAS; (b) Y. Aihara and N. Chatani, J. Am. Chem. Soc., 2013, 135, 5308 CrossRef CAS PubMed.
- S. Asako, L. Ilies and E. Nakamura, J. Am. Chem. Soc., 2013, 135, 17755 CrossRef CAS PubMed.
- X. Cong, Y. Li, Y. Wei and X. Zeng, Org. Lett., 2014, 16, 3926 CrossRef CAS PubMed.
- N. Barsu, D. Kalsi and B. Sundararaju, Chem. – Eur. J., 2015, 21, 9364 CrossRef CAS PubMed.
- (a) J. Wang, M. Sanchez-Rosello, J. L. Acena, C. Del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432 CrossRef CAS PubMed; (b) Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Acena, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422 CrossRef CAS PubMed.
- Q. Q. Min, Z. Yin, Z. Feng, W. H. Guo and X. Zhang, J. Am. Chem. Soc., 2014, 136, 1230 CrossRef CAS PubMed.
- (a) Y. Ano, M. Tobisu and N. Chatani, J. Am. Chem. Soc., 2011, 133, 12984 CrossRef CAS PubMed; (b) D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2010, 132, 3965 CrossRef CAS PubMed.
- S. Y. Zhang, G. He, W. A. Nack, Y. S. Zhao, Q. Li and G. Chen, J. Am. Chem. Soc., 2013, 135, 2124 CrossRef CAS PubMed.
- Y. Feng and G. Chen, Angew. Chem., Int. Ed., 2010, 49, 958 CrossRef CAS PubMed.
- J. Wu, J. Xiao, W. Dai and S. Cao, RSC Adv., 2015, 5, 34498 RSC.
- Y. Xiong, T. Huang, X. Ji, J. Wu and S. Cao, Org. Biomol. Chem., 2015, 13, 7389 Search PubMed.
- (a) M. Kirihara, T. Takuwa, M. Okumura, T. Wakikawa, H. Takahata, T. Momose, Y. Takeuchi and H. Nemoto, Chem. Pharm. Bull., 2000, 48, 885 CrossRef CAS PubMed; (b) S. Y. Zhang, Q. Li, G. He, W. A. Nack and G. Chen, J. Am. Chem. Soc., 2013, 135, 12135 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6qo00178e |
| ‡ These authors contributed equally. |
| This journal is © the Partner Organisations 2016 |