
Pd(II)-catalyzed β-C–H arylation of O-methyl ketoximes with iodoarenes†
Yucheng
Mu‡
a,
Xiaodong
Tan‡
ab,
Yemin
Zhang
a,
Xiaobi
Jing
b and
Zhuangzhi
Shi
*a
aState Key Laboratory of Coordination Chemistry, Collaborative Innovation Center of Chemistry for Life Sciences, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing, 210093, China. E-mail: shiz@nju.edu.cn
bCollege of Chemistry and Chemical Engineering, Yangzhou University, Yangzhou 225002, China
First published on 21st January 2016
Abstract
A Pd(II)-catalyzed selective β-arylation of O-methyl ketoximes was developed using iodoarenes as the coupling partners. This transformation has good functional group compatibility and can serve as a powerful synthetic tool for late-stage C–H arylation of complex compounds. Moreover, when employing 2-iodobenzoic acids as the substrates in this developed catalytic system, a type of unexpected five-membered lactones could be formed by tandem sp3 C–H arylation and oxygenation.
 Zhuangzhi Shi | Zhuangzhi Shi was born in Jiangsu Province in 1983. He received his B.S. and M.S. degrees in chemistry and organic chemistry with Prof. Yu Yuan from Yangzhou University in 2005 and 2008. Then he moved to Peking University and completed his Ph.D. degree in medicinal chemistry under the supervision of Prof. Ning Jiao. In 2011, he joined the group of Prof. Frank Glorius at Westfalische Wilhelms-Universitat Münster, as an Alexander von Humboldt Research Fellow studying Rh(III)-catalyzed C–H activation reactions. Since March 2014, he is working as a full professor at the Nanjing University, focusing on the development of new transition-metal catalysts and synthetic methodology. |
Introduction
Compared with traditional coupling reactions, the direct functionalization of C–H bonds exhibits more advantages due to its atom and step economy, especially when the introduction of halides in a specific synthesis intermediate is difficult.1 In particular, transition metal catalysis has emerged as a powerful tool for the functionalization of otherwise unreactive sp2 C–H in the past decades.2 The aliphatic C–H bonds, which are ubiquitous in organic molecules are the most challenging targets for effective and selective functionalization.3 Among them, notable advances have been made in the direct C–H arylation reactions through a variety of developed strategies. Initial studies focused on the direct arylation of 8-methylquinoline due to its good chelating abilities.4 Later on, external ligands were added to facilitate the Pd-catalyzed direct arylation of aliphatic acids, amides, amino acid derivatives and peptides.5 In addition, the utilization of bidentate chelating groups such as 8-aminoquinoline and picolinamide was also a powerful strategy to promote the C–H arylations.6 Despite the great progress made in this field, the current substrates are still limited to aliphatic acids, amides and the related compounds. Therefore, new catalytic systems need to be developed to expand the scope to other types of compounds.Oxime ethers as the precursor of ketones, amines, amides and cyanos are among the most useful functional building blocks available to the synthetic chemists. The β-oxygenation7 and amination8 of aliphatic C–H bonds in ketoxime derivatives have been exploited. Considering the wide existence of β-arylated ketones in natural products and pharmaceuticals,9 it is well worth developing a method to introduce a phenyl group in the β-position of oxime ethers via aliphatic C–H activation. In 2015, our group first developed a versatile method for the direct arylation of sp3 C–H bonds in O-methyl ketoximes and nitrogen-containing heterocycles.10 The key to this success depends on the appropriate choice of an Ir(III) catalyst and the use of diaryliodonium triflate salts as the coupling partners (Scheme 1a, Ir catalysis). Very recently, Chen and co-workers uncovered that the same transformation can occur in the presence of Pd catalysts (Scheme 1a, Pd catalysis).11 In view of the diaryliodonium salts, as costly and less available reagents with a low atom economy, we are encouraged to discover a milder catalytic system using a more accessible phenyl source. Herein, we report the Pd-catalyzed intermolecular direct arylation of the sp3 C–H bonds in O-methyl ketoximes with iodoarenes (Scheme 1b). Our latest method demonstrates a perfect adaptability of a wide range of substrates and proceeds smoothly in late-stage C–H functionalization, which provides a meaningful perspective in future research.
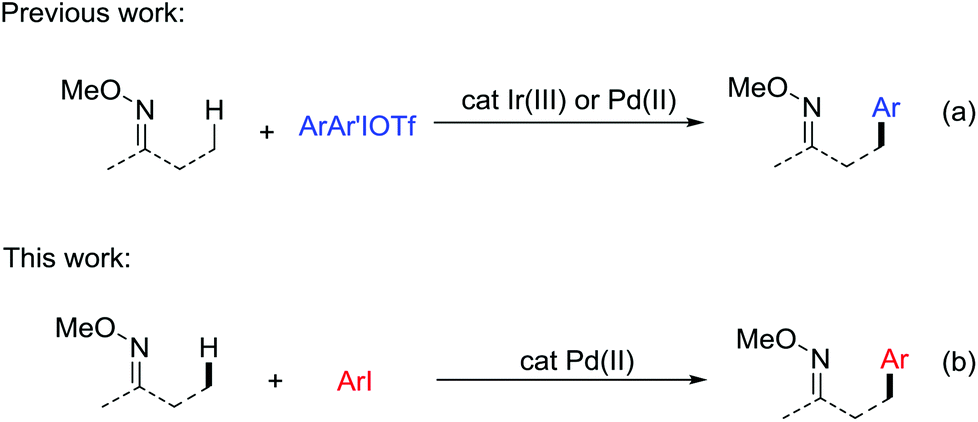 | ||
| Scheme 1 Iridium- and palladium-catalyzed direct C–H arylation of O-methyl ketoximes. | ||
Results and discussion
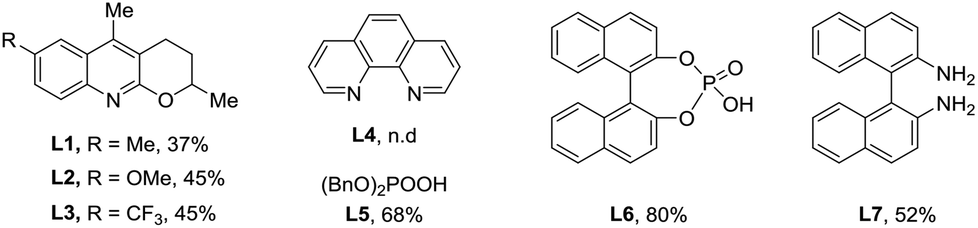
We started our research on the Pd-catalyzed C–H arylation of the readily available 2,3-dimethylcyclohex-2-en-1-one oxime (1a) and iodobenzene (2a) for the optimization of reaction conditions (Table 1). Initial studies focused on the sources of silver oxidants (entries 1–3). It was found that AgTFA was very critical to the reaction and provided the desired product 3aa in 31% yield at 100 °C using DCE as the solvent. The effects of various solvents were also examined and PhCF3 demonstrated the best effects (entry 4). Notably, lowering the temperature to 80 °C led to an improvement in the reaction outcome (entries 5 and 6). We also screened several different kinds of ligands from L1 to L7, and found that only organic phosphate L5–L6 can give positive effects (entries 7 and 8).12 Simple phosphate BINPO2H, commercially available at low cost, was the most effective affording 3aa in 80% yield. Under these conditions, reducing the amount of AgTFA or Pd(OAc)2 led to lower yields (entries 9 and 10). In addition, other palladium sources including Pd(TFA)2 and Pd2(dba)3 showed comparatively poor yields and obviously, the absence of a catalyst failed to provide the corresponding product (Table 1, entries 11–13). Based on the above results, it was revealed that entry 8 illustrated the optimal conditions for the Pd-catalyzed arylation of O-methyl ketoxime 1a.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | [Pd] (mol%) | L | Oxidant (equiv.) | Solvent | T | Yieldb (%) |
| a Reaction conditions: all the reactions were run on the 0.20 mmol scale with 1a (1.0 equiv.), 2a (2.0 equiv.), palladium catalyst (5 mol%), ligand (10 mol%) and silver oxidant (2.0 equiv.) in 1.0 mL solvent, 12 h. b Crude 1H NMR yield using CH2Br2 as the internal standard. c Isolated yield in parentheses. | ||||||
| 1 | Pd(OAc)2 (5) | — | Ag2CO3 (2.0) | DCE | 100 | Trace |
| 2 | Pd(OAc)2 (5) | — | AgOAc (2.0) | DCE | 100 | 5 |
| 3 | Pd(OAc)2 (5) | — | AgTFA (2.0) | DCE | 100 | 31 |
| 4 | Pd(OAc)2 (5) | — | AgTFA (2.0) | PhCF3 | 100 | 42 |
| 5 | Pd(OAc)2 (5) | — | AgTFA (2.0) | PhCF3 | 80 | 65 |
| 6 | Pd(OAc)2 (5) | — | AgTFA (2.0) | PhCF3 | 60 | 54 |
| 7 | Pd(OAc)2 (5) | L1 | AgTFA (2.0) | PhCF3 | 80 | 37 |
| 8 | Pd(OAc) 2 (5) | L6 | AgTFA (2.0) | PhCF 3 | 80 | 80 (73) |
| 9 | Pd(OAc)2 (5) | L6 | AgTFA (1.0) | PhCF3 | 80 | 47 |
| 10 | Pd(OAc)2 (2.5) | L6 | AgTFA (2.0) | PhCF3 | 80 | 56 |
| 11 | Pd(TFA)2 (5) | L6 | AgTFA (2.0) | PhCF3 | 80 | 63 |
| 12 | Pd(dba)2 (5) | L6 | AgTFA (2.0) | PhCF3 | 80 | 53 |
| 13 | — | L6 | AgTFA (2.0) | PhCF3 | 80 | 0 |
|
||||||
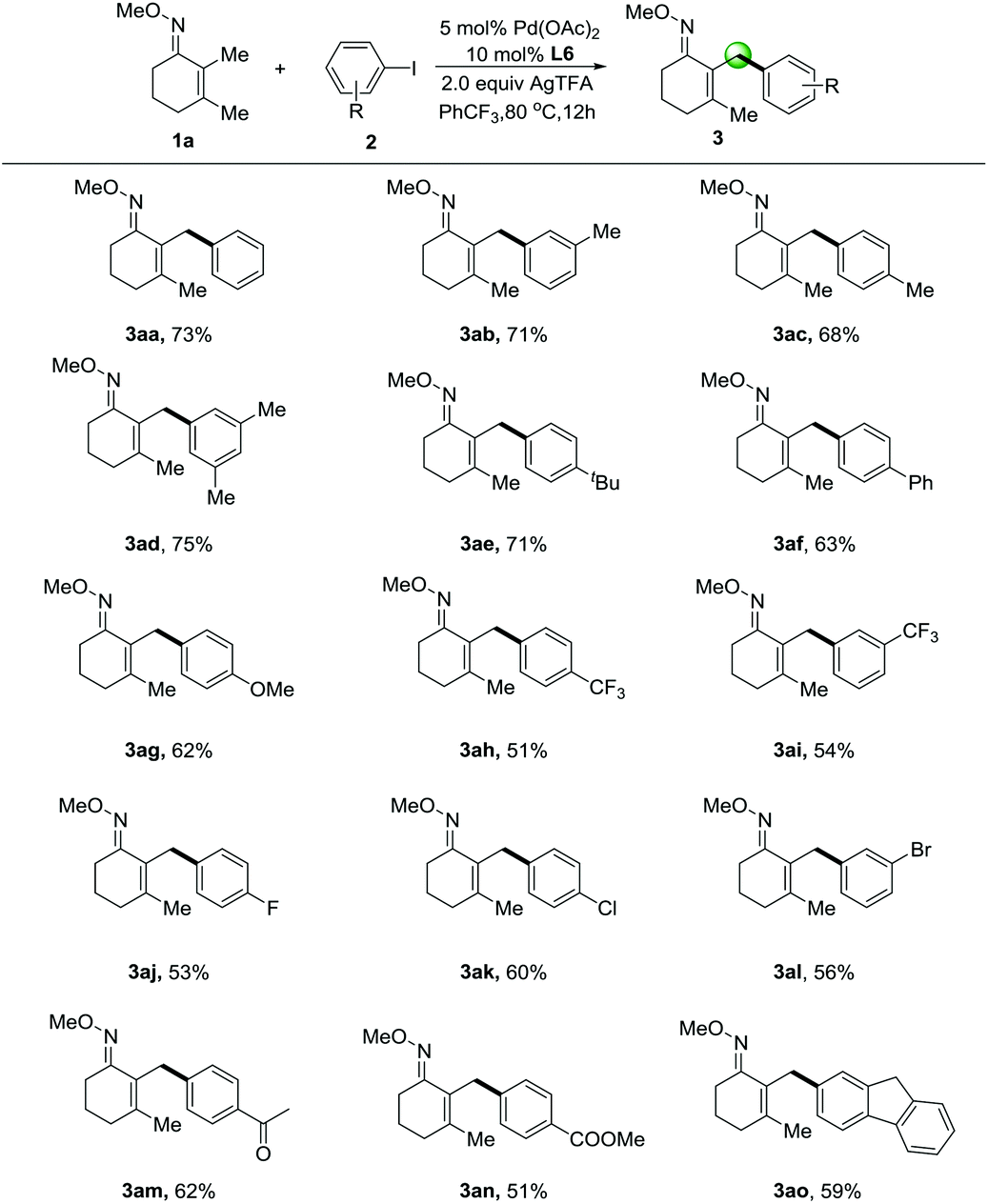
With the best conditions in hand, we sought to expand this transformation to the transfer of diverse aryl groups and we found that a range of substituted iodoarenes worked well with 2,3-dimethylcyclohex-2-en-1-one oxime (1a) (Table 2). Aromatic groups displaying electron-neutral and electron-rich substituents at the meta- and para-positions (3ab–3ad & 3ae–3ag) were transferred in particularly good yields from the corresponding iodoarenes. Useful halogenated iodoarenes such as trifluoromethyl-(3ah–3ai), fluoro-(3aj), chloro-(3ak) and even bromo-(3al) groups were accommodated, thereby providing possibilities for subsequent chemical transformations. Electron-withdrawing substituted groups such as acetyl- (3am) and methyl ester (3an) groups could be tolerated in this protocol. We were pleased that the coupling of the polycyclic aromatic motif was possible and proceeded in moderate yield (3ao), thus further enhancing the scope of our reaction.
| a Reaction conditions: all the reactions were run on the 0.20 mmol scale with 1a (1.0 equiv.), 2 (2.0 equiv.), Pd(OAc)2 (5 mol%), L6 (10 mol%) and AgTFA (2.0 equiv.) in 1.0 mL PhCF3, 12 h; isolated yields. |
|---|
|
When 2-iodobenzoic acid (2p) was employed as an ortho-substituted substrate, an unexpected five-membered lactone 3ap was observed in 62% yield under the optimized conditions (Scheme 2). Other substrates involving methyl-, chloro- and bromo-groups were well-tolerated, and the corresponding products 3aq–3as were isolated in 50%–63% yields. In addition, 2-iodo-4,5-dimethoxybenzoic acid (2t) was also compatible affording the cyclization product 3at in 54% yield. We speculated that the β-C–H arylation of O-methyl ketoximes with 2-iodobenzoic acids could afford the C–H arylation products as shown in Table 2, which caused further intramolecular C–H oxygenation in the presence of ortho-carboxylic acid groups. Although the cyclization reactions via multiple palladium-catalyzed C–H activation of arenes have been developed,13 this tandem sp3 C–H arylation and oxygenation process has seldom been reported.
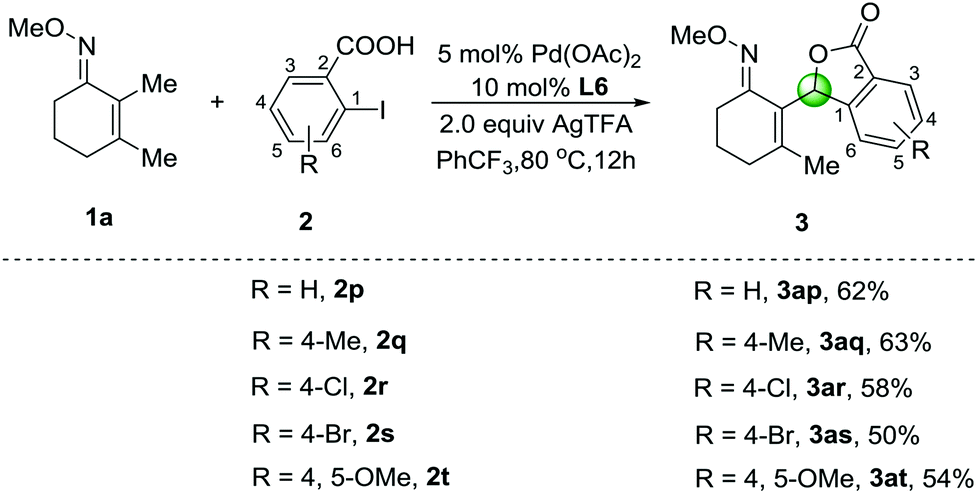 | ||
| Scheme 2 Cyclization of 2-iodobenzoic acids 2p–2t by dual C–H activation. | ||
We next investigated the substrate scope of O-methyl ketoximes with iodobenzene (2a) in Table 3. β-Arylation of ketoxime 1b, which contains a quaternary α-carbon atom, gave the monoarylated product 3ba in 77% yield. In the case of substrates 1c–1d, with the increasing of steric bulk from n-heptyl to cyclohexyl adjacent to the quaternary centers, the yield of the corresponding products gradually decreased. It should be mentioned that these substrates contain multiple possible sites for the direct arylation, showing extremely high selectivity for activation of primary β-C–H bonds in lieu of those at secondary carbon centers. Later on, we tested the substrates with several primary β-C–H bonds that can be functionalized. Gratifyingly, we were able to functionalize pinacolone derivative 1e and 1f to give a mixture of mono- and diarylated products in 73% and 77% yield, respectively. It's also noticed that the methyl groups adjacent to a quaternary center is necessary for this C–H arylation and oxime ethers with α-hydrogens having much lower reactivity, since O-methyl ketoxime 3ga only gave trace amounts of the product.
| a Reaction conditions: all the reactions were run on the 0.20 mmol scale with 1 (1.0 equiv.), 2a (2.0 equiv.), Pd(OAc)2 (5 mol%), L6 (10 mol%) and AgTFA (2.0 equiv.) in 1.0 mL PhCF3, 12 h; isolated yields. |
|---|
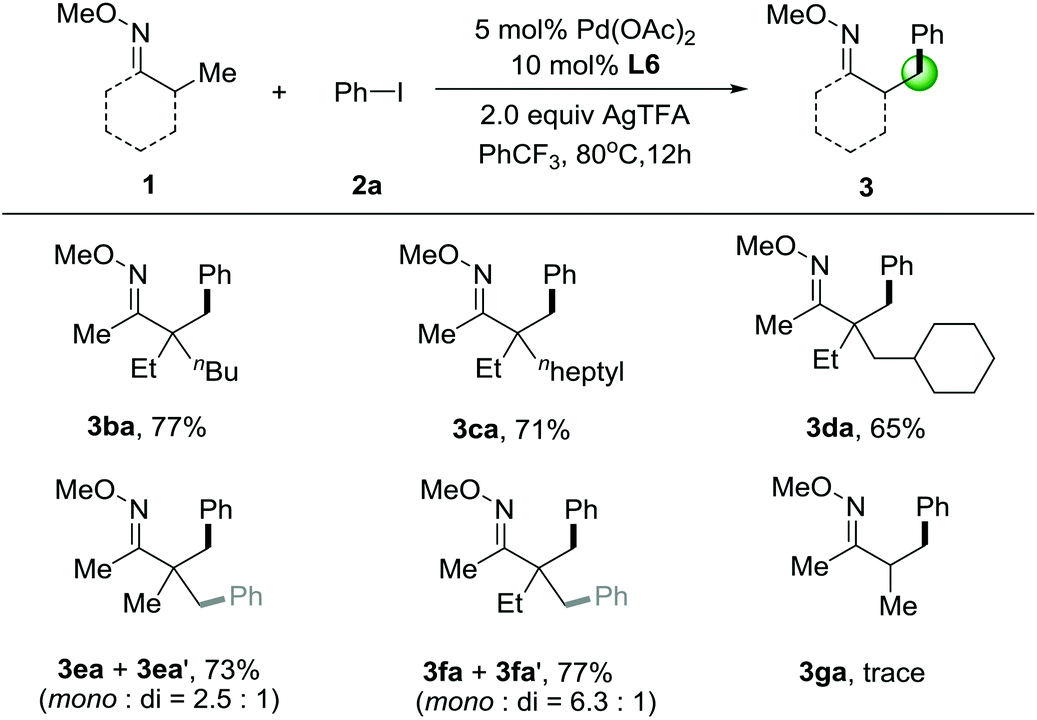
|
Late-stage modification of naturally significant molecules to access new bioactive compounds have been an emerging concept in organic chemistry.14 Encouraged by this successful sp3 C–H arylation, we also turned our attention to utilize our method as a key step for the regioselective C–H arylation of a complex molecule. O-Methyl ketoxime 5, derived from pharmaceutical compound Santonin 4, a kind of roundworm repellent, was compatible to this catalytic system affording the desired product 6 in 61% yield. The structure of 6 was further confirmed by X-ray diffraction as shown in Scheme 3.
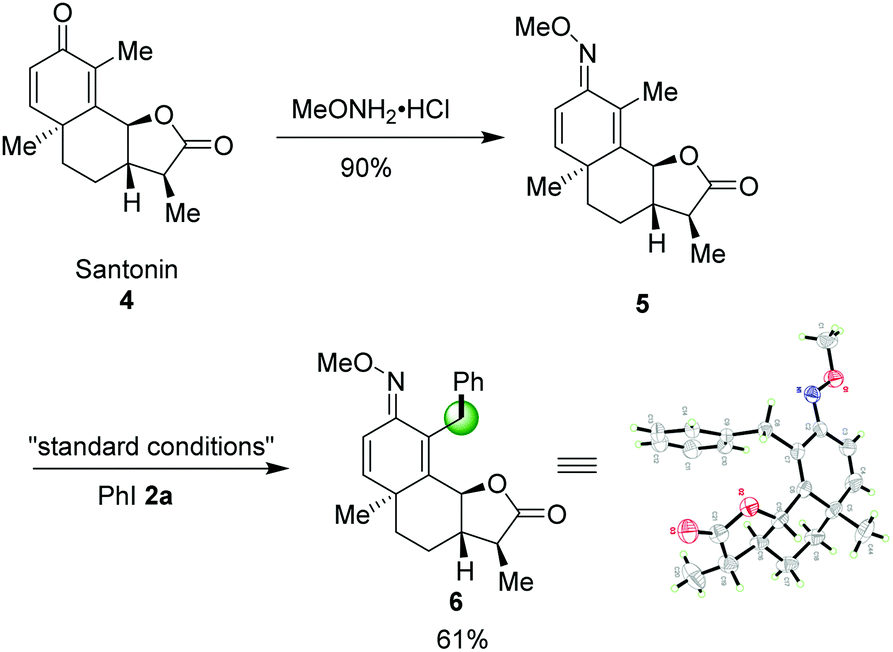 | ||
| Scheme 3 Late-stage C–H arylation of santonin. | ||
Chen and co-workers previously uncovered that Pd-catalyzed C–H arylation of O-methyl ketoximes with diaryliodonium salt triggered by the palladium species inducing C–H activation of the O-methyl ketoxime to generate a palladacycle complex.11,15 However, when the complex le′ was synthesized and treated with 2.0 equiv. of 2a and 2.0 equiv. of AgTFA, we didn't observe any corresponding arylated products (Scheme 4). We hypothesized that oxidative addition of the palladium species to iodoarenes should take place ahead of the C–H activation of O-methyl ketoximes under our conditions.
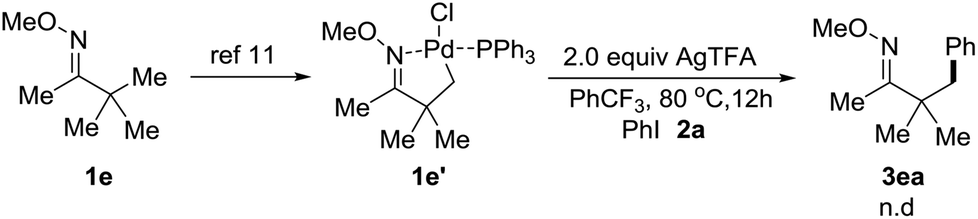 | ||
| Scheme 4 Mechanistic investigations. | ||
Conclusions
In summary, we have developed examples of the palladium-catalyzed C–H arylation of O-methyl ketoximes with iodoarenes under mild conditions. This novel method exhibited flexibility with different substrates and offered new opportunities to prepare β-arylated O-methyl ketoximes from inexpensive and available reagents. The efficient strategy not only tolerated various functional groups, but has also been applied to the modification of complex molecules. Also, a series of five-membered lactones could be generated from 2-iodobenzoic acids under standard conditions. Further studies on the mechanism and applications of this novel method are going to be researched by our group.Acknowledgements
We thank the “1000-Youth Talents Plan”, the “Jiangsu Specially-Appointed Professor Plan”, NSF of China (Grant 21402086), and NSF of Jiangsu Province (Grant BK20140594) for financial support. This work was also supported by a Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions.Notes and references
- (a) D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174 CrossRef CAS PubMed; (b) B. Li, S. Yang and Z. Shi, Synlett, 2008, 949 CAS; (c) K. Godula and D. Sames, Science, 2006, 312, 67 CrossRef CAS PubMed; (d) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094 CrossRef CAS PubMed; (e) L. Ackermann, R. Vincente and A. R. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792 CrossRef CAS PubMed; (f) O. Dougulis, H.-Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074 CrossRef PubMed; (g) G. P. McGlacken and L. M. Bateman, Chem. Soc. Rev., 2009, 38, 2447 RSC; (h) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed; (i) O. Baudoin, Chem. Soc. Rev., 2011, 40, 4902 RSC; (j) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960 CrossRef CAS PubMed; (k) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879 CrossRef CAS PubMed; (l) G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726 CrossRef CAS PubMed.
- Recent reviews on the transition-metal-catalyzed C–H activation reactions: (a) M. Lautens and P. Thansandote, Chem. – Eur. J., 2009, 15, 5874 CrossRef PubMed; (b) R. Giri, B.-F. Shi, K. M. Engle, N. Maugel and J.-Q. Yu, Chem. Soc. Rev., 2009, 38, 3242 RSC; (c) R. Jazzar, J. Hitce, A. Renaudat, J. Sofack-Kreutzer and O. Baudoin, Chem. – Eur. J., 2010, 16, 2654 CrossRef CAS PubMed; (d) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed; (e) S. H. Cho, J. Y. Kim, J. Kwak and S. Chang, Chem. Soc. Rev., 2011, 40, 5068 RSC; (f) C.-L. Sun, B.-J. Li and Z.-J. Shi, Chem. Rev., 2011, 111, 1293 CrossRef CAS PubMed; (g) J. Wencel-Delord, T. Dröge, F. Liu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740 RSC; (h) T. Newhouse and P. S. Baran, Angew. Chem., Int. Ed., 2011, 50, 3362 CrossRef CAS PubMed; (i) L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS PubMed; (j) L. McMurray, F. O'Hara and M. J. Gaunt, Chem. Soc. Rev., 2011, 40, 1885 RSC; (k) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215 CrossRef CAS PubMed; (l) Z. Shi, C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3381 RSC; (m) B.-J. Li and Z.-J. Shi, Chem. Soc. Rev., 2012, 41, 5588 RSC; (n) M. C. White, Science, 2012, 335, 807 CrossRef CAS PubMed; (o) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788 CrossRef CAS PubMed; (p) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960 CrossRef CAS PubMed; (q) S. R. Neufeldt and M. S. Sanford, Acc. Chem. Res., 2012, 45, 936 CrossRef CAS PubMed; (r) N. Kuhl, M. N. Hopkinson, J. Wencel-Delord and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 10236 CrossRef CAS PubMed; (s) J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369 CrossRef CAS PubMed; (t) X.-X. Cuo, D.-W. Gu, Z. Wu and W. Zhang, Chem. Rev., 2015, 115, 1622 CrossRef PubMed.
- Recent reviews on the transition-metal-catalyzed sp3 C–H activation reactions: (a) G. Chen and Z.-J. Shi, Natl. Sci. Rev., 2014, 1, 272 Search PubMed; (b) G. Qiu and J. Wu, Org. Chem. Front., 2015, 2, 169 RSC; (c) S.-Y. Zhang, F.-M. Zhang and Y.-Q. Tu, Chem. Soc. Rev., 2011, 40, 1937 RSC.
- (a) D. Kalyani, N. R. Deprez, L. V. Desai and M. S. Sanford, J. Am. Chem. Soc., 2005, 127, 7330 CrossRef CAS PubMed; (b) O. Daugulis and D. Shabashov, Org. Lett., 2005, 7, 3657 CrossRef PubMed; (c) K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2007, 129, 11904 CrossRef CAS PubMed; (d) W.-Y. Yu, W. Sit, Z. Zhou and A. S.-C. Chan, Org. Lett., 2009, 11, 3174 CrossRef CAS PubMed; (e) M. Kim, J. Kwak and S. Chang, Angew. Chem., Int. Ed., 2009, 48, 8935 CrossRef CAS PubMed.
- (a) R. Giri, Y. Lan, P. Liu, K. N. Houk and J.-Q. Yu, J. Am. Chem. Soc., 2012, 134, 14118 CrossRef CAS PubMed; (b) K. S. L. Chan, M. Wasa, L. Chu, B. N. Laforteza, M. Miura and J.-Q. Yu, Nat. Chem., 2014, 6, 146 CrossRef CAS PubMed; (c) J. He, S. Li, Y. Deng, H. Fu, B. N. Laforteza, J. E. Spangler, A. Homs and J.-Q. Yu, Science, 2014, 343, 1216 CrossRef CAS PubMed; (d) K.-J. Xiao, D. W. Lin, M. Miura, R.-Y. Zhu, W. Gong, M. Wasa and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 8138 CrossRef CAS PubMed; (e) Q. Deng, W. Gong, J. He and J.-Q. Yu, Angew. Chem., Int. Ed., 2014, 53, 6692 CrossRef PubMed.
- (a) V. G. Zaitsev, D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2005, 127, 13154 CrossRef CAS PubMed; (b) G. He and G. Chen, Angew. Chem., Int. Ed., 2011, 50, 5192 CrossRef CAS PubMed; (c) Q. Zhang, K. Chen, W.-H. Rao, Y. Zhang, F.-J. Chen and B.-F. Shi, Angew. Chem., Int. Ed., 2013, 52, 13588 CrossRef CAS PubMed; (d) F. Pan, P.-X. Shen, L.-S. Zhang, X. Wang and Z.-J. Shi, Org. Lett., 2013, 15, 4758 CrossRef CAS PubMed; (e) R. Shang, L. Ilies, A. Matsumoto and E. Nakamura, J. Am. Chem. Soc., 2013, 135, 6030 CrossRef CAS PubMed; (f) K. Chen and B.-F. Shi, Angew. Chem., Int. Ed., 2014, 53, 11950 CrossRef CAS PubMed; (g) Y. Aihara and N. Chatani, J. Am. Chem. Soc., 2014, 136, 898 CrossRef CAS PubMed; (h) Q. Gu, H. H. A. Mamari, K. Graczyk, E. Diers and L. Ackermann, Angew. Chem., Int. Ed., 2014, 53, 3868 CrossRef CAS PubMed.
- (a) L. V. Desai, K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2004, 126, 9542 CrossRef CAS PubMed; (b) Z. Ren, F. Mo and G. Dong, J. Am. Chem. Soc., 2012, 134, 16991 CrossRef CAS PubMed; (c) Y. Xu, G. Yan, Z. Ren and G. Dong, Nat. Chem., 2015, 7, 829 CrossRef CAS PubMed; (d) S. J. Thompson, D. Q. Thach and G. Dong, J. Am. Chem. Soc., 2015, 137, 11586 CrossRef CAS PubMed.
- (a) H. Wang, G. Tang and X. Li, Angew. Chem., Int. Ed., 2015, 54, 13049 CrossRef CAS PubMed; (b) T. Kang, Y. Kim, D. Lee, Z. Wang and S. Chang, J. Am. Chem. Soc., 2014, 136, 4141 CrossRef CAS PubMed; (c) T. Kang, H. Kim, J. G. Kim and S. Chang, Chem. Commun., 2014, 50, 12073 RSC; (d) H.-Y. Thu, W.-Y. Yu and C.-M. Che, J. Am. Chem. Soc., 2006, 128, 9048 CrossRef CAS PubMed.
- (a) U. Ryuji, U.S. Pat. Appl. Publ, US 20010034355, 2001 Search PubMed; (b) S. Wang, C. Zhang, G. Yang and Y. Yang, Nat. Prod. Commun., 2014, 9, 1027 CAS; (c) J. S. Choi, H. J. Park, H. A. Jung, H. Y. Chung, J. H. Jung and W. C. Choi, J. Nat. Prod., 2000, 63, 1705 CrossRef CAS PubMed; (d) K. Amagase, Y. Kimura, A. Wada, T. Yukishige, T. Murakami, E. Nakamura and K. Takeuchi, Curr. Pharm. Des., 2014, 20, 2783 CrossRef CAS PubMed.
- P. Gao, W. Guo, J. Xue, Y. Zhao, Y. Yuan, Y. Xia and Z. Shi, J. Am. Chem. Soc., 2015, 137, 12231 CrossRef CAS PubMed.
- J. Peng, C. Chen and C. Xi, Chem. Sci., 2015 10.1039/c5sc03903g.
- (a) S.-Y. Zhang, Q. Li, G. He, W. A. Nack and G. Chen, J. Am. Chem. Soc., 2013, 135, 12135 CrossRef CAS PubMed; (b) S.-Y. Zhang, G. He, W. A. Nack, Y. Zhao, Q. Li and G. Chen, J. Am. Chem. Soc., 2013, 135, 2124 CrossRef CAS PubMed.
- (a) V. S. Thirunavukkarasu, K. Parthasarathy and C.-H. Cheng, Angew. Chem., Int. Ed., 2008, 47, 9462 CrossRef CAS PubMed; (b) J. Karthikeyan and C.-H. Cheng, Angew. Chem., Int. Ed., 2011, 50, 9880 CrossRef CAS PubMed; (c) P. Gandeepan, K. Parthasarathy and C.-H. Cheng, J. Am. Chem. Soc., 2010, 132, 8569 CrossRef CAS PubMed.
- (a) W. R. Gutekunst, R. Gianatassio and P. S. Baran, Angew. Chem., Int. Ed., 2012, 51, 7507 CrossRef CAS PubMed; (b) H. M. Davies and J. R. Manning, Nature, 2008, 451, 417 CrossRef CAS PubMed; (c) W. R. Gutekunst and P. S. Baran, J. Am. Chem. Soc., 2011, 133, 19076 CrossRef CAS PubMed; (d) L. McMurray, F. Ohara and M. J. Gaunt, Chem. Soc. Rev., 2011, 40, 1885 RSC; (e) E. M. Simmons and J. F. Hartwig, Nature, 2012, 483, 70 CrossRef CAS PubMed; (f) J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369 CrossRef CAS PubMed.
- A. McNally, B. Haffemayer, B. S. L. Collins and M. Gaunt, Nature, 2014, 510, 129 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1444168. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qo00438a |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2016 |