
Nickel catalyzed reduction of arenols under mild conditions†
Wen-Juan
Shi
a,
Xiao-Lei
Li
a,
Zhao-Wei
Li
a and
Zhang-Jie
Shi
*ab
aBeijing National Laboratory of Molecular Sciences (BNLMS) and Key Laboratory of Bioorganic Chemistry and Molecular Engineering of Ministry of Education, College of Chemistry and Molecular Engineering and Green Chemistry Center, Peking University, Beijing, 100871, China. E-mail: zshi@pku.edu.cn
bState Key Laboratory of Organometallic Chemistry, Chinese Academy of Sciences, Shanghai 200032, China
First published on 14th January 2016
Abstract
Nickel catalyzed reduction of arenols has been developed with a mutual activation strategy under mild conditions. The reduction featured a broad substrate scope, non-sensitivity to steric hindrance and no over-reduction of the aromatic rings.
Industry has commonly focused on oil as the carbon source of chemical feedstocks.1 Compared to this non-renewable resource, the utilization of renewable and abundant biomass opens another door. Many efficient catalytic systems have been developed to degrade biomass into valuable chemicals.2 The recent flourishing developments on C–O bond activation shed light on the selective and efficient transformation of oxygen-rich lignocellulosic plant biomass to commercial chemicals.3 One of the most important achievements is the catalytic reduction of arenols and their derivatives,4–6 especially the deoxygenated analogues of phenol-based natural products.7 Traditionally, the reduction process of arenols and their derivatives includes three steps: (1) hydrolysis to their corresponding arenols, (2) conversion to the sulfonates, and (3) deoxygenation via Pd catalysis with H resources.8 Direct reduction of phenols and their derivatives is highly appealing, avoiding tedious procedures and the use of costly and unfriendly fluorinated reagents. Obviously, the desirable processes can lead to further utilization of electron-donating oxygen-based functionalities as temporary removable directing groups to achieve regioselective functionalization of arenes.9,10 In this field, heterogeneous catalysis shows its beauty and power while the reactions usually run under harsh conditions, accompanied by the over reduction of the aromatic rings.11
Recently, impressive progress in homogeneous catalysis has been made to reduce the C–O bonds of arenol derivatives, including the esters and ethers4 (Scheme 1a). To the best of our knowledge, the in situ generated arenols as byproducts have never been reduced into the desired arenes under mild conditions (under 100 °C). Under high temperature, the Ru/W bifunctional catalyst,5a the stoichiometric LiAlH4/KOtBu combination system,5b and the hydroxycyclopentadienyl iridium complexes5c could promote the reduction of arenols. However, the aromatic rings were hydrogenated to form cyclohexanes, cyclohexanol and others in many cases.
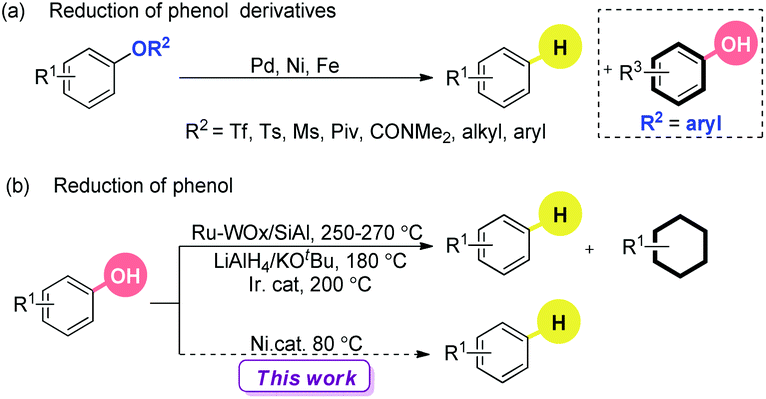 | ||
| Scheme 1 The reduction of phenol and its derivatives. | ||
The major challenges for the direct and selective reduction of arenols rely on the stability of the C–OH moiety: (1) the high bond dissociation energy (BDE) of its C–O bond; (2) the poor leaving ability of its hydroxyl group; (3) the deactivation of the catalyst by bonding with a phenolic anion; (4) and the further enhancement of the BDE by the p–π conjugative effect of its anion.3c,12,13 Based on our progress on the mutual activation of arenols,12 we envisioned that the reduction of arenols with suitable reductants assisted by an appropriate Lewis acid should be possible. Herein we describe a novel nickel catalyzed reduction of arenols under mild conditions.
We began our investigations by examining the reactivity of naphthalen-2-ol (1a) with several hydride sources in the combination of Ni(cod)2/PCy3/NaH in toluene (Table 1). The nature of the reductants was critical, and HBPin, BH3·SMe2, HSi(OEt)3, HSiEt3, iPrMgCl, LiAlH4, NaBH4, Zn, and H2, were all ineffective, even when accompanied by a stoichiometric amount of AlMe3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 4g or BEt312a (entry 1). Based on our experience in the Suzuki-type coupling of arenols,12a we considered the important interaction between arenol and boron reagents. After extensive screenings, we found that a cocktail containing Ni(cod)2/PCy3/B2Pin2/K3PO4 promoted the targeted reaction in 91% yield and other diboron reagents did not show better results (entries 2–3). The inclusion of different ligands had a profound influence on the transformation (entry 4). Strikingly, the utilization of a base with regime basicity had deleterious effects on the reactivity (entry 5). The yield slightly decreased in the absence of a base (entry 6). The air and moisture-stable nickel catalyst could also catalyze the reaction (entry 7). Moreover, a difference in the reactivity was found in the diverse solvents (entries 8–9). Amazingly, the reaction occurred at room temperature (entry 10), although the yield was sacrificed (entry 11). The addition of 1.5 equivalents of B2Pin2 delivered the product in excellent yields (entry 12). Notably, the reaction was efficient while the catalyst loading was decreased to 2.5 mol%. Indeed, this is a relatively low catalyst loading in Ni catalyzed C–O bond activations, which was considered as one of the major challenges in this field (entry 13). Importantly, no over-reduction of the aromatic ring was observed.
4g or BEt312a (entry 1). Based on our experience in the Suzuki-type coupling of arenols,12a we considered the important interaction between arenol and boron reagents. After extensive screenings, we found that a cocktail containing Ni(cod)2/PCy3/B2Pin2/K3PO4 promoted the targeted reaction in 91% yield and other diboron reagents did not show better results (entries 2–3). The inclusion of different ligands had a profound influence on the transformation (entry 4). Strikingly, the utilization of a base with regime basicity had deleterious effects on the reactivity (entry 5). The yield slightly decreased in the absence of a base (entry 6). The air and moisture-stable nickel catalyst could also catalyze the reaction (entry 7). Moreover, a difference in the reactivity was found in the diverse solvents (entries 8–9). Amazingly, the reaction occurred at room temperature (entry 10), although the yield was sacrificed (entry 11). The addition of 1.5 equivalents of B2Pin2 delivered the product in excellent yields (entry 12). Notably, the reaction was efficient while the catalyst loading was decreased to 2.5 mol%. Indeed, this is a relatively low catalyst loading in Ni catalyzed C–O bond activations, which was considered as one of the major challenges in this field (entry 13). Importantly, no over-reduction of the aromatic ring was observed.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Ligand | Reductant | Base | Solvent | Yield (%) |
| a Conditions: 1a (0.2 mmol), reductant (0.4 mmol), Ni catalyst (5 mol%), ligand (20 mol%), base (0.6 mmol), solvent (0.5 mL), 80 °C, 12 h and the yield was determined using GC analysis. b HSiEt3, HSi(OEt)3, BH3·SMe2, HBPin, Zn, iPrMgCl, NaBH4, LiAlH4, H2 were used. c Isolated yield. d Dcype, IPr·HCl, phen were used. e Cs2CO3, K2HPO4, KH2PO4, NaH, DABCO were used. f Ni(acac)2 was used. g The reaction was performed at 20 °C. h The reaction was performed at 60 °C. i B2Pin2 (1.5 equiv.)/K3PO4 (2.5 equiv.) was used. j Ni(cod)2 (2.5 mol%)/PCy3 (10 mol%) was used. k No Ni(cod)2. | |||||
| 1b | PCy3 | HSiEt3etc. | K3PO4 | Toluene | <5 |
| 2 | PCy3 | B2(nep)2 | K3PO4 | Toluene | 45 |
| 3 | PCy3 | B2Pin2 | K3PO4 | Toluene | 91 (80)c |
| 4d | Dcype etc. | B2Pin2 | K3PO4 | Toluene | <5 |
| 5e | PCy3 | B2Pin2 | Cs2CO3etc. | Toluene | <5–85 |
| 6 | PCy3 | B2Pin2 | — | Toluene | 84 |
| 7f | PCy3 | B2Pin2 | K3PO4 | Toluene | 65 |
| 8 | PCy3 | B2Pin2 | K3PO4 | THF | 53 |
| 9 | PCy3 | B2Pin2 | K3PO4 | DMF | <5 |
| 10g | PCy3 | B2Pin2 | K3PO4 | Toluene | 15 |
| 11h | PCy3 | B2Pin2 | K3PO4 | Toluene | 65 |
| 12i | PCy3 | B2Pin2 | K3PO4 | Toluene | 87 |
| 13j | PCy3 | B2Pin2 | K3PO4 | Toluene | 72 |
| 14k | PCy3 | B2Pin2 | K3PO4 | Toluene | <5 |
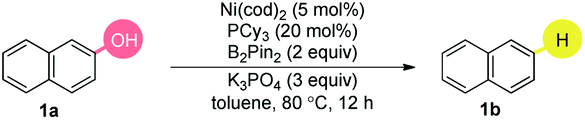
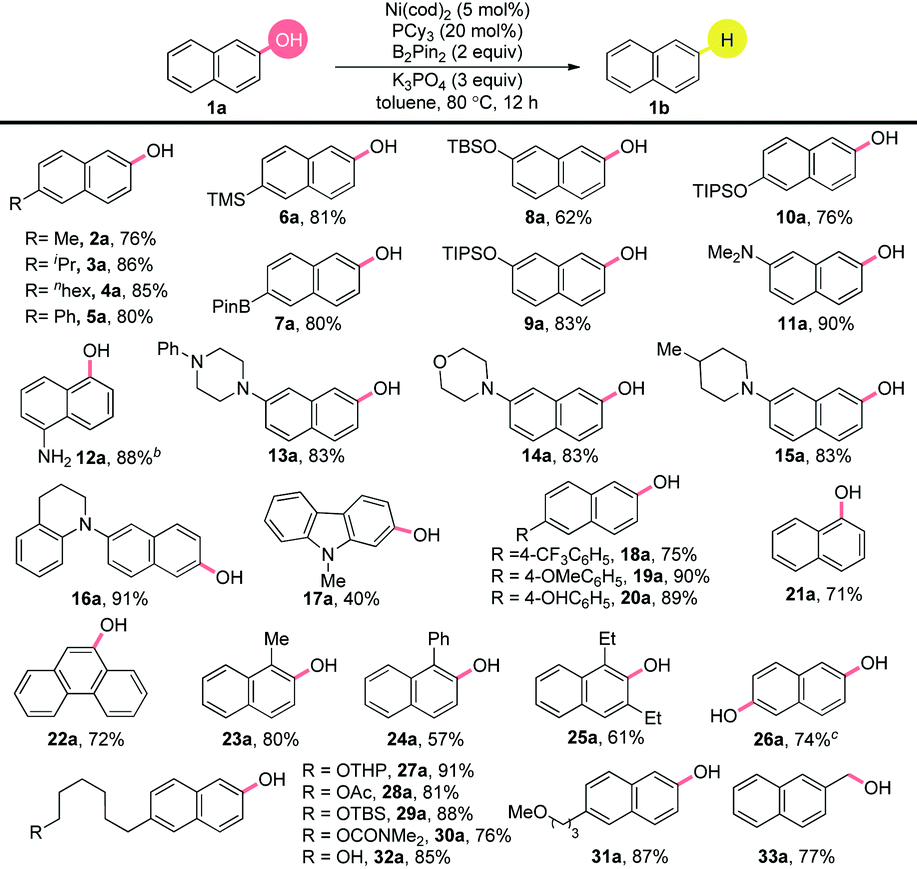
With the optimized conditions in hand, we found that the substrate scope encompassed electron-neutral (2a–4a), electron-poor (18a) and electron-rich naphthols (19a). The chemoselective character of this reduction was nicely illustrated by the good tolerance to diverse substituents in good yields (Table 2). Notably, an excellent yield was obtained with the silyl group (6a),14 which could be easily converted to other functionalities.15–18 Importantly, the boronic ester group survived well (7a).19 Silyloxyl groups in different positions on the aromatic ring showed competing reactivities (8–10a). N,N-Dimethylamino and free amine functionalities were tolerated well (11–12a). Substrates with nitrogen containing heterocycles, such as piperazinyl (13a), morpholinyl (14a), piperidinyl (15a) and tetrahydroquinolinyl (16a) produced the corresponding arenes in excellent yields. A worthy reactivity was found for the carbazole structural unit, a novel building block in pharmaceuticals, natural products and materials science (17a).20 The reduction took place smoothly for the substrate bearing trifluoromethyl group, which is highly valuable for wide application in material and pharmaceutical molecules (18a).21 The survival of the methoxyl and hydroxyl groups on the phenyl ring was achieved by the control of the amount of B2Pin2 (19–20a). The reaction proceeded efficiently with α-naphthol (21a) and fused-aromatic substrates (22a). Notably, the ortho substituted groups did not have an obvious effect on the efficiency, as exemplified by 23–24a giving good yields. Most importantly, the highly sterically hindered 1,3-diethylnaphthalen-2-ol (25a) showed great reactivity. In addition, the dihydroxyl groups gave efficient conversion to the product of the mono reduction (26a). Luckily, the successful preparation of 27–32a illustrated the selectivity profile among the different C–O bonds, showing the practical utility of this method for further transformations in the late-stage modification of biological compounds. Unfortunately, cyano, ester, halide and ether groups on the naphthyl ring could not be retained. Luckily, the benzyl hydroxyl group could also be deoxygenized in a high yield (33a). It is noteworthy that, over-reduction products of the aromatic ring were not observed.
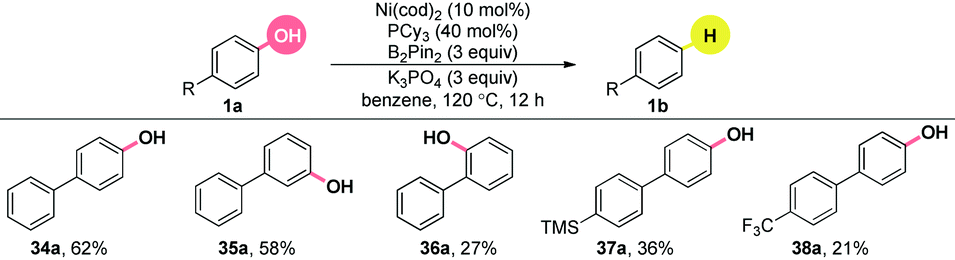
A closer look into the literature indicated that the inclusion of π-extended systems strongly enhanced the reactivity of a C–O bond.22 Encouraged by the results in Table 2, we extended the substrate scope to the simple, yet, challenging phenols without the utilization of ortho-directing groups under the finely modified conditions (Table 3). The substrate bearing an ortho phenyl group generated the product, albeit in a relatively lower yield, compared to its meta and para analogues (34–36a). Both electron-rich (37a) and electron-poor (38a) groups were tolerated. Unfortunately, other π-extended systems, such as (E)-4-styrylphenol were fully recovered. Further efforts to extend this chemistry to general phenols are underway.
| a Conditions: 1 (0.2 mmol), B2Pin2 (0.6 mmol), Ni(cod)2 (0.02 mmol), PCy3 (0.08 mmol), K3PO4 (0.6 mmol), benzene (0.5 mL) at 120 °C, 12 h. |
|---|
|
To further understand this reduction, we conducted preliminary mechanistic experiments (Scheme 2). Firstly, when the deuterated naphthalen-2-ol (1aa) was subjected to the standard conditions (eqn (1)), no deuterium incorporation in the product ruled out the direct deoxygenation pathway. The use of benzene-d6 or toluene together with 1 equivalent of D2O only afforded 52% and 68% deuterium incorporation of the products (eqn (2) and (3)). Those results indicated that the solvent was at least one of the hydrogen atom sources in accordance with the literature report.23 An unsymmetrical substrate was subjected to the standard conditions with benzene-d6 as the solvent. And a H/D exchange occurred because the deuterium incorporated was observed at different aryl C–H bonds in the final product (eqn (4)). This result indicated that the C–H bonds in the naphthalene rings could be another source of the hydrogen atoms, since the C–H bonds could be cleaved by low valent metal catalysts with boron reagents,24 which made the labeling experiments complicated. Moreover, according to the result that the reduction was very sensitive to the basicity of the base, we speculated whether the reduction product could originate from the aryl boronic pinacol ester since the arylboronate product in the borylation of C(sp2)–H was reported to be possibly decomposed in the presence of K3PO4.25 The starting material was recovered when we subjected 7b to the standard reaction conditions (eqn (5)), indicating that the borylated product was not the precursor in this transformation. Finally, we found that the catalytic reduction product of 1a was generated in 80% GC yield by the addition of 20 mol% TEMPO (eqn (6)). This observation indicated that a single electron transfer process might not come into play. Finally, the unique effect of B2Pin2 stimulated us to investigate its role in this reduction under such mild conditions. From the 11B NMR spectra in Scheme S1,† no doublet peaks of HBPin were detected,26 when a mixture of 1a, B2Pin2, Ni(cod)2 and PCy3 in toluene was stirred at 80 °C for 30 min, while a new singlet peak at δ = 22.50 ppm27 indicated that a Nap-OBPin intermediate was formed. And in this reaction, a very low conversion to naphthalene was observed. Since Np-OBPin was very prone to being hydrolyzed, we failed to isolate this intermediate, but it was confirmed by GC-MS (Scheme S2†). When B2Pin2 was replaced by 4 equivalents of HBPin under the standard conditions, 1a was converted into trace amounts of 1b and the major product was Nap-OBPin, which was confirmed by GC-MS (Scheme S3†).
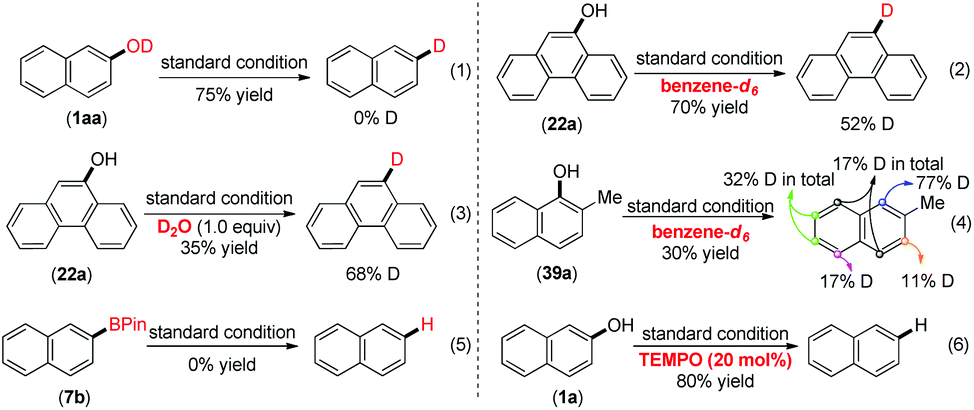 | ||
| Scheme 2 Investigation of the mechanism. | ||
Based on the literature report and current mechanism studies, we proposed the catalytic cycle as shown in Scheme 3. The arenol was coordinated with B2Pin2 to form the key intermediate A. With a mutual activation strategy, the phenolic oxygen atom worked as a Lewis base to weaken the B–B bond, thus enhancing the transmetallation of B2Pin2. Meanwhile, the Lewis acidic B atoms decreased the electron density of the C–O bond, reducing the energy barrier of the oxidation addition step. After the oxidative addition of the B–B bond to the low-valent Ni species,28 the intermediate B was transformed in situ to the nickel species D and a relatively active aryl boronic ester C, which was oxidized to generate the nickel species E. The reductive elimination of F generated from the transmetallation between the two nickel species D and E afforded the final product and regenerated the nickel catalyst.29
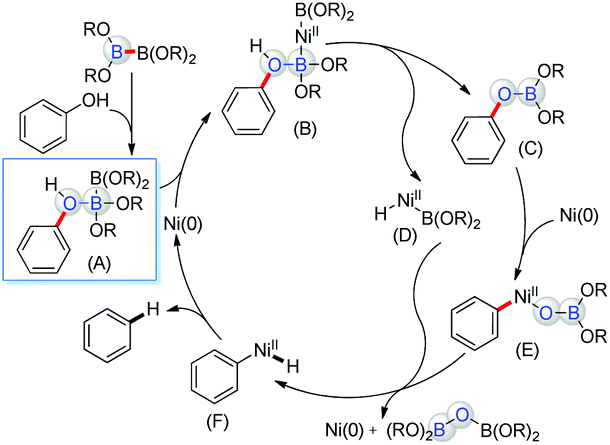 | ||
| Scheme 3 Proposed mechanism. | ||
In summary, the efficient nickel catalyzed reduction of the C–O bonds of arenols under mild conditions with a mutual activation strategy was reported. This new transformation featured a good group tolerance and non-sensitivity to the steric hindrance. Over-reduction of the arene ring was not observed. The vital role of B2Pin2 was the key to this successful transformation of arenols, which could be utilized for the further design of interesting transformations of C–O bonds.
Experimental section
A representative procedure (Table 2)
To an oven-dried Schlenk tube with a stirring bar were added 2-naphthol (1a) (28.8 mg, 0.20 mmol) and B2Pin2 (101.6 mg, 0.40 mmol) in air, and then K3PO4 (127.2 mg, 0.60 mmol), PCy3 (11.2 mg, 0.04 mmol) and Ni(cod)2 (2.8 mg, 0.01 mmol) were added, followed by the injection of toluene (0.50 mL) in a glove box. The tube was sealed up and the mixture was stirred at 80 °C for 12 h. The mixture was then cooled to room temperature and directly purified using column chromatography to afford compound 1b as a white solid (20.5 mg, 80%).Acknowledgements
Support of this work by the “973” Project from the MOST of China (2015CB856600, 2013CB228102) and NSFC (no. 20925207 and 21002001) is gratefully acknowledged.Notes and references
- (a) J. D. Nguyen, B. S. Matsuura and C. R. J. Stephenson, J. Am. Chem. Soc., 2014, 136, 1218–1221 CrossRef CAS PubMed; (b) J. Mottweiler, T. Rinesch, C. Besson, J. Buendia and C. Bolm, Green Chem., 2015, 17, 5001–5008 RSC.
- (a) A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed; (b) J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed; (c) F.-X. Luo, T.-G. Zhou, X. Li, Y.-L. Luo and Z.-J. Shi, Org. Chem. Front., 2015, 2, 1066–1070 RSC; (d) T. vom Stein, T. den Hartog, J. Buendia, S. Stoychev, J. Mottweiler, C. Bolm, J. Klankermayer and W. Leitner, Angew. Chem., 2015, 127, 5957–5961 ( Angew. Chem., Int. Ed. , 2015 , 54 , 5859–5863 ) CrossRef.
- For selected reviews on C–O bonds activation: (a) B. M. Rosen, K. W. Quasdorf, D. A. Wilson, N. Zhang, A.-M. Resmerita, N. K. Garg and V. Percec, Chem. Rev., 2011, 111, 1346–1416 CrossRef CAS PubMed; (b) B.-J. Li, D.-G. Yu, C.-L. Sun and Z.-J. Shi, Chem. – Eur. J., 2011, 17, 1728–1759 CrossRef CAS PubMed; (c) D.-G. Yu, B.-J. Li and Z.-J. Shi, Acc. Chem. Res., 2010, 43, 1486–1495 CrossRef CAS PubMed; (d) D.-G. Yu, F. de Azambuja and F. Glorius, Angew. Chem., 2014, 126, 7842–7845 ( Angew. Chem., Int. Ed. , 2014 , 53 , 7710–7712 ) CrossRef; (e) S. I. Kozhushkov, H. K. Potukuchi and L. Ackermann, Catal. Sci. Technol., 2013, 3, 562–571 RSC; (f) F.-S. Han, Chem. Soc. Rev., 2013, 42, 5270–5298 RSC; (g) B. Su, Z.-C. Cao and Z.-J. Shi, Acc. Chem. Res., 2015, 48, 886–896 CrossRef CAS PubMed; (h) S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299–309 CrossRef CAS PubMed; (i) E. J. Tollefson, L. E. Hanna and E. R. Jarvo, Acc. Chem. Res., 2015, 48, 2344–2353 CrossRef CAS PubMed; (j) M. Tobisu and N. Chatani, Acc. Chem. Res., 2015, 48, 1717–1726 CrossRef CAS PubMed.
- For selected examples on the reduction reactions of C–O bonds: (a) H. Sajiki, A. Mori, T. Mizusaki, T. Ikawa, T. Maegawa and K. Hirota, Org. Lett., 2006, 8, 987–990 CrossRef CAS PubMed; (b) V. Kogan, Tetrahedron Lett., 2006, 47, 7515–7518 CrossRef CAS; (c) B. H. Lipshutz, B. A. Frieman, T. Butler and V. Kogan, Angew. Chem., 2006, 118, 814–817 ( Angew. Chem., Int. Ed. , 2006 , 45 , 800–803 ) CrossRef; (d) W. K. Chow, C. M. So, C. P. Lau and F. Y. Kwong, Org. Chem. Front., 2014, 1, 464–467 RSC; (e) P. Álvarez-Bercedo and R. Martin, J. Am. Chem. Soc., 2010, 132, 17352–17353 CrossRef PubMed; (f) M. Tobisu, K. Yamakawa, T. Shimasaki and N. Chatani, Chem. Commun., 2011, 47, 2946–2948 RSC; (g) A. G. Sergeev and J. F. Hartwig, Science, 2011, 332, 439–443 CrossRef CAS PubMed; (h) A. G. Sergeev, J. D. Webb and J. F. Hartwig, J. Am. Chem. Soc., 2012, 134, 20226–20229 CrossRef CAS PubMed; (i) Y. Ren, M. Yan, J. Wang, Z. C. Zhang and K. Yao, Angew. Chem., 2013, 125, 12906–12910 ( Angew. Chem., Int. Ed. , 2013 , 52 , 12674–12678 ) CrossRef; (j) A. Fedorov, A. A. Toutov, N. A. Swisher and R. H. Grubbs, Chem. Sci., 2013, 4, 1640–1645 RSC; (k) D.-G. Yu, X. Wang, R.-Y. Zhu, S. Luo, X.-B. Zhang, B.-Q. Wang, L. Wang and Z.-J. Shi, J. Am. Chem. Soc., 2012, 134, 14638–14641 CrossRef CAS PubMed; (l) M. Tobisu, T. Morioka, A. Ohtsuki and N. Chatani, Chem. Sci., 2015, 6, 3410–3414 RSC; (m) C. Zarate, R. Manzano and R. Martin, J. Am. Chem. Soc., 2015, 137, 6754–6757 CrossRef CAS PubMed; (n) T. Mesganaw, N. F. Fine Nathel and N. K. Garg, Org. Lett., 2012, 14, 2918–2921 CrossRef CAS PubMed.
- (a) Y.-B. Huang, L. Yan, M.-Y. Chen, Q.-X. Guo and Y. Fu, Green Chem., 2015, 17, 3010–3017 RSC; (b) H. Xu, B. Yu, H. Zhang, Y. Zhao, Z. Yang, J. Xu, B. Han and Z. Liu, Chem. Commun., 2015, 51, 12212–12215 RSC; (c) S. Kusumoto and K. Nozaki, Nat. Commun., 2015, 6, 6296 CrossRef PubMed; (d) When we are preparing for the submission of this manuscript, another beautiful example was reported via Ni-catalysis at 120 °C: A. Ohgi and Y. Nakao, Chem. Lett., 2016, 45, 45–47 CrossRef.
- (a) D. Cavagnat, T. Buffeteau and T. Brotin, J. Org. Chem., 2008, 73, 66–75 CrossRef CAS PubMed; (b) Y.-G. Si, M. P. Gardner, F. I. Tarazi, R. J. Baldessarini and J. L. Neumeyer, J. Med. Chem., 2008, 51, 983–987 CrossRef CAS PubMed.
- (a) D. A. Evans, M. R. Wood, B. W. Trotter, T. I. Richardson, J. C. Barrow and J. L. Katz, Angew. Chem., 1998, 110, 2868–2872 ( Angew. Chem., Int. Ed. , 1998 , 37 , 2700–2704 ) CrossRef; (b) J. Cianci, J. B. Baell, B. L. Flynn, R. W. Gable, J. A. Mould, D. Paul and A. J. Harvey, Bioorg. Med. Chem. Lett., 2008, 18, 2055–2061 CrossRef CAS PubMed; (c) S. E. Shockley, J. C. Holder and B. M. Stoltz, Org. Lett., 2014, 16, 6362–6365 CrossRef CAS PubMed; (d) N. Z. Burns, I. N. Krylova, R. N. Hannoush and P. S. Baran, J. Am. Chem. Soc., 2009, 131, 9172–9173 CrossRef CAS PubMed.
- (a) A. Mori, T. Mizusaki, T. Ikawa, T. Maegawa, Y. Monguchi and H. Sajiki, Chem. – Eur. J., 2007, 13, 1432–1441 CrossRef CAS PubMed; (b) S. Cacchi, P. G. Ciattini, E. Morera and G. Ortar, Tetrahedron Lett., 1986, 27, 5541–5544 CrossRef CAS.
- G. Arnott, H. Brice, J. Clayden and E. Blaney, Org. Lett., 2008, 10, 3089–3092 CrossRef CAS PubMed.
- (a) R. Zhu, J. Wei and Z. Shi, Chem. Sci., 2013, 4, 3706–3711 RSC; (b) S. Luo, F.-X. Luo, X.-S. Zhang and Z.-J. Shi, Angew. Chem., 2013, 125, 10792–10795 ( Angew. Chem., Int. Ed. , 2013 , 52 , 10598–10601 ) CrossRef; (c) X.-S. Zhang, Z.-W. Li and Z.-J. Shi, Org. Chem. Front., 2014, 1, 44–49 RSC; (d) B. Xiao, T.-J. Gong, Z.-J. Liu, J.-H. Liu, D.-F. Luo, J. Xu and L. Liu, J. Am. Chem. Soc., 2011, 133, 9250–9253 CrossRef CAS PubMed.
- A.-L. Marshall and P. J. Alaimo, Chem. – Eur. J., 2010, 16, 4970–4980 CrossRef CAS PubMed.
- (a) D.-G. Yu and Z.-J. Shi, Angew. Chem., 2011, 123, 7235–7238 ( Angew. Chem., Int. Ed. , 2011 , 50 , 7097–7100 ) CrossRef; (b) D.-G. Yu, B.-J. Li, S.-F. Zheng, B.-T. Guan, B.-Q. Wang and Z.-J. Shi, Angew. Chem., 2010, 122, 4670–4674 ( Angew. Chem., Int. Ed. , 2010 , 49 , 4566–4570 ) CrossRef; (c) Z.-C. Cao, D.-G. Yu, R.-Y. Zhu, J.-B. Wei and Z.-J. Shi, Chem. Commun., 2015, 51, 2683–2686 RSC; (d) Z.-C. Cao, F.-X. Luo, W.-J. Shi and Z.-J. Shi, Org. Chem. Front., 2015, 2, 1505–1510 RSC.
- Z. Chen, H. Zeng, S. A. Girard, F. Wang, N. Chen and C.-J. Li, Angew. Chem., 2015, 127, 14695–14699 ( Angew. Chem., Int. Ed. , 2015 , 54 , 14487–14491 ) CrossRef.
- C. Cheng and J. F. Hartwig, Chem. Rev., 2015, 115, 8946–8975 CrossRef CAS PubMed.
- (a) L. T. Ball, G. C. Lloyd-Jones and C. A. Russell, Science, 2012, 337, 1644–1648 CrossRef CAS PubMed; (b) K. Funaki, T. Sato and S. Oi, Org. Lett., 2012, 14, 6186–6189 CrossRef CAS PubMed.
- C. J. Moody and P. Shah, J. Chem. Soc., Perkin Trans. 1, 1989, 2463–2471 RSC.
- (a) C. Zarate and R. Martin, J. Am. Chem. Soc., 2014, 136, 2236–2239 CrossRef CAS PubMed; (b) S. R. Wilson and L. A. Jacob, J. Org. Chem., 1986, 51, 4833–4836 CrossRef CAS.
- N. Chernyak, A. S. Dudnik, C. Huang and V. Gevorgyan, J. Am. Chem. Soc., 2010, 132, 8270–8272 CrossRef CAS PubMed.
- (a) J. F. Hartwig, Acc. Chem. Res., 2012, 45, 864–873 CrossRef CAS PubMed; (b) H. Kinuta, M. Tobisu and N. Chatani, J. Am. Chem. Soc., 2015, 137, 1593–1600 CrossRef CAS PubMed; (c) X.-W. Liu, J. Echavarren, C. Zarate and R. Martin, J. Am. Chem. Soc., 2015, 137, 12470–12473 CrossRef CAS PubMed; (d) T. Niwa, H. Ochiai, Y. Watanabe and T. Hosoya, J. Am. Chem. Soc., 2015, 137, 14313–14318 CrossRef CAS PubMed.
- (a) M. Tobisu, Y. Kita, Y. Ano and N. Chatani, J. Am. Chem. Soc., 2008, 130, 15982–15989 CrossRef CAS PubMed; (b) H. Gao, Q.-L. Xu, M. Yousufuddin, D. H. Ess and L. Kürti, Angew. Chem., 2014, 126, 2739–2743 ( Angew. Chem., Int. Ed. , 2014 , 53 , 2701–2705 ) CrossRef.
- T. A. S. R. T. Furuya, Nature, 2011, 473, 470–477 CrossRef CAS PubMed.
- (a) C. Zarate, R. Manzano and R. Martin, J. Am. Chem. Soc., 2015, 137, 6754–6757 CrossRef CAS PubMed; (b) M. Tobisu, A. Yasutome, H. Kinuta, K. Nakamura and N. Chatani, Org. Lett., 2014, 16, 5572–5575 CrossRef CAS PubMed; (c) A. Correa and R. Martin, J. Am. Chem. Soc., 2014, 136, 7253–7256 CrossRef CAS PubMed; (d) C. Zarate and R. Martin, J. Am. Chem. Soc., 2014, 136, 2236–2239 CrossRef CAS PubMed; (e) A. Correa, T. León and R. Martin, J. Am. Chem. Soc., 2014, 136, 1062–1069 CrossRef CAS PubMed; (f) J. Yang, T. Chen and L.-B. Han, J. Am. Chem. Soc., 2015, 137, 1782–1785 CrossRef CAS PubMed; (g) M. Tobisu, T. Takahira, A. Ohtsuki and N. Chatani, Org. Lett., 2015, 17, 680–683 CrossRef CAS PubMed; (h) M. Tobisu, T. Takahira and N. Chatani, Org. Lett., 2015, 17, 4352–4355 CrossRef CAS PubMed; (i) M. A. Greene, I. M. Yonova, F. J. Williams and E. R. Jarvo, Org. Lett., 2012, 14, 4293–4296 CrossRef CAS PubMed.
- M. Tobisu, K. Nakamura and N. Chatani, J. Am. Chem. Soc., 2014, 136, 5587–5590 CrossRef CAS PubMed.
- (a) T. Furukawa, M. Tobisu and N. Chatani, Chem. Commun., 2015, 51, 6508–6511 RSC; (b) T. Furukawa, M. Tobisu and N. Chatani, J. Am. Chem. Soc., 2015, 137, 12211–12214 CrossRef CAS PubMed.
- H.-X. Dai and J.-Q. Yu, J. Am. Chem. Soc., 2012, 134, 134–137 CrossRef CAS PubMed.
- For HBPin, 11B NMR (128.4 MHz, C6D6): δ = 28.4 ppm (d, 1J = 173 Hz), see: H. Braunschweig, F. Guethlein, L. Mailänder and T. B. Marder, Chem. – Eur. J., 2013, 19, 14831–14835 CrossRef CAS PubMed.
- For PhO-BPin, 11B NMR (128.4 MHz, C6D6): δ = 22.9 ppm (s), see: K.-T. Lai, W.-C. Ho, T.-W. Chiou and W.-F. Liaw, Inorg. Chem., 2013, 52, 4151–4153 CrossRef CAS PubMed.
- (a) T. Ishiyama, N. Matsuda, N. Miyaura and A. Suzuki, J. Am. Chem. Soc., 1993, 115, 11018–11019 CrossRef CAS; (b) T. Ishiyama, N. Matsuda, M. Murata, F. Ozawa, A. Suzuki and N. Miyaura, Organometallics, 1996, 15, 713–720 CrossRef CAS.
- A bimetallic/transmetallation mechanism: D. Wang, Y. Izawa and S. S. Stahl, J. Am. Chem. Soc., 2014, 136, 9914–9917 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5qo00395d |
| This journal is © the Partner Organisations 2016 |