
Morphology–activity correlation in hydrogen evolution catalyzed by cobalt sulfides†
Bo
You
,
Nan
Jiang
and
Yujie
Sun
*
Department of Chemistry & Biochemistry, Utah State University, Logan, Utah 84322-0300, USA. E-mail: yujie.sun@usu.edu; Fax: +1-435-797-3390; Tel: +1-435-797-7608
First published on 7th December 2015
Abstract
Transition metal chalcogenides such as cobalt sulfides (CoS) have recently attracted significant interest in electrocatalytic hydrogen evolution reaction (HER). In addition to the constituent elements and hence intrinsic activity, the morphology, porosity, and specific surface area of a nanostructured catalyst would substantially impact its overall electrocatalytic performance. In this paper, we report a facile and rapid two-step microwave-assisted anion-exchange route to prepare nanostructured CoS. By simply controlling the microwave sulfurization time, CoS of various morphologies such as hollow prisms, broken prisms, and nanoparticles could be obtained. Importantly, the correlation between morphology and HER activity of CoS in neutral water was systematically studied through a set of material characterization and electrochemical techniques. It's revealed that the morphology of CoS changed from hollow nanoprisms to 3D nanoparticles when increasing the microwave sulfurization time from 5 to 60 min. The results demonstrated that CoS with 3D nanoparticle morphology, prepared by microwave sulfurization of 30 min, possessed the largest specific surface area and electrochemically active surface area. These nanostructured features resulted in the promoted accessibility of active sites, enhanced mass/charge transport and easier release of hydrogen bubbles, rendering its highest HER activity and excellent stability and showing small overpotentials of 233, 314, and 364 mV to achieve current densities of 10, 50, and 100 mA cm−2, respectively, in neutral water.
 Yujie Sun | Yujie Sun received a B.S. degree in Chemistry from Fudan University in 2005 and conducted undergraduate research under Prof. Heyong He. He then pursued graduate studies in inorganic photochemistry with Prof. Claudia Turro at The Ohio State University and was awarded a Ph.D. degree in 2010. Subsequently, he joined the group of Prof. Christopher J. Chang at the University of California, Berkeley and Lawrence Berkeley National Laboratory, working on electrocatalytic hydrogen evolution. In 2013, Yujie started his independent career as an assistant professor at Utah State University. His group is interested in developing and understanding inexpensive materials and complexes for energy catalysis and biomedical applications. |
Introduction
The increasing energy demands as well as the environmental concerns have stimulated considerable interest in alternative clean and sustainable energy sources.1,2 Molecular hydrogen, as a clean renewable energy carrier, has become one of the promising alternatives to fossil fuels.3–8 In the pursuit of hydrogen production, electro-catalytic water splitting is an appealing solution.3–8 The hydrogen evolution reaction (HER) is effectively promoted by precious metals such as Pt or its alloys.8 However, the high cost and scarcity of these noble metals hinder the large-scale commercialization.8 Therefore, considerable efforts have been aimed at searching for earth-abundant, inexpensive, and robust electrocatalysts with comparable HER performance as Pt replacements, including metal alloys,3 chalcogenides,4 nitrides,5 borides,6 carbides,5b,6,7 and phosphides.8 Nevertheless, most of these electrocatalysts function in strongly acidic media, which renders difficulty in the integration with oxygen evolution reaction (OER) to realize overall water splitting, because most OER electrocatalysts are vulnerable in electrolytes of low pH.9 In this regard, it is highly desirable to develop high performance nonprecious HER electrocatalysts in neutral water, which is the ultimate ideal solvent for water splitting.4g In addition, an extra benefit of HER electrocatalysts functioning at pH 7 is the potential coupling with biological species for biofuel production.10Previously, we reported that an electrodeposited cobalt sulfide film on fluorine-doped tin oxide (FTO) showed excellent electrochemical and photoelectrochemical hydrogen generation from neutral pH water.4g However, the preparation of electrodeposited catalysts is largely limited by the conductive substrates. Recently, our group also demonstrated that hollow cobalt sulfide nanoprisms which combine the unique features of a hollow nanostructure such as low density, high surface–volume ratio, and kinetically favourable open structure exhibited improved HER activity in neutral water.4h In order to be applicable on an industrial scale, further improvement in the efficiency and stability of HER electrocatalysts is still needed. It is well known that besides elemental composition, the morphology, specific surface area, and the porous structure of an electrocatalyst will dramatically govern its overall electrocatalytic performance.11 With these considerations in mind, we synthesized nanostructured CoS with various morphologies including hollow prisms, broken prisms, and 3D nanoparticles by simply controlling the microwave sulfurization time and comprehensively studied the morphology–activity correlation for HER in neutral water. A variety of material characterization and electrochemical techniques were employed. Our results demonstrate that by increasing the microwave sulfurization time from 5 to 60 min, the resulting CoS evolved from hollow nanoprisms, broken prisms, to 3D nanoparticles. Concomitantly, the specific surface area, pore size, and electrochemical double-layer capacitance showed volcano-shape dependence on the microwave sulfurization time. The CoS after 30 min sulfurization exhibited the highest HER catalytic activity in neutral pH water and only needed overpotentials of 233, 314, and 364 mV to afford current densities of 10, 50, and 100 mA cm−2, respectively. Nitrogen sorption measurement and electrochemical double-layer capacitance characterization implied that the superior HER activity originated from the high specific surface area, large pore size and electrochemically active surface area, which resulted in excellent accessibility of catalytically active sites, enhanced mass and charge transport, and easy release of hydrogen bubbles.
Experimental
Chemicals
Polyvinylpyrrolidone (PVP, MW – 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) was purchased from TCI. Thioacetamide (TAA) was purchased from Alfa Aesar. Cobalt acetate tetrahydrate, monobasic dihydrogen phosphate, and dibasic monohydrogen phosphate, and Nafion (5%) were purchased from Sigma-Aldrich. Ethanol was purchased from Decon Laboratories. All chemicals were used as received without any further purification. Deionized water (18 MΩ) via a Barnstead E-Pure system was used in all experiments.
000) was purchased from TCI. Thioacetamide (TAA) was purchased from Alfa Aesar. Cobalt acetate tetrahydrate, monobasic dihydrogen phosphate, and dibasic monohydrogen phosphate, and Nafion (5%) were purchased from Sigma-Aldrich. Ethanol was purchased from Decon Laboratories. All chemicals were used as received without any further purification. Deionized water (18 MΩ) via a Barnstead E-Pure system was used in all experiments.
Microwave-assisted synthesis of cobalt sulfides
Cobalt acetate hydroxide precursors were prepared according to the published method.4h Briefly, 4.5 g PVP and 1.92 g cobalt acetate tetrahydrate were dissolved in 300 mL ethanol at room temperature to afford a clear pink solution. The resulting pink solution was subjected to microwave treatment at 85 °C for 10 min. After centrifugation, the solid cobalt acetate hydroxide precursors were rinsed with ethanol and dried under vacuum at room temperature.For the synthesis of CoS, 80 mg of the above cobalt acetate hydroxide precursors and 112.5 mg thioacetamide were dispersed into 40 mL ethanol. The resulting mixture was subjected to microwave-assisted sulfurization at 120 °C for a certain period of time, 5, 10, 20, 30, or 60 min. After sulfurization, the final products were collected via centrifugation, washed with ethanol, and dried under vacuum at room temperature. Hereafter cobalt sulfides with different sulfurization times were named as CoS-x for short, where x represents the microwave time.
Characterization
Scanning electron microscopy measurements were conducted on a FEI QUANTA FEG 650 (FEI, USA). X-ray diffraction (XRD) patterns were recorded on a Rigaku MiniFex II Desktop X-ray diffractometer. Nitrogen sorption isotherms were collected at 77 K with an autosorb iQ automated gas sorption analyzer (Quantachrome Instruments, USA). Before testing, each sample was degassed under vacuum at 200 °C for 6 h. The Brunauer–Emmett–Teller (BET) method and the Barrett–Joyner–Halenda (BJH) model were utilized to calculate the specific surface area (BET) and pore size distribution, respectively. X-ray photoelectron spectroscopy analyses were performed using a Kratos Axis Ultra instrument (Chestnut Ridge, NY) at the Surface Analysis Laboratory of Nanofab, University of Utah. The samples were affixed on a stainless steel Kratos sample bar, loaded into the instrument's load lock chamber, and evacuated to 5 × 10−8 Torr before transferred into the sample analysis chamber under ultrahigh vacuum conditions (∼10−10 Torr). X-ray photoelectron spectra were collected using the monochromatic Al Kα source (1486.7 eV) at a 300 × 700 μm spot size. Low resolution survey and high resolution region scans at the binding energy of interest were measured for each sample. To minimize charging, each sample was flooded with low-energy electrons and ions from the instrument's built-in charge neutralizer. The samples were also sputter cleaned inside the analysis chamber with 1 keV Ar+ ions for 30 seconds to remove adventitious contaminants and surface oxides. Data were analysed using CASA XPS software, and energy corrections on high resolution scans were conducted by referencing the C 1s peak of adventitious carbon to 284.5 eV.Electrochemical measurements
Each CoS sample ink was prepared by ultrasonically mixing 4 mg of the catalyst powder with a mixture of 1160 μL ethanol, 800 μL H2O, and 40 μL 5% Nafion solution for 20 min to form a homogeneous catalyst ink. Subsequently, 8.5 μL of the catalyst ink was carefully dropcast onto the polished glassy carbon rotating disk electrode with a mass loading of 0.24 mg cm−2. The catalyst ink-loaded electrodes were dried at room temperature prior to electrochemical experiments.Cyclic voltammetry of each catalyst was performed by a computer-controlled Gamry Interface 1000 electrochemical workstation with a traditional three-electrode cell system. A CoS-x loaded glassy carbon electrode (RRDE-3A, d = 3 mm, S = 0.07065 cm2) was used as the working electrode, a Ag/AgCl (sat. KCl) electrode as the reference electrode, and a Pt wire as the counter electrode. All electrochemical experiments were conducted in N2 saturated 1.0 M phosphate buffer of pH 7 at room temperature. The potential range is cyclically scanned from open-circuit voltage to −1.1 V vs. Ag/AgCl at a scan rate of 2 mV s−1 and a rotating speed of 2000 rpm. The reference Ag/AgCl (sat. KCl) electrode in neutral aqueous media was calibrated with ferrocenecarboxylic acid whose Fe3+/2+ couple is 0.284 V vs. SCE.4k iR (current time internal resistance) compensation was applied in polarization experiments to account for the voltage drop between the reference and working electrodes using Gamry Framework™ Data Acquisition Software 6.11. All potentials reported herein were converted from vs. Ag/AgCl to vs. SHE (standard hydrogen electrode) by adding 0.197 V. Electrochemical impedance spectroscopy measurements (EIS) in N2 saturated 1.0 M phosphate buffer of pH 7 were carried out in the same configuration at −0.650 V vs. SHE from 105 to 0.1 Hz with an AC potential amplitude of 30 mV. The durability of the CoS-30 sample in neutral water was assessed by consecutive potential cycling, in which the polarization curves before and after 1000 potential cycles were collected.
Results and discussion
The morphologies of the as-prepared cobalt acetate hydroxide precursors (CAHs) and CoS-x samples were first examined by field emission scanning electron microscopy (SEM). As shown in Fig. 1a, the uniform CAH nanoprisms with a size of roughly 100 × 300 nm were successfully synthesized after the first-step microwave treatment. Its XRD pattern confirmed the formation of a tetragonal cobalt acetate hydroxide phase (Fig. S1,† JCPDS card no. 22-0582). After the second-step microwave-assisted sulfurization for 5 to 60 min, the resulting CoS-x exhibited dramatically different morphologies. For instance, CoS-5 maintained the overall nanoprism morphology of the mother CAHs. The presence of several broken nanoprisms of CoS-5 indicated the formation of hollow nanostructures (Fig. 1b), similar to our previous report.4h Prolonging the sulfurization time to 10 min, the regular nanoprism structure of the resulting CoS-10 samples was severely destroyed and an open half-nanoprism morphology was observed (Fig. 1c). Further increasing the microwave sulfurization time to 20, 30, and 60 min resulted in 3D nanoparticles for CoS-20, CoS-30, and CoS-60, implying the entire destruction of the original prism-like morphology. | ||
| Fig. 1 SEM images of (a) cobalt acetate hydroxide precursors, (b) CoS-5, (c) CoS-10, (d) CoS-20, (e) CoS-30, and (f) CoS-60 samples. | ||
In order to confirm the same crystal structures of each CoS-x sample, their powder XRD patterns were collected. All the XRD patterns of CoS-x samples showed four pronounced reflection peaks (Fig. 2a and S2†) at 2θ of 31.4°, 37.2°, 47.8°, and 54.8°, assignable to the (100), (101), (102), and (110) planes of hexagonal CoS (JCPDS card no. 65-8977). These results clearly demonstrate that varying microwave-assisted sulfurization time did not change the crystal structure of the resulting cobalt sulfides.
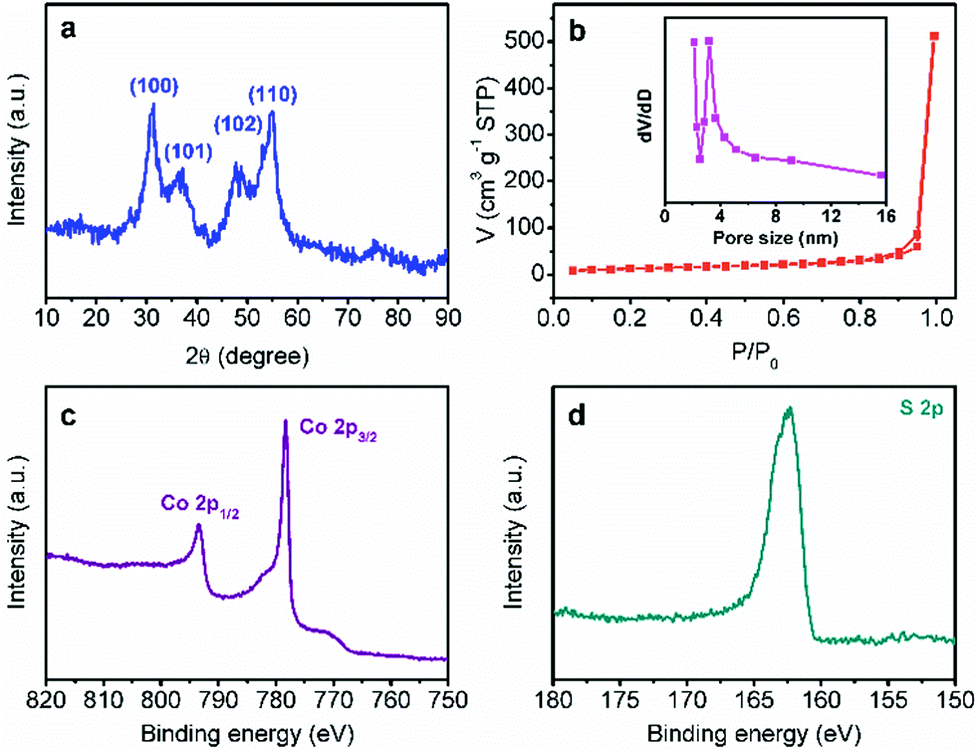 | ||
| Fig. 2 (a) XRD pattern, (b) N2 sorption isotherms, and high-resolution Co 2p (c) and S 2p (d) XPS spectra of CoS-30. The inset in (b) shows the corresponding pore size distribution curve. | ||
The porosity of each CoS-x sample was also measured by the nitrogen adsorption–desorption technique. As plotted in Fig. 2b and S3,† all the CoS-x samples exhibited a type-IV isotherm with a sharp capillary condensation step in the relative pressure (P/P0) range from 0.8 to 1.0 and an obvious H2-type hysteresis loop,12 corresponding to a narrow pore size distribution of large mesopores (>2 nm). Based on the standard BET method, the specific surface area of the CoS-5, CoS-10, CoS-20, CoS-30, and CoS-60 samples were calculated to be 30.1, 32.8, 45.0, 45.3, and 33.9 m2 g−1, respectively (Table 1). The pore size distribution curves derived from the adsorption branches of the isotherms by using the Barrett–Joyner–Halenda method implied that the main pore size (D) for CoS-5, CoS-10, CoS-20, and CoS-30 was in the range of 4.7 to 3.2 nm (Fig. 2b and S3a–c insets†). However, the main pore size of CoS-60 decreased to roughly 2.4 nm (Fig. S3d inset†), implying severe structure collapse due to the long-standing sulfurization (60 min).
| Catalysts | S BET (m2 g−1) | D (nm) | C dl (mF cm−2) | η 10 (mV) | η 50 (mV) | η 100 (mV) | Tafel slop (mV dec−1) |
|---|---|---|---|---|---|---|---|
| CoS-5 | 30.1 | 4.7 | 4.6 | 276 | 376 | 455 | 75 |
| CoS-10 | 32.8 | 4.7 | 5.6 | 249 | 356 | 435 | 77 |
| CoS-20 | 45.0 | 3.8 | 6.9 | 247 | 326 | 385 | 76 |
| CoS-30 | 45.3 | 3.2 | 8.6 | 233 | 314 | 364 | 71 |
| CoS-60 | 33.9 | 2.4 | 5.8 | 249 | 348 | 415 | 80 |
X-ray photoelectron spectroscopic (XPS) analysis was further conducted for CoS-30 to reveal its surface composition and valence states of the constituent elements. As shown in Fig. 2c, d and S4,† the XPS spectrum of CoS-30 exhibited two peaks for the Co 2p spectrum at binding energies of 793.2 and 778.3 eV, which can be attributed to Co 2p1/2 and Co 2p3/2, respectively. The peaks at 778.3 eV in the Co 2p region and 163.2 eV in the S 2p region agreed well with the XPS spectral characteristics of CoS reported in the literature.13
Overall, the same crystal structure while varying morphologies of these CoS samples enable us to investigate electrocatalytic HER activity–morphology correlation via a set of electrochemical techniques.11 Linear sweep voltammetry using a rotating disk electrode at 2000 rpm in N2-saturated pH 7 phosphate buffer was conducted with a standard three-electrode configuration. The slow scan rate of 2 mV s−1 was employed to minimize capacitive current. As shown in Fig. 3a, the polarization curve comparison undoubtedly manifested that the morphology of these CoS samples played a critical role in their electrocatalytic HER performance. For instance, CoS-5 with the hollow nanoprism morphology (Fig. 1b) showed the smallest geometric current density at a higher overpotential (η), affording current densities of 10, 50, and 100 mA cm−2 at η10 = 276, η50 = 376, and η100 = 455 mV, respectively (Table 1). However, the broken nanoprisms of CoS-10 achieved the same current densities at overpotentials of 249, 356, and 435 mV, respectively. Increasing the microwave-assisted sulfurization time to 20 and 30 min, the required overpotentials could be further decreased (Table 1). Specifically, the overpotentials that were required to drive current densities of 10, 50, and 100 mA cm−2 for CoS-30 were as low as 233, 314, and 364 mV, respectively, which were significantly smaller than those of other CoS-x catalysts and reported HER electrocatalysts, including MoS2.7@NPG (η10 > 350 mV),14 Cu/Cu2O (η10 > 300 mV),15 Co-NRCNT (η10 = 540 mV),16 and Fe1−xS (η10 > 780 mV).17 The detailed performance comparison between CoS-30 and representative earth-abundant HER catalysts at pH 7 is listed in Table S1.† Interestingly, prolonging the sulfurization time to 60 min resulted in much poorer electrocatalytic HER activity (Fig. 3a and Table 1) compared to that of CoS-30. Consequently, the trend of HER activity of these CoS-x catalysts is CoS-30 > CoS-20 > CoS-60 > CoS-10 > CoS-5, which is in good agreement with the results of their electrical impedance spectra (EIS). It is well known that the semi-circular diameter in EIS is related to the contact and charge transport impedance.4h A smaller diameter indicates smaller contact and charge transfer resistance. As shown in Fig. 3b, the EIS-derived resistance of CoS-30 is smaller than that of CoS-20, followed by CoS-60 and CoS-10. The resistance of CoS-5 as hollow nanoprisms displayed the largest resistance, consistent with its lowest electrocatalytic performance for HER among these samples.
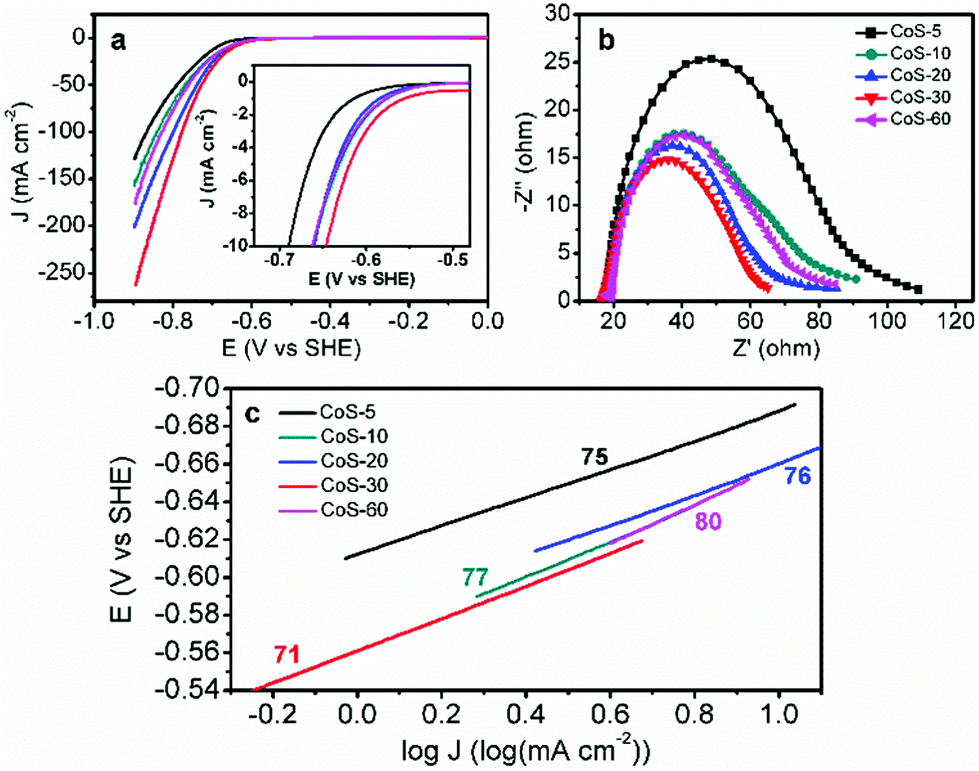 | ||
| Fig. 3 (a) Polarization curves, (b) electrical impedance spectra measured at −650 mV vs. SHE, and (c) Tafel plots of the CoS-x samples. The inset in (a) shows the expanded region around the catalytic onsets of those polarization curves. The mass loadings for all CoS-x were 0.24 mg cm−2. | ||
In order to evaluate the HER kinetics of these CoS catalysts, their Tafel plots were also drawn as shown in Fig. 3c. It is interesting to observe that all the Tafel slopes of the five CoS-x catalysts are in the range of 71–80 mV dec−1 (Fig. 3c). These values compare quite favourably with those of other recently reported high-performance earth-abundant HER electrocatalysts in neutral electrolytes (Table S1†). The similar Tafel slopes for these CoS-x catalysts suggested that the disparity in their HER performance was not due to the different intrinsic activities of these CoS catalysts, instead it is more likely resulting from their drastically different morphologies. For instance, the inner surface of the hollow nanoprisms of CoS-5 is not favourable for the diffusion of electrolytes and the accessibility of catalytically active sites. Increasing the sulfurization time from 10 to 30 min gradually destroyed the prism-like nanostructure and gave rise to an open architecture and large specific surface area, which would facilitate the accessibility of active sites and the release of H2 bubbles.12a–c However, too long sulfurization time would lead to severe structure collapse and aggregations as well as small pore size and low specific surface area (Table 1), which are detrimental to the overall HER performance.
To verify the above speculation, the effective electrochemically active surface area (ECSA) of all the CoS-x catalysts was determined by cyclic voltammetry (CV) in a non-Faradaic potential region from −0.15 to −0.25 V vs. SHE. It is widely accepted that the ECSA of electrocatalysts of similar compositions is proportional to the electrochemical double-layer capacitance (Cdl), which can be derived from the slope of a linear plot of current density vs. scan rate.18Fig. 4a exhibits the CV curves of CoS-30 at varying scan rates from 10 to 60 mV s−1. The typical nearly rectangular shape of all the CV curves implied its excellent conductivity and superior ion transport properties.12d All the five CoS-x samples showed good linearity of current density vs. scan rate (Fig. 4b). The Cdl of each CoS sample was calculated to be 4.6, 5.6, 6.9, 8.6, and 5.8 mF cm−2 for CoS-5, CoS-10, CoS-20, CoS-30, and CoS-60 sample, respectively. Therefore, the trend of Cdl matches well that of the HER performance of these five CoS samples, further corroborating the vital role of morphology in HER electrocatalysis (Fig. 4c).
 | ||
| Fig. 4 (a) Cyclic voltammetry curves of CoS-30 catalyst at different scan rates ranging from 10 to 60 mV s−1. (b) Scan rate dependence of the current densities of CoS-30 at −0.20 V vs. SHE. (c) Comparison of the electrochemically active surface area (ECSA) and overpotentials at 50 mA cm−2 for CoS-x samples. | ||
Another important criterion of HER electrocatalysts for practical application is long-term stability. In addition to the superior HER activity of CoS-30 at pH 7, it also possessed robust durability. As shown in Fig. 5, the nearly overlap of the two polarization curves collected prior to and post 1000 continuous potential cycles unambiguously proved its strong robustness for electrocatalytic HER in neutral electrolytes.
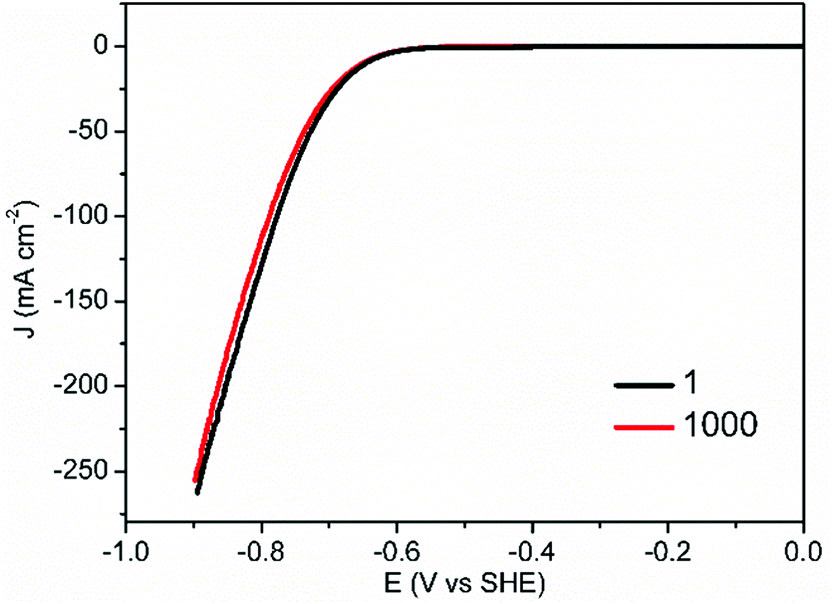 | ||
| Fig. 5 Stability tests of CoS-30 through consecutive potential cycling in 1.0 M KPi at pH 7, in which the polarization curves before and after 1000 potential cycles are displayed (scan rate: 2 mV s−1). | ||
Conclusions
In summary, we have reported a facile and rapid two-step microwave-assisted synthesis of nanostructured CoS with different morphologies (hollow nanoprism, broken nanoprism, and nanoparticle) by simply controlling the sulfurization time. Importantly, the correlation between morphology and HER activity in neutral water was systematically studied through a suite of physical characterization and electrochemical techniques. Our results suggested that the morphology of cobalt sulfide changed from hollow nanoprism to 3D nanoparticle when increasing the microwave-assisted sulfurization time from 5 to 60 min. The CoS sample with 3D nanoparticle morphology, prepared by sulfurization of 30 min, exhibited the largest electrochemically active surface area. Its tailored nanoarchitecture resulted in high accessibility of active sites, enhanced mass/charge transport capability, and facile release of hydrogen bubbles, rendering its superior HER activity (showing a Tafel slope of 71 mV dec−1 and requiring overpotentials of 233, 314, and 364 mV to afford current densities of 10, 50, and 100 mA cm−2, respectively) and excellent stability in neutral media.Acknowledgements
This work was supported by Utah State University (USU) and Ralph E. Powe Junior Faculty Enhancement Award (ORAU). N. J. acknowledges the Governor's Energy Leadership Scholars grant program of Utah Energy Research Triangle and Y. S. thanks the Microscopy Core Facility at USU.Notes and references
- (a) S. Chu and A. Majumdar, Nature, 2012, 484, 294 CrossRef PubMed; (b) T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474 CrossRef CAS PubMed; (c) S. Cobo, J. Heidkamp, P. A. Jacques, J. Fize, V. Fourmond, L. Guetaz, B. Jousselme, V. Ivanova, H. Dau, S. Palacin, M. Fontecave and V. Artero, Nat. Mater., 2012, 11, 802 CrossRef CAS PubMed.
- (a) M. G. Walter, E. L. Warren, J. R. Mckone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446 CrossRef CAS PubMed; (b) C. C. L. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347 CrossRef CAS PubMed; (c) F. Song and X. Hu, Nat. Commun., 2014, 5, 4477 CAS.
- (a) Q. Li, G. S. Hutchings, W. Yu, Y. Zhou, R. V. Forest, R. Tao, J. Rosen, B. T. Yonemoto, Z. Cao, H. Zheng, J. Q. Xiao, F. Jiao and J. G. Chen, Nat. Commun., 2015, 6, 6567 CrossRef PubMed; (b) J. R. McKone, B. F. Sadtler, C. A. Werlang, N. S. Lewis and H. B. Gray, ACS Catal., 2013, 3, 166 CrossRef CAS; (c) X. Wang, R. Su, H. Aslan, J. Kibsgaard, S. Wendt, L. Meng, M. Dong, Y. Huang and F. Besenbacher, Nano Energy, 2015, 12, 9 CrossRef CAS.
- (a) T. F. Jaramillo, K. P. Jorgensen, J. Bonde, J. H. Nielsen, S. Horch and I. I. Chorkendorff, Science, 2007, 317, 100 CrossRef CAS PubMed; (b) J. Kibsgaard, Z. B. Chen, B. N. Reinecke and T. F. Jaramillo, Nat. Mater., 2012, 11, 963 CrossRef CAS PubMed; (c) Y. Li, H. Wang, L. Xie, Y. Liang, G. Hong and H. Dai, J. Am. Chem. Soc., 2011, 133, 7296 CrossRef CAS PubMed; (d) M. A. Lukowski, A. S. Daniel, F. Meng, A. Forticaux, L. Li and S. Jin, J. Am. Chem. Soc., 2013, 135, 10274 CrossRef CAS PubMed; (e) C. G. Morales-Guio and X. Hu, Acc. Chem. Res., 2014, 47, 2671 CrossRef CAS PubMed; (f) B. Lassalle-Kaiser, D. Merki, H. Vrubel, S. Gul, V. K. Yachandra, X. Hu and J. Yano, J. Am. Chem. Soc., 2015, 137, 314 CrossRef CAS PubMed; (g) Y. Sun, C. Liu, D. C. Grauer, J. Yano, J. R. Long, P. Yang and C. J. Chang, J. Am. Chem. Soc., 2013, 135, 17699 CrossRef CAS PubMed; (h) B. You, N. Jiang, M. Sheng and Y. Sun, Chem. Commun., 2015, 51, 4252 RSC; (i) D. Wang, M. Gong, H. Chou, C. Pan, H. Chen, Y. Wu, M. Lin, M. Guan, J. Yang, C. Chen, Y. Wang, B. Hwang, C. Chen and H. Dai, J. Am. Chem. Soc., 2015, 137, 1587 CrossRef CAS PubMed; (j) X. Yu, L. Yu, H. Wu and X. W. Lou, Angew. Chem., Int. Ed., 2015, 54, 5331 CrossRef CAS PubMed; (k) N. Jiang, L. Bogoev, M. Popova, S. Gul, J. Yano and Y. Sun, J. Mater. Chem. A, 2014, 2, 19407 RSC; (l) N. Jiang, Q. Tang, M. Sheng, B. You, D. E. Jiang and Y. Sun, Catal. Sci. Technol., 2016 10.1039/C5CY01111F; (m) D. Merki, H. Vrubel, L. Rovelli, S. Fierro and X. Hu, Chem. Sci., 2012, 3, 2515 RSC; (n) H. Vrubel, T. Moehl, M. Grätzel and X. Hu, Chem. Commun., 2013, 49, 8985 RSC.
- (a) B. Cao, G. M. Veith, J. C. Neuefeind, R. R. Adzic and P. G. Khalifah, J. Am. Chem. Soc., 2013, 135, 19186 CrossRef CAS PubMed; (b) W. F. Chen, J. T. Muckerman and E. Fujita, Chem. Commun., 2013, 49, 8896 RSC.
- H. Vrubel and X. Hu, Angew. Chem., Int. Ed., 2012, 51, 12703 CrossRef CAS PubMed.
- (a) H. B. Wu, B. Y. Xia, L. Yu, X. Y. Yu and X. W. Lou, Nat. Commun., 2015, 6, 6512 CrossRef CAS PubMed; (b) R. Michalsky, Y. J. Zhang and A. A. Peterson, ACS Catal., 2014, 4, 1274 CrossRef CAS; (c) Y. Zhao, K. Kamiya, K. Hashimoto and S. Nakanishi, J. Am. Chem. Soc., 2015, 137, 110 CrossRef CAS PubMed; (d) W. Cui, N. Cheng, Q. Liu, C. Ge, A. M. Asiri and X. Sun, ACS Catal., 2014, 4, 2658 CrossRef CAS; (e) J. Zhu, K. Sakaushi, G. Clavel, M. Shalom, M. Antonietti and T. P. Fellinger, J. Am. Chem. Soc., 2015, 137, 5480 CrossRef CAS PubMed; (f) X. Fan, H. Zhou and X. Guo, ACS Nano, 2015, 9, 5125 CrossRef CAS PubMed.
- (a) S. Carenco, D. Portehault, C. Boissière, N. Mézailles and C. Sanchez, Chem. Rev., 2013, 113, 7981 CrossRef CAS PubMed; (b) C. G. Morales-Guio, L. A. Stern and X. Hu, Chem. Soc. Rev., 2014, 43, 6555 RSC; (c) Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2015, 44, 2060 RSC; (d) B. You, N. Jiang, M. Sheng, S. Gul, J. Yano and Y. Sun, Chem. Mater., 2015, 27, 7636 CrossRef; (e) N. Jiang, B. You, M. Sheng and Y. Sun, ChemCatChem, 2016 DOI:10.1002/cctc.201501150.
- (a) N. Jiang, B. You, M. Sheng and Y. Sun, Angew. Chem., Int. Ed., 2015, 54, 6251 CrossRef CAS PubMed; (b) Y. Leng, G. Chen, A. J. Mendoza, T. B. Tighe, M. A. Hickner and C. Y. Wang, J. Am. Chem. Soc., 2012, 134, 9054 CrossRef CAS PubMed.
- (a) E. M. Nichols, J. J. Gallagher, C. Liu, Y. Su, J. Resasco, Y. Yu, Y. Sun, P. Yang, M. C. Y. Chang and C. J. Chang, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 11461 CrossRef CAS PubMed; (b) C. Liu, J. J. Gallagher, K. K. Sakimoto, E. M. Nichols, C. J. Chang, M. C. Y. Chang and P. Yang, Nano Lett., 2015, 15, 3634 CrossRef CAS PubMed.
- (a) M. S. Faber, R. Dziedzic, M. A. Lukowski, N. S. Kaiser, Q. Ding and S. Jing, J. Am. Chem. Soc., 2014, 136, 10053 CrossRef CAS PubMed; (b) L. A. Stern, L. Feng and X. Hu, Energy Environ. Sci., 2015, 8, 2347 RSC.
- (a) B. You, P. Yin, J. Zhang, D. He, G. Chen, F. Kang, H. Wang, Z. Deng and Y. Li, Sci. Rep., 2015, 5, 11739 CrossRef PubMed; (b) J. Zhang, G. Chen, Q. Zhang, F. Kang and B. You, ACS Appl. Mater. Interfaces, 2015, 7, 12760 CrossRef CAS PubMed; (c) B. You, P. Yin and L. An, Small, 2014, 10, 4352 CAS; (d) B. You, L. Wang, N. Li and C. Zheng, ChemElectroChem, 2014, 1, 772 CrossRef CAS; (e) B. You, J. Jiang and S. Fan, ACS Appl. Mater. Interfaces, 2014, 6, 15302 CAS; (f) B. You, N. Jiang, M. Sheng, W. S. Drisdell, J. Yano and Y. Sun, ACS Catal., 2015, 5, 7068 CrossRef CAS.
- C. W. Kung, H. W. Chen, C. Y. Lin, K. C. Huang, R. Vittal and K. C. Ho, ACS Nano, 2012, 6, 7016 CrossRef CAS PubMed.
- X. Ge, L. Chen, L. Zhang, Y. Wen, A. Hirata and M. Chen, Adv. Mater., 2014, 26, 3100 CrossRef CAS PubMed.
- J. Zhao, P. D. Tran, Y. Chen, J. S. C. Loo, J. Barber and Z. J. Xu, ACS Catal., 2015, 5, 4115 CrossRef CAS.
- X. Zou, X. Huang, A. Goswami, R. Silva, B. R. Sathe, E. Mikmeková and T. Asefa, Angew. Chem., Int. Ed., 2014, 53, 4372 CrossRef CAS PubMed.
- C. D. Giovanni, W. A. Wang, S. Nowak, J. M. Grenèche, H. Lecoq, L. Mouton, M. Giraud and C. Tard, ACS Catal., 2014, 4, 681 CrossRef.
- E. Gileadi, Physical Electrochemistry, Wiley-VCH, Weinheim, 2011 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional characterization data and electrocatalytic results. See DOI: 10.1039/c5qi00196j |
| This journal is © the Partner Organisations 2016 |