
A terahertz absorption spectroscopy study of structural changes in D-penicillaminato CuI8CuII6 clusters induced by water desorption†
Hal
Suzuki
*a,
Chiko
Otani
a,
Nobuto
Yoshinari
b and
Takumi
Konno
b
aRIKEN, 519-1399 Aramaki-Aoba, Aoba-ku, Sendai, Miyagi 980-0845, Japan. E-mail: h_suzuki@riken.jp; Fax: +81-22-228-2128; Tel: +81-22-228-2124
bOsaka University, 1-1 Machikaneyama-cho, Toyonaka, Osaka 560-0043, Japan
First published on 7th December 2015
Abstract
Structural changes induced by water desorption from D-penicillaminato (D-pen) CuI8CuII6 clusters aggregated into three polymorphic crystal structures ((1) 1D helical, (2) 2D hexagonal sheet, and (3) discrete cubic) were investigated by terahertz (THz) absorption spectroscopy and X-ray diffractometry (XRD), which provided information on the skeletal and crystal structures of the clusters, respectively. The water molecules incorporated into the crystals were removed by evacuation and heating to 100 °C. In the case of crystal 1, the helical structure collapsed upon water evacuation, whereas the skeletal structure of the D-pen CuI8CuII6 clusters did not change. However, the skeletal structures were distorted by the heating process. For crystals 2 and 3, water evacuation did not affect either the skeletal or the sheet and cubic crystal structures of the clusters, which, however, collapsed upon heating. The mechanisms driving these structural changes were discussed in relation to the type and amount of water molecules removed in each process.
Introduction
Supramolecular chemistry of metal cluster complexes plays an important role in nanoscale science and technology.1 Many interesting physical and chemical properties of these clusters have been reported, such as molecular nanomagnetism,2 catalytic activity,3 or the capability of encapsulating small molecules,4 which could be exploited for developing new functional materials. In order to understand these properties in detail, structural information on coordination environments and cluster aggregation is very important. Recently, several multinuclear metal cluster complexes were reported to show extraordinary aggregation,5,6 which is expected to open up a new field of coordination chemistry.The structures of metal cluster complexes and their aggregates have previously been investigated by X-ray diffractometry (XRD),7 mid-infrared (MIR) spectroscopy,8 and nuclear magnetic resonance (NMR) spectroscopy.9 XRD is primarily used when the clusters are aggregated in a crystalline form. However, XRD is not particularly useful when the clusters are randomly arranged, and MIR and NMR spectroscopy are often used in these cases. The structural information provided by these spectroscopic techniques is limited to the local structure: in particular, MIR provides information on the functional groups present, whereas NMR is used to characterize the local environments of the nuclei. Different methods are thus needed for a deeper understanding of cluster structures. In this work, we employed terahertz (THz) spectroscopy for this purpose.
THz (also known as far-infrared (FIR)) spectroscopy is a low-frequency vibrational technique (0.1–10 THz; 3–300 cm−1). With the dramatic developments in techniques for the generation and detection of THz waves during the last two decades,10 THz spectroscopy has recently recaptured the spotlight not only as an analytical tool for materials science,11–13 but also as a sensing and imaging tool for industrial purposes.14,15 The frequency of a vibrational mode is known to decrease with decreasing force constant and with the increasing mass of the vibrator. As the THz frequency range is 10 to 100 times lower than the MIR one (300–3000 cm−1), THz spectroscopy can detect very “soft” vibrational modes such as intermolecular vibrations or collective vibrations such as lattice vibrations in crystals. As these vibrational modes are associated with molecular arrangements, THz spectroscopy is expected to provide information on the geometric structure of molecular aggregates. In fact, the THz spectra of low-molecular-weight molecules have been successfully interpreted in relation to their crystal structures.16–18
In this study, we used THz spectroscopy to investigate structural changes in D-penicillaminato (D-pen) CuI8CuII6 clusters, and in their polymorphic crystals, induced by water desorption. We were interested in the type of structural information we could obtain from the THz spectra of multinuclear metal cluster complexes and their aggregates. Since these clusters are larger in size and more complex in structure, the information provided by THz spectroscopy was expected to be different from that of low-molecular-weight molecules. The recently synthesized D-pen CuI8CuII6 cluster is a multinuclear copper complex.19 The cluster unit, [CuI8CuII6(D-pen)12Cl]5−, is about one nanometer in size, and consists of five atomic layers20 (Fig. 1a): (1) a central Cl atom, (2) eight cubic CuI atoms, (3) twelve icosahedral thiolate S atoms belonging to the D-pen ligands, (4) six octahedral CuII atoms, and (5) the remaining atoms of the D-pen ligands at the outermost edges. The Cu atoms are coordinated to the S and N atoms of the D-pen ligands. The key feature of the D-pen CuI8CuII6 cluster is that, by slightly changing the pH in the buffer solution used in the redox reaction, three different polymorphic crystal structures can be formed: (1) a 1D helical structure, (2) a 2D hexagonal sheet structure, and (3) a discrete cubic structure (see Fig. 1b).
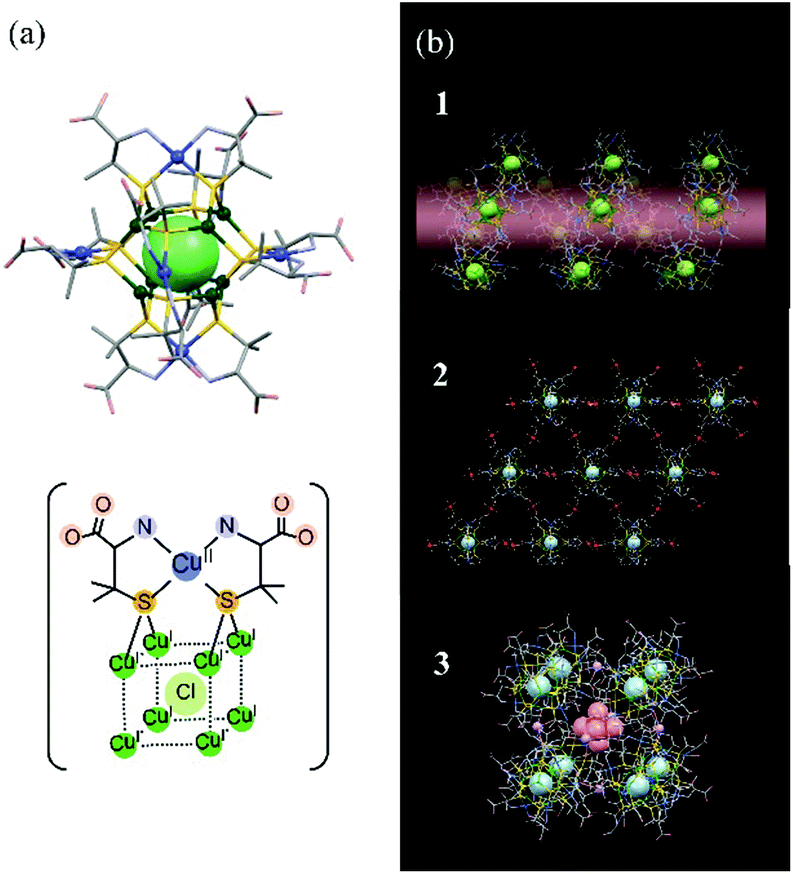 | ||
| Fig. 1 Models of (a) [CuI8CuII6(D-pen)12Cl]5− cluster, and (b) the three types of crystal structures (labeled 1, 2, and 3 in the figure) discussed in the text. | ||
Crystal 1 (CoK3[Cu14(D-pen)12Cl]·nH2O) is obtained when K[Co(D-pen)2]·1.5H2O21 is treated with CuICl in a potassium acetate (OAc) buffer solution at pH 5.5 (HOAc/KOAc = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6). [CuI8CuII6(D-pen)12Cl]5− anionic clusters are connected to each other through N–H⋯O hydrogen bonds to form a 1D helical structure. The counter cations (Co2+ and K+) are also aggregated into a 1D helical structure, and the two helices are connected to each other in parallel to each other, forming a hexagonal columnar crystal structure (see Fig. S1a†). Crystal 2 (Co3[Cu14(D-pen)12Cl]Cl·nH2O) is prepared in a buffer solution at pH 5.0 (HOAc/KOAc = 1:2). Six carboxylate groups belonging to D-pen in each [CuI8CuII6(D-pen)12Cl]5− cluster anion are coordinated to four trans-[Co(H2O)4]2+ cationic moieties to form a 2D hexagonal sheet structure, and the sheets are linked by cis-[Co(H2O)4]2+ moieties through Co–OOC bonds to form an (ABC)n stacking structure (see Fig. S1b†). Crystal 3 (Co2.125K0.75[Cu14(D-pen)12Cl]·nH2O) is prepared in a buffer solution at pH 4.5 (HOAc/KOAc = 3:2). Eight [CuI8CuII6(D-pen)12Cl]5− cluster anions are bridged by six K+ ions to form a discrete cubic arrangement. One [Co(H2O)4]2+ cation is encapsulated in a cube, and 16 [Co(H2O)4]2+ cations are located outside the cube to balance the total charge. The discrete cubes are packed in a face-centered cubic lattice by NH2⋯OOC hydrogen bonds, as well as OH2⋯OOC hydrogen bonds with the [Co(H2O)4]2+ cations (see Fig. S1c†).
:6). [CuI8CuII6(D-pen)12Cl]5− anionic clusters are connected to each other through N–H⋯O hydrogen bonds to form a 1D helical structure. The counter cations (Co2+ and K+) are also aggregated into a 1D helical structure, and the two helices are connected to each other in parallel to each other, forming a hexagonal columnar crystal structure (see Fig. S1a†). Crystal 2 (Co3[Cu14(D-pen)12Cl]Cl·nH2O) is prepared in a buffer solution at pH 5.0 (HOAc/KOAc = 1:2). Six carboxylate groups belonging to D-pen in each [CuI8CuII6(D-pen)12Cl]5− cluster anion are coordinated to four trans-[Co(H2O)4]2+ cationic moieties to form a 2D hexagonal sheet structure, and the sheets are linked by cis-[Co(H2O)4]2+ moieties through Co–OOC bonds to form an (ABC)n stacking structure (see Fig. S1b†). Crystal 3 (Co2.125K0.75[Cu14(D-pen)12Cl]·nH2O) is prepared in a buffer solution at pH 4.5 (HOAc/KOAc = 3:2). Eight [CuI8CuII6(D-pen)12Cl]5− cluster anions are bridged by six K+ ions to form a discrete cubic arrangement. One [Co(H2O)4]2+ cation is encapsulated in a cube, and 16 [Co(H2O)4]2+ cations are located outside the cube to balance the total charge. The discrete cubes are packed in a face-centered cubic lattice by NH2⋯OOC hydrogen bonds, as well as OH2⋯OOC hydrogen bonds with the [Co(H2O)4]2+ cations (see Fig. S1c†).
Each of the three crystal structures contains a large number of voids, which can host numerous water molecules. The hydration numbers determined by elemental analysis were n = 51, 39, and 42 for 1, 2, and 3, respectively.19 Among the hydration water molecules, crystallographic positions were determined for 19, 39, and 35 molecules in 1, 2, and 3, respectively, by means of single crystal XRD measurements. The positions and orientations of the other water molecules are disordered, indicating that these molecules are loosely bound in the voids and can thus be removed by evacuation or heating. As self-assembled crystal structures strongly depend on inter-molecular (inter-cluster) interactions, desorption of the loosely bound water molecules is expected to affect the above structures.
In this study, we investigated the structural changes in D-pen CuI8CuII6 clusters and their aggregates by THz spectroscopy and XRD with dual aims. The first aim was to verify the usefulness of THz spectroscopy as a structural tool for multinuclear metal cluster complexes. The second one was to reveal the interrelation between the crystal framework and the water molecules incorporated into the crystals of the multinuclear metal complexes. The D-pen CuI8CuII6 clusters are ideal investigational targets for achieving these goals, since the crystal structures can be changed by slight variations of pH and probably by water desorption.
Experimental
Crystals 1, 2, and 3 were prepared according to methods described in the literature.19 The crystals were dried first by evacuation and then by heating to 100 °C. High-quality powder XRD patterns were recorded in transmission mode using a diffractometer equipped with a blue imaging plate detector at the SPring-8 BL02B2 beam line in Japan (synchrotron radiation λ = 1.0 Å; 2θ range = 0–78°; step width = 0.01°; data collection time = 5 min; Debye–Scherrer camera22). THz spectra (1–11 THz) were recorded by Fourier-transform far-infrared (FT-FIR) spectroscopy with a frequency resolution of 0.06 THz. The measurements were made using a JASCO FARIS instrument equipped with a high-pressure mercury lamp, a Mylar beam splitter, and a Si bolometer. Since D-pen CuI8CuII6 clusters show high absorbance in the THz region, the crystals were diluted with dried polyethylene (PE) powder (14.2, 12.6, and 14.0 wt% for 1, 2, and 3, respectively) in order to avoid signal saturation.Results and discussion
Fig. 2 shows the results of the XRD measurements together with the simulated patterns calculated from the single-crystal XRD data using the Mercury 3.0 program.23 The XRD patterns of the evacuated samples of crystals 2 and 3 are almost identical to the simulated patterns of the single crystals, indicating that the crystal structures do not change upon evacuation. For crystal 1, on the other hand, the XRD pattern after evacuation is considerably different from the simulated one; in particular, the XRD pattern shows a very broad peak centered at around 2θ = 5°, showing that upon evacuation the crystal structure of 1 collapses into an amorphous-like disordered structure. The XRD patterns of crystals 2 and 3 after heating to 100 °C are also different from the simulated ones; in particular, the post-heating peaks are significantly broadened, showing that the crystal structures break down upon heating. The small and broadened peaks between 4° and 6° indicate the short-range order of the cluster arrangements. By simply applying Bragg's law, the angle between 4° and 6° corresponds to a d-spacing of between 10 and 14 Å. These values are comparable with the diameter of the Cu14S12 core in the [CuI8CuII6(D-pen)12Cl]5− cluster (10 Å), as well as the distance between the neighboring clusters (14 Å). Thus, the clusters are equally spaced even after heating. The XRD pattern of crystal 1 at 100 °C is similar to that before heating, indicating that heating does not further change the amorphous-like structure.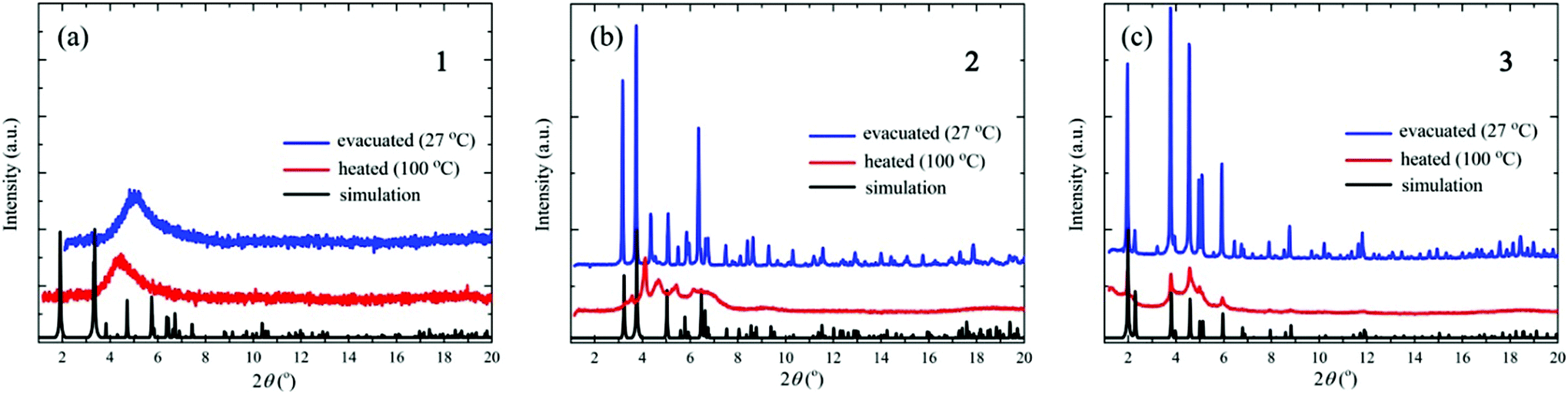 | ||
| Fig. 2 XRD patterns of crystals (a) 1, (b) 2, and (c) 3 after evacuation (at 27 °C) and heating (to 100 °C). The simulated patterns calculated from the single-crystal XRD data are also shown. | ||
The THz spectroscopy results are in marked contrast to those obtained from the XRD measurements. Fig. 3 shows the THz spectra obtained after evacuation and after heating to 100 °C. The spectral signals from pure PE, also shown in Fig. 3, are of very low intensity compared with the signals from the crystals. The spectra of the three crystals after evacuation are rather similar to each other, with absorption peaks centered at around 2.5, 3.0, 5.5, 6.0, 7.8, 9.0, and 10.5 THz. The similarities between the THz spectra are remarkable, considering that the XRD patterns of the three crystals are completely different from each other. This result indicates that the THz spectra of the D-pen CuI8CuII6 clusters are not correlated with their crystal structures, at variance with what is observed for low-molecular-weight molecular crystals. This difference may occur because the frequencies of the lattice vibrations for the three crystals are shifted out of the THz range due to the extremely large weight of the vibrator, i.e., the [CuI8CuII6(D-pen)12Cl]5− cluster, whose resonance peaks may thus fall at a much lower frequency range. The THz spectra observed in Fig. 3 are, therefore, related to the internal structure of the [CuI8CuII6(D-pen)12Cl]5− clusters, which is the same for the three crystals. In order to eliminate the possible contribution from the ancillary groups, we also measured the THz spectra of [Co(H2O)6]Cl2, D-penicillamine, and K[Co(D-pen)2]·1.5H2O, which showed significantly different spectra from those of the three crystals (Fig. S2†). The lack of a sharp peak for [Co(H2O)6]Cl2 indicates that the contribution of the [Co(H2O)6]2+ ion in Fig. 3 is negligible. D-Penicillamine and K[Co(D-pen)2]·1.5H2O show many peaks but the spectral shapes are different from each other. Both compounds contain D-penicillamine, so that the peaks observed are not from the intramolecular vibrations of D-penicillamine. The peaks observed may be of the lattice mode of D-penicillamine and K[Co(D-pen)2]·1.5H2O crystals. These results support the interpretation that the THz peaks observed for the three crystals in Fig. 3 are characteristic of the [CuI8CuII6(D-pen)12Cl]5− clusters. The observed peaks in Fig. 3 may be related to the coupled vibrations of Cu–S and Cu–Cu stretching modes, and of other vibrational modes within the D-pen ligands. The coordination structure of the [CuI8CuII6(D-pen)12Cl]5− cluster entails that these vibrational modes are very likely to be coupled with each other, and their vibrational frequencies closely depend on the extent of the coupling, i.e., on the skeletal structure of the cluster. Therefore, the vibrational modes observed can be interpreted as “skeletal vibrations of the cluster complex”.
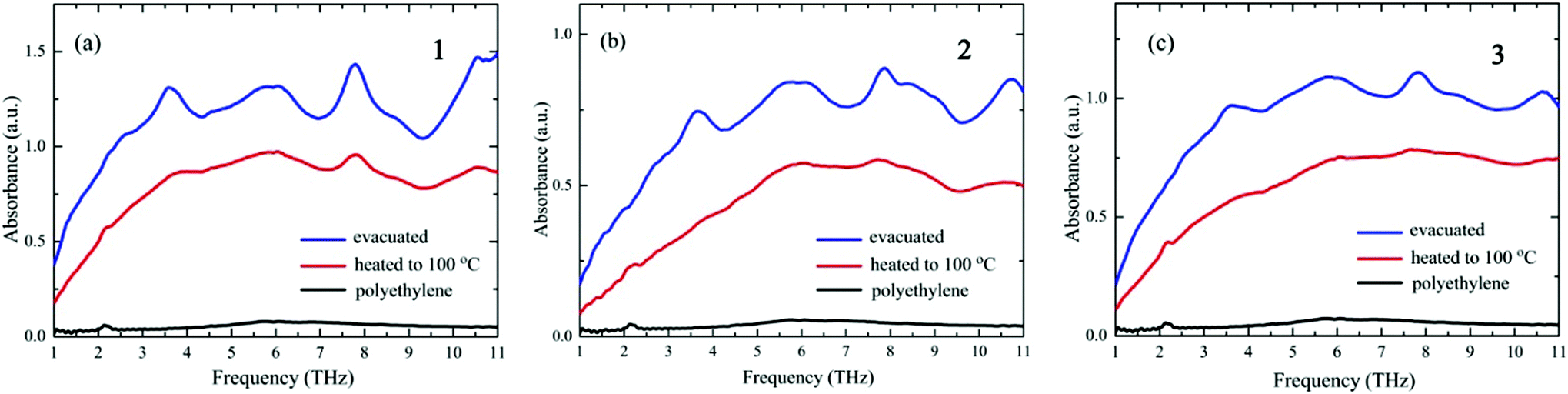 | ||
| Fig. 3 THz spectra of (a) 1, (b) 2, and (c) 3 after evacuation and after heating to 100 °C. The spectrum of the diluent polyethylene powder is also shown. | ||
The spectra of all three crystals are altered by the heating process; the absorption intensities are reduced over the whole frequency range and the peaks are significantly broadened. The decrease in spectral intensities may be due to the reduction in the number of water molecules upon evaporation. Liquid water is known to exhibit a broad peak ranging from 2 to 9 THz.24,25 The broadening of the spectral peaks by heating indicates that the skeletal structures of D-pen CuI8CuII6 clusters are distorted in a random way due to water desorption. We also confirmed that the MIR spectra (absorption spectroscopy: JASCO FT/IR-4100; range: 500–4000 cm−1) and UV-vis spectra (diffuse reflection spectroscopy: JASCO V-670; range: 300–800 nm) do not change upon heating to 100 °C. The MIR spectroscopy result indicates that the heating process does not affect the structure of the functional groups, such as carboxyl groups, whereas the UV-vis results indicate that the CuI–CuII and S–CuII coordination bonds are not broken upon heating.
The amount of water desorbed with increasing temperature was also monitored by thermogravimetry (TG) (Shimadzu DTG-60 DTA/TG thermogravimetric analyzer; dT·dt−1 = 5 °C min−1; N2 gas flow (50 mL min−1)). Two stages of significant weight loss were observed; the first gradually took place between 30 and 100 °C and was due to water desorption, whereas the second occurred at around 150 °C and may be due to chemical decomposition. The percent weight losses up to 100 °C were 16, 17, and 21 (±1) wt% for 1, 2, and 3, respectively. If all these losses are attributed to water desorption, these amounts correspond to hydration numbers n = 31, 34, and 42 (±2). For crystals 2 and 3, these values are comparable to those determined by elemental analysis (n = 39, 42), indicating that most water molecules were removed by heating. For crystal 1, on the other hand, the value estimated from TG (n = 31) is lower than that obtained from elemental analysis (n = 51), which may reflect the fact that some water molecules had already been removed through the evacuation process before heating.
Combining the THz spectroscopy results with those of the XRD and TG measurements, the structural changes of the D-pen CuI8CuII6 clusters by water desorption can be understood as follows. For crystal 1, a significant amount of loosely bound water molecules is removed by evacuation, leading to the collapse of the 1D helical crystal structure, as indicated by the XRD pattern showing only one broad peak. The remaining tightly bound water molecules maintain the skeletal structure of the D-pen CuI8CuII6 clusters as unchanged, which results in a THz spectrum with sharp peaks, as in crystalline phases. When crystal 1 is heated to 100 °C, the tightly bound water molecules are removed, and the skeletal structure of the D-pen CuI8CuII6 clusters becomes distorted. Each cluster undergoes a different distortion, so that the peaks in the spectrum are significantly broadened. For crystals 2 and 3, on the other hand, most water molecules are tightly bound and remain even after evacuation, so that the crystal structures (XRD patterns) as well as the skeletal structures of the clusters (THz spectra) never change. When crystals 2 and 3 are heated to 100 °C, the tightly bound water molecules are removed, and both the crystal and skeletal structures collapse at the same time. Therefore, the observed different behaviors of crystal 1 and crystals 2 and 3 arise from their different binding strengths for water molecules.
Conclusions
In conclusion, we have shown that THz spectroscopy can be used to obtain information on the skeletal structure of multinuclear metal cluster complexes, as exemplified by D-pen CuI8CuII6 clusters. When all clusters adopt the same skeletal structure, the vibrational modes over the coordinated metal network have a narrow distribution of frequencies, and sharp peaks are observed in the THz spectrum. When the skeletal structures are distorted in a random way, the skeletal vibrational modes show a wide frequency distribution, and the corresponding peaks become significantly broadened. Considering that XRD provides information on the crystal structure, i.e., on the long-range periodic arrangements of atoms, whereas MIR spectroscopy probes the local molecular structure, the structural information provided by THz spectroscopy is complementary to that obtained from XRD and MIR methods.Several recent investigations on metal clusters26 and metal-doped silicon clusters27 have also shown that THz spectroscopy represents a useful tool for the geometrical analysis of clusters. Our results extend the range of the THz spectroscopy targets, from covalently or metallically bonded clusters to coordinatively bonded clusters. Because coordination bonds are weaker than covalent or metallic bonds, the cluster structure can be changed with relative ease by changing the external conditions. THz spectroscopy is useful for monitoring such structural changes, as was demonstrated in this study for D-pen CuI8CuII6 clusters, whose skeletal structures were modified by removing tightly bound water molecules through heating to 100 °C.
Acknowledgements
This work was supported by CREST, JST, and the Japan Society for the Promotion of Science (JSPS) Grant-in-Aid for Scientific Research (KAKENHI), grant numbers 15K05404 and 25870387. The synchrotron radiation experiments were performed at the BL02B2 beamline of SPring-8 (Japan) with the approval of the Japan Synchrotron Radiation Research Institute (JASRI) (proposal no. 2015A1506 and 2015A1520).Notes and references
- C. Femoni, M. C. Iapalucci, F. Kaswalder, G. Longoni and S. Zacchini, Coord. Chem. Rev., 2006, 250, 1580–1604 CrossRef CAS.
- D. Gatteschi, R. Sessoli and J. Villain, Molecular Nanomagnets, Oxford University Press, Oxford, 2011 Search PubMed.
- C. J. Brown, F. D. Toste, R. G. Bergman and K. N. Raymond, Chem. Rev., 2015, 115, 3012–3035 CrossRef CAS PubMed.
- F. Hof, S. L. Craig, C. Nuckolls and J. J. Rebek, Angew. Chem., Int. Ed., 2002, 41, 1488–1508 CrossRef CAS.
- R. Lee, A. Igashira-Kamiyama, M. Okumura and T. Konno, Bull. Chem. Soc. Jpn., 2013, 86, 908–920 CrossRef CAS.
- N. Yoshinari, U. Yamashita and T. Konno, CrystEngComm, 2013, 15, 10016–10019 RSC.
- The Chemistry of Metal Cluster Complexes, ed. D. F. Shriver, H. D. Kaesz and R. D. Adams, Wiley-VCH, New York, 1990 Search PubMed.
- J. A. Fournier, C. J. Johnson, C. T. Wolke, G. H. Weddle, A. B. Wolk and M. A. Johnson, Science, 2014, 344, 1009–1012 CrossRef CAS PubMed.
- C. Bonhomme, C. Gervais and D. Laurencin, Prog. Nucl. Magn. Reson. Spectrosc., 2014, 77, 1–48 CrossRef CAS PubMed.
- M. Tonouchi, Nat. Photonics, 2007, 1, 97–105 CrossRef CAS.
- S. Wietzke, C. Jansen, T. Jung, M. Reuter, B. Baudrit, M. Bastian, S. Chatterjee and M. Koch, Opt. Express, 2009, 17, 19006–19014 CrossRef CAS PubMed.
- H. Suzuki, S. Ishii, C. Otani and H. Hoshina, Eur. Polym. J., 2015, 67, 284–291 CrossRef CAS.
- H. Hoshina, Y. Morisawa, H. Sato, H. Minamide, I. Noda, Y. Ozaki and C. Otani, Phys. Chem. Chem. Phys., 2011, 13, 9173–9179 RSC.
- H. Hoshina, Y. Sasaki, A. Hayashi, C. Otani and K. Kawase, Appl. Spectrosc., 2009, 63, 81–86 CrossRef CAS PubMed.
- K. Kawase, Y. Ogawa, Y. Watanabe and H. Inoue, Opt. Express, 2003, 11, 2549–2554 CrossRef CAS PubMed.
- H. Suzuki, H. Hoshina and C. Otani, Cryst. Growth Des., 2014, 14, 4087–4093 CAS.
- P. U. Jepsen and S. J. Clark, Chem. Phys. Lett., 2007, 442, 275–280 CrossRef CAS.
- O. Kambara, C. S. Ponseca, K. Tominaga, J. Nishizawa, T. Sasaki, H. W. Wang and M. Hayashi, Bull. Chem. Soc. Jpn., 2013, 86, 714–720 CrossRef CAS.
- N. Yoshinari, K. Tatsumi, A. Igashira-Kamiyama and T. Konno, Chem. – Eur. J., 2010, 16, 14252–14255 CrossRef CAS PubMed.
- P. J. M. W. L. Birker and H. C. Freeman, J. Am. Chem. Soc., 1977, 99, 6890–6899 CrossRef CAS PubMed.
- K. Okamoto, K. Wakayama, H. Einaga, S. Yamada and J. Hidaka, Bull. Chem. Soc. Jpn., 1983, 56, 165–170 CrossRef CAS.
- E. Nishibori, M. Takata, K. Kato, M. Sakata, Y. Kubota, S. Aoyagi, Y. Kuroiwa, M. Yamakata and N. Ikeda, J. Phys. Chem. Solids, 2001, 62, 2095–2098 CrossRef CAS.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van de Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS.
- M. N. Afsar and J. B. Hasted, J. Opt. Soc. Am., 1977, 67, 902–904 CrossRef CAS.
- H. R. Zelsmann, J. Mol. Struct., 1995, 350, 95–114 CrossRef CAS.
- A. Fielicke, A. Kirilyuk, C. Ratsch, J. Behler, M. Scheffler, G. von Helden and G. Meijer, Phys. Rev. Lett., 2004, 93, 023401 CrossRef PubMed.
- Y. Li, J. T. Lyon, A. P. Woodham, P. Lievens, A. Fielicke and E. Janssens, J. Phys. Chem. C, 2015, 119, 10896–10903 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Crystal structures of 1, 2, and 3; THz spectra of [Co(H2O)6]Cl2, D-penicillamine, and K[Co(D-pen)]·1.5H2O. See DOI: 10.1039/c5qi00207a |
| This journal is © the Partner Organisations 2016 |