
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePoly(thioacrylate)s: expanding the monomer toolbox of functional polymers†
Suzan
Aksakal
and
C.
Remzi Becer
 *
*
Polymer Chemistry Laboratory, School of Engineering and Materials Science, Queen Mary University of London, E1 4 NS, UK. E-mail: r.becer@qmul.ac.uk
First published on 25th October 2016
Abstract
Radically polymerizable monomers such as acrylates, acrylamides, and styrenes have been investigated because of their corresponding polymers for many years. However, thioacrylates are a forgotten class of monomers and have not been studied for decades. Herein, we present a simple synthetic approach to prepare various thioacrylate monomers as well as their controlled radical polymerization for the first time. Four different thioacrylate (TA) monomers namely, ethyl thioacrylate, phenyl thioacrylate, n-propyl thioacrylate and isopropyl thioacrylate have been polymerized via RAFT polymerization. A trithiocarbonate chain transfer agent was utilized to control the molar mass and dispersity of thioacrylate polymers. Homopolymers of thioacrylates with different degrees of polymerizations were prepared and their block copolymerizations were studied with ethylacrylate and thioacrylates. All obtained polymers were analyzed in detail using GPC, NMR, TGA, DSC and water contact angle.
Introduction
Sulphur-containing polymers are attractive materials within the polymer chemistry field due to their remarkable features that determine their outstanding properties. The presence of sulphur in polymer structures enhances a wide variety of material properties such as mechanical, electrical, optical, adhesion, heat resistance, and biocompatibility.1–4 Thiols are known as softer nucleophiles in comparison with alcohols and amines and are able to form disulphide bonds, which can be further used as a reactive group.5 Due to their numerous advantages over their nonfunctionalized counterparts, thiol-containing polymers open new avenues for advanced functional materials. Detailed studies on various molecular designs including pendant groups, chain end functionalization, graft and hyperbranched polymers have been reported to fine-tune their physical properties. For instance, much effort has been devoted for the preparation of thiol-containing monomers that consist of either free thiols on the side chain or in heterocyclic systems (e.g. thiomers,6 thiazoline,7 thiazole,8 thiophene9,10 or thiolactones3).Sulphur containing polymers are hence of significant interest to researchers and form the basis of an emerging platform in materials science. Unfortunately, due to their relatively high reactivity, thiol containing monomers are known to be incompatible with most polymerization techniques as they are prone to undergo chain transfer reactions with monomers, active species or even catalysts. Protection and deprotection of thiol groups becomes therefore necessary in order to not induce transfer reactions during polymerization and also to improve their storage, as they are not stable under oxidative conditions. Mostly because of these reasons very few examples have been reported to date. Three main synthetic routes can be listed for the preparation of thiol containing polymers. The first one is by polymerization of monomers with protected thiols as pendant groups.11,12 Polymerization of thiol monomers, which do not have to be protected under specific conditions, can also be carried out, especially with polycondensation. As the second route, linear polymers with secondary thiols as the pendant group have been synthesized by Matsumura et al. by taking the advantage of highly selective biological processes between hydroxyl and mercapto groups using a lipase enzyme.13 Additionally, linear polythiols have been prepared by metal catalysis of polyesters with pendant mercapto groups from the reaction of diols and thiomalic acid in the presence of scandium(III) triflate. Subsequent functionalization by thiol–ene glycosidation or cross-linking with thiocarbonyldiimidazole leads to linear polythiols.14 The final route is based on the sol–gel process that was also successfully used for the reaction of dimethoxysiloxane with free thiols as the pendant group in acidic medium, to synthesize co(polymers) carrying thiol moieties.15 The most widely applied method for the preparation of polythiols is based on the post-polymerization modification, which has been demonstrated for thiolated polymers (thiomers) for mucoadhesive drug delivery, generally carried out via thiol Michael addition.16
The synthesis and free radical polymerization of a couple of thioacrylate monomers were first reported in 1956, when Marvel prepared a range of thioacrylates by the reaction of α,β-dibromopropionyl chloride with thiols and subsequent treatment with sodium iodine for dehalogenation.17 In this way, methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl and tert-butyl thioacrylates with an over-all yield of 25–45% were obtained. These monomers were further analysed via IR. Free radical polymerization was carried out in bulk using benzoyl peroxide as the initiator. Conversion was calculated to be between 33–73%. Another example for the preparation of thio(meth)acrylates was published in 1977 by Hadjichristidis.18,19 This article elucidated the higher chain flexibility of polythiomethacrylates compared to their polymethacrylates. Monomers were obtained with methacryloylchloride by treatment with thiols in aqueous sodium hydroxide solution and repeated distillation, and free radical polymerization with azobisisobutyronitrile (AIBN) was followed. Furthermore, copolymerization with styrene via free radical polymerization has been investigated by Otsu et al.20 Relative reactivity values for different ester alkyl groups for their copolymerization with styrene have to be higher for thioacrylates compared to copolymerization with similar alkylacrylates. Even polymerization with some anionic catalysts (e.g. n-BuLi, PhMgBr, and LiAlEt3Bu) has been studied for ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec- and tert-butyl thioacrylates.21 However, neither their controlled polymerization nor their complete physical properties have been studied so far. The fundamental understanding of the synthesis and polymerization of thioacrylate monomers and their physical properties is still very limited, although the introduction of thioesters or thiol groups brings unique functionalities to the macromolecules. Transformation of the thioester group via post-polymerization modification, allows selective functionalization into other functional moieties.
For instance, one of the main versatilities of thioesters is the reversible trans thioesterification reaction for the generation of higher order structures in peptides or proteins. In consideration of post polymerization functionalization, a thioester containing peptide can participate in a native chemical ligation (NCL) reaction to generate a peptide bond of an N-terminal cysteinyl peptide under mild conditions in the presence of a thiol.22 These promising properties of thioesters point to their use in novel unexplored applications in materials science and for this reason there has been a drive towards the development of a synthetic strategy for the syntheses of thioacrylate monomers.
Recent progress in living radical polymerization has started a new era of well-defined polymers via well-known controlled radical polymerization techniques such as single electron transfer living radical polymerization (SET-LRP),23–25 nitroxide mediated polymerization (NMP),26–28 atom-transfer radical polymerization (ATRP)29–31 and RAFT polymerization.32–34 The latter was introduced in 1998 and has revolutionized the field of polymer synthesis since then.35 The main advantage of RAFT polymerization is the possibility of polymerizing a wide range of monomer classes by choosing the right chain transfer agent. As this process enables access to polymerize a wide range of monomers and tolerates many functional groups, RAFT polymerization is preferred to prepare well-defined poly(thioacrylate)s in this study.
Herein, we report an easy and efficient method for the preparation of four different thioacrylate monomers that will potentially generate a high level of interest and novel applications in materials science. Additionally, the polymerization of these monomers via a controlled radical polymerization technique is investigated for the first time. Detailed kinetic studies under different reaction conditions have been performed and the obtained poly(thioacrylate)s are characterized in detail by 1H NMR, GPC, water contact angle, as well as their thermal stability and transitions are determined using TGA and DSC.
Results and discussion
The synthesis route of thioacrylate monomers is depicted in Scheme 1. Synthesis of thioester was initially achieved by a typical base catalyzed esterification of bromoacetylbromide (1.00 equiv.), in the presence of 2.00 equiv. of triethylamine (TEA) and 1.30 equiv. of the respective thiol to yield 54% of the desired thioester. However, a more elegant way of incorporating a thioester function to a thiol was reported by Steglich using a DCC/DMAP catalyst system.33 This method was used for further esterification reactions as it provided higher yields and easier work up. Therefore, thioesters with different thiol-groups were prepared from the coupling reaction of bromoacetic acid and the respective thiol. The thioester formation was verified by 1H NMR spectra which are shown in Fig. S2.† In the 1H NMR spectrum, there are two characteristic resonances at 11.5 ppm (Br–CH2–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O–OH) and 1.5 ppm of the respective thiol (R-SH), which have to disappear throughout the coupling reaction. The formation of the phosphonium bromide was accelerated, when the thioester and triphenylphosphine were heated to reflux under nitrogen in DCM for an hour. For the formation of phosphorane, Na2CO3 was initially used as a base according to a literature procedure.16 It was proposed that the yield of the obtained phosphorene could be increased by varying the size of the alkali metal cation, and the yield was increased when Na2CO3 was replaced with K2CO3. Therefore, the choice of K2CO3 as a base led to the necessity of using this base for all Wittig reactions. The characteristic hydrogen signal of the phosphorane was observed as a doublet at ∼3.5–3.6 ppm. Cross metathesis of the Wittig reagent and paraformaldehyde, followed by flash chromatography and distillation was carried out for thioacrylates according to the literature. Typical silica column chromatography was performed using a cold diethyl ether/hexane mixture (1
O–OH) and 1.5 ppm of the respective thiol (R-SH), which have to disappear throughout the coupling reaction. The formation of the phosphonium bromide was accelerated, when the thioester and triphenylphosphine were heated to reflux under nitrogen in DCM for an hour. For the formation of phosphorane, Na2CO3 was initially used as a base according to a literature procedure.16 It was proposed that the yield of the obtained phosphorene could be increased by varying the size of the alkali metal cation, and the yield was increased when Na2CO3 was replaced with K2CO3. Therefore, the choice of K2CO3 as a base led to the necessity of using this base for all Wittig reactions. The characteristic hydrogen signal of the phosphorane was observed as a doublet at ∼3.5–3.6 ppm. Cross metathesis of the Wittig reagent and paraformaldehyde, followed by flash chromatography and distillation was carried out for thioacrylates according to the literature. Typical silica column chromatography was performed using a cold diethyl ether/hexane mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9), identification of compounds was performed using Thin Layer Chromatography (TLC), and Rf values were calculated. No further distillation was necessary and pure monomers were obtained following the evaporation of the solvent. Hydroquinone was added to prevent auto-polymerization and was removed before polymerization by passing through a short column of aluminium oxide. The characteristic vinyl protons are observed at ∼5.5–6.5 ppm (Fig. 1).
:9), identification of compounds was performed using Thin Layer Chromatography (TLC), and Rf values were calculated. No further distillation was necessary and pure monomers were obtained following the evaporation of the solvent. Hydroquinone was added to prevent auto-polymerization and was removed before polymerization by passing through a short column of aluminium oxide. The characteristic vinyl protons are observed at ∼5.5–6.5 ppm (Fig. 1).
 | ||
| Scheme 1 Synthesis route for the preparation of thioacrylate monomers used in this work: (i) 1.05 equiv. DCC, 0.10 equiv. DMAP, DCM, inert atmosphere, overnight; Ra: EtSH (1.30 equiv.), Rb: n-PrSH (1.30 equiv.), Rc: i-PrSH (1.30 equiv.), Rd: PhSH (1.30 equiv.); (ii) 2.00 equiv. triphenylphosphine, benzene, 1 h reflux, (iii) 10 wt% K2CO3, DCM, 30 min, ambient temperature; (iv) 5.00 equiv. paraformaldehyde, pentane, 1 h reflux. | ||
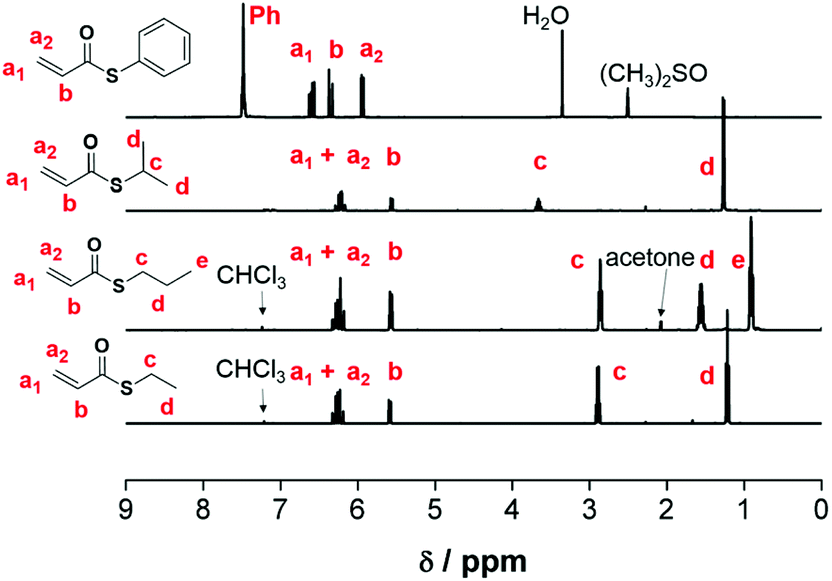 | ||
| Fig. 1 1H NMR analysis of 5a, 5b, and 5c (400 MHz, CDCl3, 25 °C) and 5d (400 MHz, (CH3)2SO-d6, 25 °C) with peak assignments. | ||
RAFT polymerization of thioacrylate monomers
The polymerization of thioacrylates has been investigated using a trithiocarbonate ester chain transfer agent (CTA), namely butyl 2-(((dodecylthio)carbonothioyl)thio)2-methyl propanoate (BDTMP), which enables the polymerization of a wide variety of functional monomers, including acrylates and acrylamides. We have studied the polymerization kinetics of ETA, PhTA, n-PTA and i-PTA using BDTMP as the CTA, examining the influence of several experimental parameters on the rate of propagation. To identify optimized conditions for the preparation of well-defined polymers containing thioacrylates using the RAFT technique, various parameters have been investigated therefore; the influences of the initiator concentration and degree of polymerization on the rate of propagation were assessed. RAFT homopolymerization of thioacrylates is shown in Scheme 2.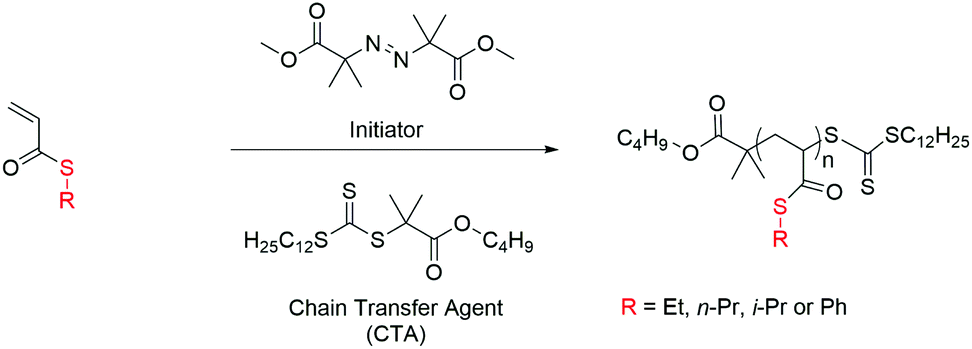 | ||
| Scheme 2 Schematic of the synthetic pathway for the RAFT polymerization of thioacrylate monomers: [CTA]/[I] = 10, toluene, 70 °C. | ||
The performance of the radical polymerization with BDTMP for all thioacrylate monomers was explored at 70 °C and the results are listed in Table 1. The number average of molecular weight (Mn) vs. conversion and the semi-logarithmic first-order kinetic plots are shown in Fig. 2. Molecular weight distributions (Đ) determined by GPC were found to be below 1.2 for all poly(thioacrylate)s indicative of a well-controlled polymerization and a high chain transfer efficiency of the used CTA under the conditions investigated.
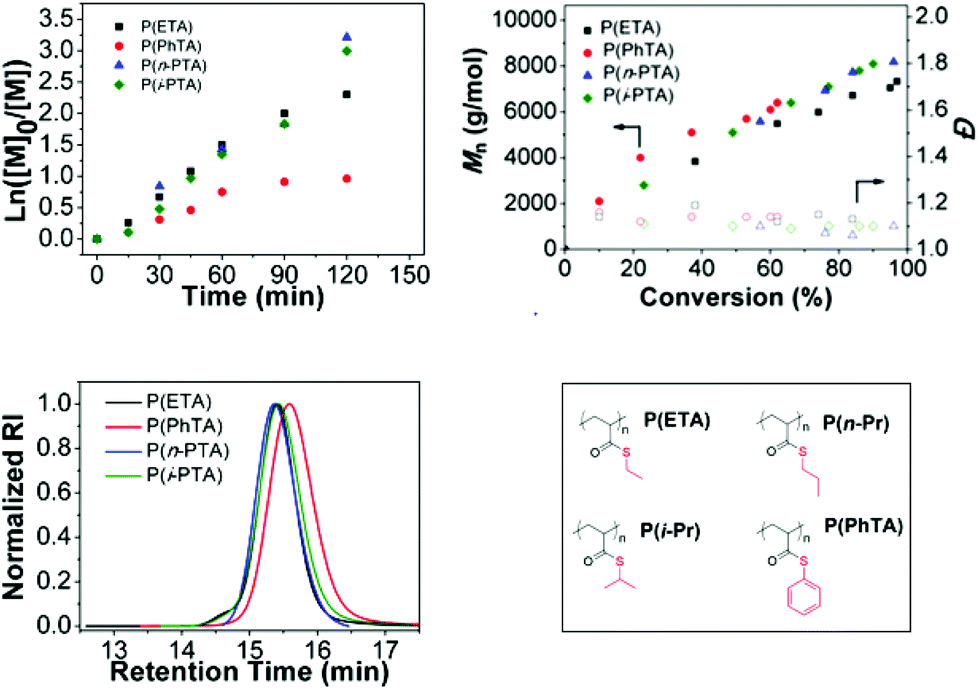 | ||
| Fig. 2 Investigation of the homopolymerization of different thioacrylates via RAFT. Ln([M]0/[M]) vs. time plot for P(ETA), P(nPTA) and P(i-PTA) and P(PhTA) (top left); Mnvs. conversion plot for P(ETA), P(nPTA) and P(i-PTA) and P(PhTA) (top right). Colored filled symbols represent Mn,GPC and blank symbols represent their Đ. GPC traces of the obtained poly(thioacrylate)s with DP = 60 in toluene at 70 °C for ETA, n-PTA, i-PTA, PhTA (bottom left); the chemical structures of the thioacrylate monomers (bottom right). | ||
:[CTA]:[I] = 60:1:0.1 at 70 °C, reaction time = 60 min
| Run | Monomer | [M]/[CTA] | Conv.a (%) | M n,theo (g mol−1) | M n,GPC (g mol−1)b | Đ | k app (s−1) |
|---|---|---|---|---|---|---|---|
| a Conversion measured by 1H NMR spectroscopy. b THF eluent, linear PMMA standard. | |||||||
| 1 | EA | 60 | 78 | 5110 | 5300 | 1.11 | 3.57 × 10−4 |
| 2 | ETA | 60 | 77 | 5790 | 7060 | 1.10 | 3.37 × 10−4 |
| 3 | ETA | 120 | 78 | 11300 |
12700 |
1.12 | 3.57 × 10−4 |
| 4 | ETA | 180 | 56 | 12110 |
12100 |
1.11 | 2.29 × 10−4 |
| 5 | PhTA | 30 | 60 | 3370 | 2870 | 1.19 | 1.90 × 10−4 |
| 6 | PhTA | 60 | 53 | 5640 | 5700 | 1.14 | 2.06 × 10−4 |
| 7 | n-PTA | 60 | 66 | 5570 | 6930 | 1.07 | 4.11 × 10−4 |
| 8 | i-PTA | 30 | 87 | 3800 | 3770 | 1.13 | 3.92 × 10−4 |
| 9 | i-PTA | 60 | 76 | 6350 | 6930 | 1.07 | 3.95 × 10−4 |
For ETA, n-PTA, i-PTA and PhTA, a linear relationship between the molecular weight and conversion indicates the controlled nature of the polymerization (Fig. 2). The Mnvs. conversion plots show similar deviations at low and high monomer conversions compared to the theoretical values (Mn,theo = MWRAFT agent) + (MWmonomer × DP × Convmonomer). Nevertheless, the measured average molecular weights of poly(thioacrylate)s against linear poly(methyl methacrylate) (PMMA) standards showed higher Mn than the theoretical values. There may be two reasons that led to this observation. The first possible reason is that the transfer between the RAFT agent and a propagating radical is less efficient than the subsequent transfer between a dormant chain and a propagating radical. The second possible reason is that in this study the obtained Mn values are calculated using PMMA calibration standards and the discrepancy may be attributed to the inadequacy of PMMA standards to approximate the hydrodynamic volume of the obtained poly(thioacrylate)s in THF. We believe that the latter is more likely as the polymers display a narrow Đ. The linear increase of Mn values with the increase in conversion and the low dispersities strongly indicate the controlled nature of the reactions and confirm that the polymerization of thioacrylates by the RAFT process follows virtually a “living” polymerization mechanism. The semi-logarithmic kinetic plot of homopolymerizations of ETA, n-PTA, i-PTA and PhTA is shown in Fig. 2. The linear relationship indicates a stable radical concentration throughout the polymerization.
The apparent propagation rates (kapp) can be estimated from the slopes of the semi-logarithmic kinetic plots and are listed in Table 1. In the case of PhTA, a slight deviation from the linear relationship was observed (Fig. 2) and a maximum conversion of 62% was obtained after 120 minutes. The low propagation rate indicates a slow initiation although appearance of an uncontrolled period in the early stage is evident, due to equilibrium between active radical-containing chains and dormant species, which potentially leads to early termination events.
Whereas, the kapp for the homopolymerization of ETA shows a two-fold increase (3.37 × 10−4 s−1), isomeric monomers, n-PTA (4.11 × 10−4 s−1) and i-PTA (3.95 × 10−4 s −1) show very similar propagation rates, but are relatively higher than those of ETA and PhTA. Furthermore, the 1H NMR spectra of the purified polymers are provided in Fig. 3.
 | ||
| Fig. 3 1H NMR spectra (CDCl3, (CH3)2SO) of purified PETA, PnPTA, PiPTA and PPhTA. | ||
The effect of reduction of the initiator concentration
In all of the above experiments 0.1 molar ratio of the initiator was used, resulting in a [CTA]/[initiator] ratio of 10. It is known that, in the RAFT process, the fractions of living chains at the end of the polymerization, and thus the obtained degree of control, are strongly dependent on this ratio. Therefore, the concentration of RAFT agent to initiator was optimized by changing the ratio of RAFT agent to initiator (V-601) while maintaining a constant monomer-to-RAFT molar ratio of 60 in toluene at 70 °C. The concentration of the initiator with respect to the chain transfer agent (BDTMP) was reduced from 0.1 to 0.01 molar ratios. An increase in the amount of V-601 resulted in an increase in the rate of polymerization as the propagation rate of polymerization is proportional to [I]1/2. Both polymerization reactions exhibited linear kinetics (Fig. 4), and provided Mnvs. conversion plots that were linear. Polymerization with 0.01 mol% V-601 to BDTMP showed a decrease in the rate of polymerization in time from 3.95 × 10−4 s−1 to 2.28 × 10−4 s−1. Furthermore, the dispersity decreased slightly as the initiator concentration was decreased. | ||
| Fig. 4 Influence of the monomer-to-initiator molar ratio on the homopolymerization of i-PTA via RAFT with BDTMP as the CTA agent. Ln([M]0/[M]) vs. time plot for P(i-PTA) (left); Mnvs. conversion plot for P(i-PTA) (right). Diamond shaped symbols represent Mn,GPC and circles represent their corresponding dispersity. | ||
Investigation of the relative concentration of the monomer to the chain transfer agent
The influence of the relative concentrations of the monomer on the chain transfer agent was evaluated by a series of experiments that involved variations of the [monomer]/[BDTMP] values from 60, 120 and 180 in toluene (Fig. 5). The ratio of the initiator and CTA was held constant at 0.10 mol% and the temperature was kept at 70 °C. Increase of the targeted DP corresponded to dilution of radicals and therefore a decrease in the rate of propagation. Interestingly, conversion values were similar for targeted DPs of 60 and 120, in comparison with low conversion values obtained with a DP of 180. All polymers showed a narrow Đ in the range of 1.09 to 1.12. | ||
| Fig. 5 Influence of the ETA-to-CTA molar ratio on the polymerization of ETA via RAFT. Ln(([M]0/[M])) vs. time plot for P(ETA) with different DPs (left), Mnvs. conversion plot for P(ETA) (right). Black and red symbols represent Mn,GPC and Đ, respectively. | ||
Block copolymerizations of thioacrylate and ethyl acrylate
The living nature of polymers was further verified by block copolymerization. In order to assess the end-group fidelity, the chain extension of P(ETA) or P(EA) was attempted with the addition of EA or ETA (DP = 60), respectively. The first block copolymerization was initiated with EA, as it showed higher transfer ability for the CTA selected. Therefore, the block copolymerization was initiated with EA and followed by sequential addition of ETA as the second monomer without performing any purification step in between. The conversion of the first block was higher than 94% with the GPC revealing a monomodal peak, which suggests the absence of termination reactions. The second block of poly(ETA) was grown in a controlled fashion as a good correlation between the theoretical and the experimental values and narrow Đ (1.12–1.15) was achieved throughout the chain extensions, further confirming the controlled/living character of the polymerization (Fig. 6). The reverse strategy of starting first with poly(ETA) and growing a second block of poly(EA) was also investigated. As expected the block copolymers were grown to satisfactory molecular weights, however the Đ slightly increased (Đ ∼ 1.20–1.23), whereby the theoretical and experimental Mn values were still in good correlation. GPC traces, as shown in Fig. 6E have a similar shape with shoulders indicating some termination by coupling reactions. In summary, for a block copolymer formation for which an acrylate and a thioacrylate with similar propagation rates, the order of the blocks does not have a significant difference in the preparation of block copolymers.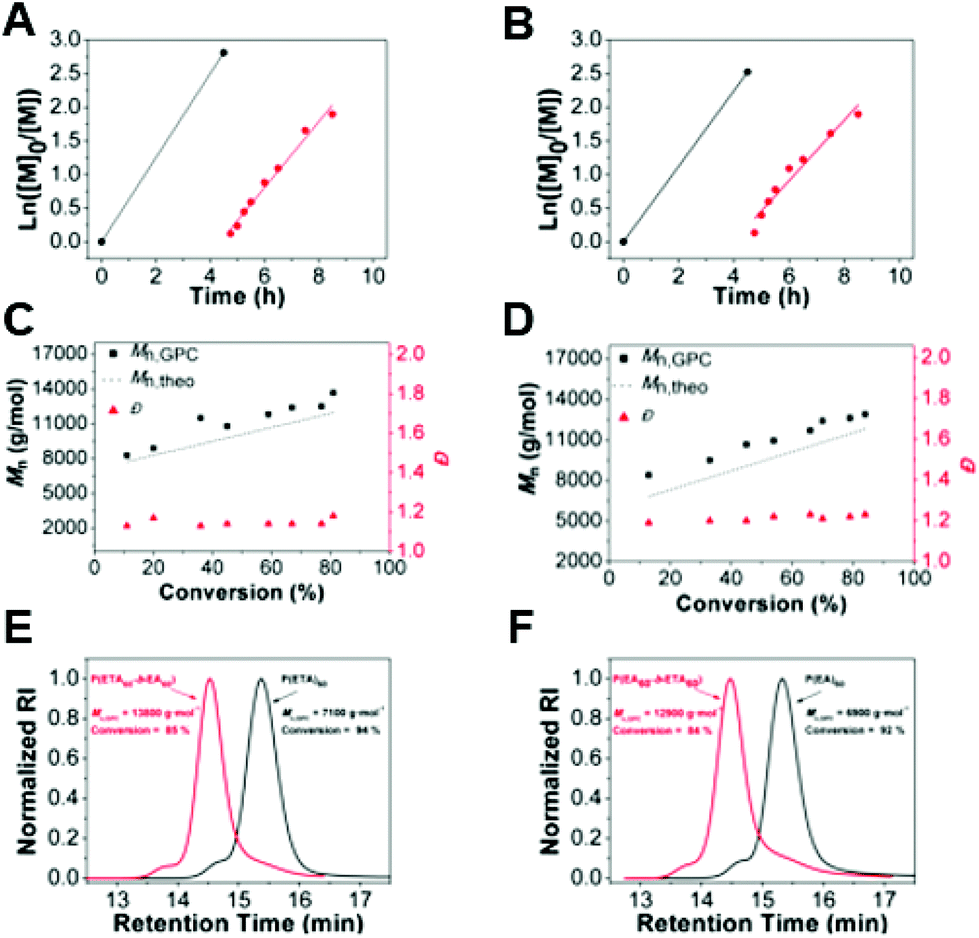 | ||
| Fig. 6 Chain extension experiment of thioacrylates via RAFT. (A) Ln(([M]0/[M])) vs. time for the first block of P(ETA), presented in black and the chain extended second block of P(EA) in red, carried out in toluene at 70 °C. (B) Ln([M]0/[M]) vs. time for the first block of P(EA), presented in black and the chain extended second block of P(ETA) in red, carried out in toluene at 70 °C. (C) Mnvs. conversion plot for P(ETA60-b-EA60), black squares represent the Mn,GPC and the red up-pointed triangles their Đ. (D) Mnvs. conversion plot for P(EA60-b-ETA60), black squares represent the Mn,GPC and the red up-pointed triangles their Đ. (E) GPC traces of the first block of P(ETA) in black and the second block in red. (F) GPC traces of the first block of P(EA) in black and the second block in red. | ||
Physical properties of poly(thio acrylates)
Thermal properties of the obtained polymers were investigated using a thermal gravimetric analyzer (TGA) and a differential scanning calorimeter (DSC) (Fig. 7). P(ETA) exhibited thermal properties with 5% mass loss at T > 316 °C and a glass transition temperature (Tg) of 12 °C. The weight loss of the polymers occurring above 300 °C was ascribed to the decomposition of the polymers. Poly(thioacrylate)s have a relatively higher Tg than the corresponding acrylate polymers P(EA) (Tg = −21 °C) possibly due to the replacement of the thioester with an ester in the polymer side chain. The 5% mass loss for P(EA) occurs at a lower temperature (293 °C).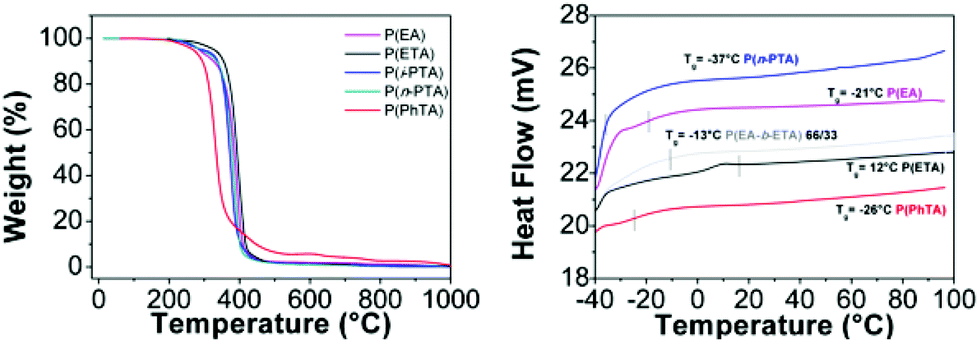 | ||
| Fig. 7 TGA thermogram of poly(ethyl acrylate) P(EA), poly(thioacrylate)s: P(ETA), P(PhTA), P(n-PTA) and P(i-PTA) (left); DSC thermograms of poly(ethyl acrylate): P(EA), poly(thioacrylate)s: P(ETA), P(PhTA), P(n-PTA) and block copolymer P(EA-b-ETA) (right). | ||
After an initial weight loss, associated with some humidity absorbed by the environment, the main loss occurs in the range between 199 °C and 269 °C for poly(thioacrylate)s. The derivative curves of heat flow indicate the temperature for the maximum weight loss (Tmax). Thioacrylate groups provide more stable homopolymers, as the degradation of P(EA) starts at 247.2 °C and shows 50% weight loss at 385.6 °C, whereas thiol-analogues are stable up to 269.7 °C, with a weight loss of 50% at a higher temperature (T50 = 393.6 °C). Poly(ethyl thioacrylate) shows the highest thermal stability for all poly(thioacrylate)s as well as in comparison with poly(ethyl acrylate). A large solid residue at 600 °C of the homopolymer of the thiophenolacrylate is worth noticing (5.9% residue) as for all other samples it has been found to be between 1.2% and 1.9%, including P(EA).
The calorimetric behaviour of polymers was studied by differential scanning calorimetry (DSC). The only thermal event that has been detected in the DSC curve is an exothermal baseline shift, associated with the glass transition temperature (Tg), which is listed in Table 2. In addition, the absence of crystallization and also melting transitions indicates that the obtained polymers are amorphous. As Tg depends primarily on the flexibility of the polymer backbone, the exchange of an oxygen with a bigger sulphur atom in the side-group possibly restricts the flexibility and therefore leads to an increase of the Tg. Varying the length of the side-group results in a change of the movement of the individual repeating units, which can lead to an inefficient packing and therefore higher fragility. This effect can be seen by varying the side-groups of thioacrylates for P(n-PTA) (Tg = −37 °C) and P(ETA) (Tg = 12 °C) due to the increase of the side group flexibility. As the thiophenol side group in P(PhTA) presents a bulky and stiff group, the barrier for segmental motion is higher than that for the alkyl chain. The Tg of this polymer increases to −37 °C. Homopolymerized i-PTA was expected to have a related value to its isomeric analogue poly(n-PTA). As the thiophenol side group in P(PhTA) presents a bulky and stiff group, the barrier for segmental motion is higher than that for the alkyl chain. The Tg of this polymer increases to −37 °C. The homopolymer of the isomeric analogue of n-PTA, i-PTA is assumed to be related to the value of poly(n-PTA).
| Run | Polymer | T d (°C) | T max (°C) | Residue at 600 °C (%) | T g (°C) | WCA (°) |
|---|---|---|---|---|---|---|
| 1 | P(EA) | 247.2 | 385.6 | 1.925 | −21 | 71.2 ± 0.26 |
| 2 | P(ETA) | 269.7 | 393.6 | 1.507 | 12 | 109.1 ± 0.44 |
| 3 | P(i-PTA) | 242.9 | 370.2 | 1.211 | N/A | 100.10 ± 0.03 |
| 4 | P(n-PTA) | 211.3 | 381.7 | 1.210 | −37 | 95.45 ± 0.2 |
| 5 | P(PhTA) | 199.6 | 332.7 | 5.904 | −26 | N/A |
Furthermore, water contact angles of poly(thioacrylate)s were measured. Water contact angle (WCA, θ) measurements were performed on the homopolymers and on selected block copolymers. Compared to P(EA) (θ = 71.2°), P(ETA) is much more hydrophobic (θ = 109.1°). Values for water contact angles are also highlighted in Table 2 for other poly(thioacrylate)s. Their hydrophobicity also corresponds to the insolubility of obtained poly(thioacrylate)s in water. In Fig. 8 the experimental water contact angle values for P(ETA), P(EA-b-ETA), P(n-PTA) and P(i-PTA) are shown. The hydrophilicity of P(EA) can be modified by copolymerization with more hydrophilic monomers such as ETA. For example, the addition of 66% ETA increased the contact angle value by around 30%.
 | ||
| Fig. 8 Contact angle images for droplets of water on P(ETA), P(ETA-b-EA), P(n-PTA) and P(i-PTA) surfaces. | ||
Experimental
Materials
All chemicals were purchased from Sigma-Aldrich (UK) and used as received at the highest purity available. Dichloromethane (DCM, HPLC grade), benzene (HPLC grade), pentane (HPLC grade), methanol (HPLC grade), tetrahydrofuran (THF, HPLC grade) and toluene (HPLC grade) were used as received. Ethyl acrylate (EA, Aldrich, 99%) was destabilized by passing through a short column of basic aluminium oxide prior to polymerization. All other chemicals and solvents were purchased from Sigma-Aldrich (UK) at the highest purity available and used as received unless mentioned otherwise.Instruments and analysis
1H NMR spectra were acquired in deuterated dimethyl sulfoxide ((CD3)2SO) or deuterated chloroform (CDCl3-d) on a Bruker AV-400 at 303 K at a proton frequency of 400.13 MHz. Once the medium was homogeneous, three drops of the solution were transferred to an NMR tube at ambient temperature, time t = 0 and measured. A portion of the aliquot was taken out at desired times and placed directly into (CD3)2SO or CDCl3 and analyzed by 1H NMR. Conversion was determined by 1H NMR, by integration of the vinyl peaks of a monomer peak relative to the residual solvent peak of mesitylene as an internal standard at 6.7 ppm.The molecular weight distributions of the polymers were characterized using GPC on an Agilent 390-LC system equipped with a PL-AS RT autosampler, 2 PLgel 5 μm mixed-C columns (300 × 7.5 mm), a PLgel 5 mm guard column (50 × 7.5 mm), and a differential refractive index (DRI). The system was eluted with THF containing 2% triethylamine (TEA) at a flow rate of 1 mL min−1 and the differential refractive index (DRI) was calibrated with linear narrow poly(methyl methacrylate) (PMMA) standards.
TGA experiments were conducted with a TA Instruments TGA Q500 thermogravimetric analyzer (TA Instruments, USA) using 7–13 mg of respective samples for analysis. The samples were heated from 30 °C to 1000 °C at a ramp rate of 10 °C min−1 in a nitrogen environment (the balance nitrogen purge flow was 40 mL min−1 and the sample purge flow was 60 mL min−1).
The thermal properties of the polymers were analyzed using a PerkinElmer DSC 4000 under an N2 atmosphere (flow rate of 60 mL min−1). Calibration was conducted with an indium standard. Thermograms of the specimen ranging from 5 to 10 mg in platinum crucibles were recorded. The system was equilibrated at 25 °C, cooled to −40 °C at a rate of 10 °C min−1 and heated to 100 °C at a rate of 10 °C min−1 under a flushing nitrogen atmosphere. The heating and cooling scans were repeated twice to erase the effect of the previous thermal history of the samples. From DSC curves, glass transitions (Tg) were determined from the inflection point temperature.
Differences in the water contact angles of the polymers were determined using the DSA 100 Krüss. The static contact angle of a 5 μl drop of water deposited on a spin-coated solid surface was measured; the results are the average of six measurements.
Thioacrylate monomer synthesis
:90 Et2O/pentane) and identification of the compounds was followed by Thin Layer Chromatography (TLC) to yield the desired monomers. Hydroquinone was added to the solution to prevent auto-polymerization and was removed before polymerization by passing through a short column of aluminium oxide.
General procedure for polymerization of thioacrylates
In a typical polymerization, (0.629 g, 5.414 mmol) ETA, BDTMP (33.9 mg, 0.081 mmol), V-601 (1.92 mg, 0.008 mmol), 5% v/v mesitylene and 50% v/v toluene were charged into a Schlenk tube and degassed by gentle bubbling of N2 gas for 30 minutes. The Schlenk tube was then submerged into an oil bath at 70 °C. Samples were taken via degassed syringe at desired time points. The samples were then analyzed by gas chromatography (GC), 1H NMR and gel-permeation chromatography (GPC). Polymers were isolated by precipitation in ethanol as a yellowish sticky solid for P(ETA), P(n-PTA) and P(i-PTA) and a pale yellowish powder for P(PhTA).General procedure for chain extension polymerizations
ETA (0.58 g, 5 mmol), BDTMP (0.034 g, 0.08 mmol), V-601 (2 mg, 0.008 mmol), 0.6 mL of mesitylene and 0.58 mL of toluene were purged with nitrogen gas and transferred via purged syringes to a dry Schlenk flask. The resulting mixture was degassed for 30 min, and then submerged into an oil bath at 70 °C to initiate polymerization. An initial sample was taken via a purged syringe. The first block reached 94% monomer conversion within 4.5 h where after the chain extension with EA (0.49 g, 4.9 mmol in 0.5 mL toluene) was carried out. Samples were taken at timed intervals and analyzed by GPC, GC and 1H NMR to follow the kinetics of the reaction. The polymerization was terminated by the removal of the Schlenk tube from the oil bath and exposing the tube to air.Conclusions
In summary, a straight-forward strategy for the synthesis of various thioacrylate monomers has been developed and four different thioacrylate monomers were successfully polymerized via RAFT polymerization. The synthesis of monomers was proved to be efficient for different thiols as starting materials. Thereby, the kinetics of the homo- and block polymerizations were followed by NMR and GPC analysis. The obtained polymers were further analyzed by TGA and DSC, which showed degradation temperatures around 332 °C and glass transition temperatures in the range of −37 °C–12 °C. Finally, poly(thio acrylate)s resulted in a significant increase of water contact angles compared to P(EA). Poly(thioacrylates) are shown to be easily accessible and can be adapted in most of the controlled radical polymerization techniques. Last but not least, the thio ester group has the potential to be used in thio-exchange reactions, which can open new avenues for the preparation of polymer–peptide bioconjugates.Acknowledgements
This work was supported by the European Commission Horizon2020 programme (EU-ITN EuroSequences Proposal no. 642083).References
- M. Le Neindre and R. Nicolay, Polym. Chem., 2014, 5, 4601 RSC.
- R. K. Iha, K. L. Wooley, A. M. Nyström, D. J. Burke, M. J. Kade and C. Hawker, J. Chem. Rev., 2009, 109, 5620 CrossRef CAS PubMed.
- S. Reinicke, P. Espeel, M. M. Stamenovic and F. E. Du Prez, Polym. Chem., 2014, 5, 5461 RSC.
- G.-Z. Li, R. K. Randev, A. H. Soeriyadi, G. Rees, C. Boyer, Z. Tong, T. P. Davis, C. R. Becer and D. M. Haddleton, Polym. Chem., 2010, 1, 1196 RSC.
- I. V. Koval, Russ. J. Org. Chem., 2005, 41, 631 CrossRef CAS.
- S. Bonengel and A. Bernkop-Schnuerch, J. Controlled Release, 2014, 195, 120 CrossRef CAS PubMed.
- Y. Iwakura, K. Kurita and F. Hayano, J. Polym. Sci., Part A1, 1969, 7, 3075 CrossRef CAS.
- K. Nakabayashi, A. Matsumura, Y. Abiko and H. Mori, Macromolecules, 2016, 49, 1616 CrossRef CAS.
- Y. Qiao, X. Yin and C. Tang, Sci. China: Chem., 2015, 58, 1641 CrossRef CAS.
- C. Zhao, Y. Zhang, S. Pan, L. Rothberg and M.-K. Ng, Macromolecules, 2007, 40, 1816 CrossRef CAS.
- C. G. Overberger and H. Aschkenasy, J. Am. Chem. Soc., 1960, 82, 4357 CrossRef CAS.
- C. G. Overberger and H. Aschkenasy, J. Org. Chem., 1960, 25, 1648 CrossRef CAS.
- M. Kato, K. Toshima and S. Matsumura, Biomacromolecules, 2009, 10, 366 CrossRef CAS PubMed.
- K. Yamamoto and A. Takasu, Macromolecules, 2010, 43, 8519 CrossRef CAS.
- P. J. Evans, R. C. T. Slade, J. R. Varcoe and K. E. Young, J. Mater. Chem., 1999, 9, 3015 RSC.
- A. Bernkop-Schnürch, A. H. Krauland, V. M. Leitner and T. Palmberger, Eur. J. Pharm. Biopharm., 2004, 58, 253 CrossRef PubMed.
- C. S. Marvel, S. L. Jacobs, W. K. Taft and B. G. Labbe, J. Polym. Sci., 1956, 19, 59 CrossRef CAS.
- N. Hadjichristidis, Macromolecules, 1981, 14, 128 CrossRef CAS.
- N. Hadjichristidis, Polymer, 1981, 22, 63 CrossRef.
- T. Otsu, K. Tsuda and T. Fukumizu, Die Makromol. Chem., 1968, 119, 140 CrossRef CAS.
- Y. Nakayama, T. Tsuruta, J. Furukawa, A. Kawasaki and G. Wasai, Die Makromol. Chem., 1961, 43, 76 CrossRef CAS.
- P. Thapa, R.-Y. Zhang, V. Menon and J.-P. Bingham, Molecules, 2014, 14461 CrossRef PubMed.
- A. Anastasaki, V. Nikolaou and D. M. Haddleton, Polym. Chem., 2016, 7, 1002 RSC.
- Q. Zhang, L. Su, J. Collins, G. Chen, R. Wallis, D. A. Mitchell, D. M. Haddleton and C. R. Becer, J. Am. Chem. Soc., 2014, 139, 4325–4332 CrossRef PubMed.
- Q. Zhang, J. Collins, A. Anastasaki, R. Wallis, D. A. Mitchell, C. R. Becer and D. M. Haddleton, Angew. Chem., Int. Ed., 2013, 25, 4435–4439 CrossRef PubMed.
- R. B. Grubbs, Polym. Rev., 2011, 51, 104 CrossRef CAS.
- J. Nicolas, Y. Guillaneuf, C. Lefay, D. Bertin, D. Gigmes and B. Charleux, Prog. Polym. Sci., 2013, 38, 63–235 CrossRef CAS.
- E. L. Malins, S. Amabilino, G. Yilmaz, F. H. Iskigor, B. M. Gridley and C. R. Becer, Eur. Polym. J., 2015, 62, 347 CrossRef CAS.
- C. Boyer, N. A. Corrigan, K. Jung, D. Nguyen, T.-K. Nguyen, N. N. M. Adnan, S. Oliver, S. Shanmugam and J. Yeow, Chem. Rev., 2016, 116, 1803 CrossRef CAS PubMed.
- V. Coessens, T. Pintauer and K. Matyjaszewski, Prog. Polym. Sci., 2001, 26, 337 CrossRef CAS.
- C. R. Becer, R. M. Paulus, S. Höppener, R. Hoogenboom, C.-A. Fustin, J.-F. Gohy and U. Schubert, Macromolecules, 2008, 41(14), 5210–5215 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Polymer, 2008, 49, 1079 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2009, 1402 CrossRef CAS.
- K. Kempe, A. Krieg, C. R. Becer and U. S. Schubert, Chem. Soc. Rev., 2012, 41, 176 RSC.
- G. Moad and D. H. Solomon, The Chemistry of Radical Polymerization, Elsevier, Oxford, 2nd edn, 2006 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of monomers and polymers are presented in the ESI. See DOI: 10.1039/c6py01721e |
| This journal is © The Royal Society of Chemistry 2016 |