
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of well-defined catechol polymers for surface functionalization of magnetic nanoparticles†
Qiang
Zhang
a,
Gabit
Nurumbetov
a,
Alexandre
Simula
a,
Chongyu
Zhu
a,
Muxiu
Li
a,
Paul
Wilson
 a,
Kristian
Kempe
a,
Bin
Yang
b,
Lei
Tao
b and
David M.
Haddleton
*a
a,
Kristian
Kempe
a,
Bin
Yang
b,
Lei
Tao
b and
David M.
Haddleton
*a
aDepartment of Chemistry, University of Warwick, CV4 7AL, Coventry, UK. E-mail: D.M.Haddleton@warwick.ac.uk
bThe Key Laboratory of Bioorganic Phosphorus Chemistry & Chemical Biology (Ministry of Education), Department of Chemistry, Tsinghua University, Beijing 100084, P. R. China
First published on 28th October 2016
Abstract
In order to obtain dual-modal fluorescent magnetic nanoparticles, well-defined fluorescent functional polymers with terminal catechol groups were synthesized by single electron transfer living radical polymerization (SET-LRP) under aqueous conditions for “grafting to” modification of iron oxide nanoparticles. Acrylamide, N-isopropylacrylamide, poly(ethylene glycol) methyl ether acrylate, 2-hydroxyethyl acrylate, glycomonomer and rhodamine B piperazine acrylamide were homo-polymerized or block-copolymerized directly from an unprotected dopamine-functionalized initiator in an ice-water bath. The Cu-LRP tolerated the presence of catechol groups leading to polymers with narrow molecular weight distributions (Mw/Mn < 1.2) and high or full conversion obtained in a few minutes. Subsequent immobilization of dopamine-terminal copolymers on an iron oxide surface were successful as demonstrated by Fourier transform infrared spectroscopy (FTIR), dynamic light scattering (DLS), transition electron microscopy (TEM) and thermogravimetric analysis (TGA), generating stable polymer-coated fluorescent magnetic nanoparticles. The nanoparticles coated with hydrophilic polymers showed no significant cytotoxicity when compared with unmodified particles and the cellular-uptake of fluorescent nanoparticles by A549 cells was very efficient, which also indicated the potential application of these advanced nano materials for bio-imaging.
Introduction
Nanoparticles decorated with biofunctional polymers have received increasing attention in the last two decades due to both their ability to simulate the endocytosis process and to their potential in medical diagnostics and treatment.1–3 Uptake of engineered nanoparticles into cells tends to be size, shape and charge-dependent.2,4,5 The properties of surface materials are also of importance for the targeted delivery of particles into specific cells, or even defined organelles.5,6 Immobilization of carbohydrates, especially glycopolymers, onto magnetic or fluorescent nanoparticles has previously been exploited for pathogen detection and cell imaging.7,8Dopamine has been frequently used as a robust anchor to immobilize functional molecules, e.g. metalated complexes, monosaccharides as well as functional polymers to the surface of metal oxide materials, including iron oxide, titanium oxide and aluminum oxide etc.4,9–14 Controlled radical polymerizations initiated or mediated by dopamine-functionalized initiators or chain transfer agents (CTA), typically transition metal mediated living radical polymerization (TMM-LRP) and reversible addition–fragmentation chain transfer (RAFT) polymerization, have shown promising applications in obtaining well-defined catechol polymers. RAFT polymerization shows excellent tolerance to catechol groups and various well-defined polymers could be successfully synthesized from dopamine-functionalized CTA.15–18 These polymers had relatively narrow molecular weight distribution (Mw/Mn < 1.20) and no significant radical coupling side reactions were observed, which indicated that the RAFT system could effectively control the concentration and present time of active radicals during polymerization and the effect of radical scavenge caused by catechol group to the polymerization was not obvious.15–18 However, when using TMM-LRP to synthesize catechol-containing polymers the interaction between catechol groups and transition metal catalysts can be problematic. Any chelates formed by catechol groups and metal ions have been shown to have very high stability constants and can also be pH-dependent, typically shown as the form, break and reform of mono-, bis- or tris catechol-Fe3+ complexes, which have been used to induce cross-linked self-healing gels.19 Copper(II) complexes could potentially act as effective catalysts to accelerate the oxidation of catechol groups.20
In order to avoid the interference from catechol groups, protected dopamine-functionalized initiators were successfully used for atom transfer radical polymerization (ATRP) of zwitterionic monomers yet the final polymers possessed relatively broad MW distribution (Mw/Mn = 1.5–1.8), indicating the polymerization needed further optimization.21–23 Although ATRP of methacrylate monomers using unprotected dopamine-functionalized initiator could occur, no detailed polymerization kinetic plots or clear MW distribution data were reported.24 Hydrophobic polymers with terminal catechol groups have been synthesized by ATRP and single electron transfer living radical polymerization (SET-LRP) in organic solvents utilizing unprotected dopamine-functionalized initiators, which showed slow polymerization rate and low monomer conversion in high DP polymerization yet the final polymers showed relatively broad MW distribution.25 Control experiments by copper wire mediated SET-LRP using a protected initiator in toluene/alcohol mixed solvents lead to polymers with much narrower MW distribution (dispersity, Đ) as compared to those prepared with the protected initiator, suggesting the side reactions of catechol groups cannot be overlooked in this system. Our central hypothesis was that well defined hydrophilic polymers with terminal catechol anchors could be synthesized by our recently developed SET-LRP strategy in an aqueous solvent system.26,27
In this present work, several fluorescent (co)polymers were synthesized directly from unprotected a dopamine-functionalized initiator by SET-LRP in an aqueous solvent and were subsequently used for surface modification of iron oxide leading to dual-modal fluorescent magnetic nanoparticles. The cell uptake behaviours of these functional polymer-coated nanoparticles were studied, which also proved these particles are useful tools for efficient cellular uptake and biomedical imaging.
Results and discussion
Synthesis and characterization of dopamine-terminal hydrophilic/fluorescent polymers
Dopamine, and certain derivatives, have shown high activities in free radical scavenging thus the catechol groups are often protected when dopamine-functionalized monomers are used in free radical polymerizations.28,29 In order for successful polymerization of unprotected dopamine-functionalized monomers reactions were often performed in high-polar aprotic solvents such as N,N-dimethylformamide, dimethyl sulfoxide or acetonitrile, in which case the catechol groups were believed to possibly form non-covalent bonds with the solvent thus limiting the interaction of catechol with propagating radicals.30–32 Previous research has also suggested to avoid the use of protic polar solvents e.g. alcohols as in these solvents the hydrogen donating behavior of catechols to scavenge free radicals was not deduced.30,33However, recent developments in SET-LRP have shown that SET-LRP is suitable for polymerization of many monomers under conditions that used to be thought as a challenge for ATRP; especially for the polymerization of water-soluble monomers when using water as the only solvent.26,34–36 Thus taking acrylamide (AM) as an example, reports on the successful ATRP of this hydrophilic monomer in aqueous media have been limited, which generally reflected difficult control in polymerization such as significantly differed apparent molecular weight, relatively high MW dispersities and failed chain extension reactions due to the loss of terminal groups etc.27,37–43 It has been reported that the competitive coordination of solvent, monomer and polymer to the transition metal may cause reversible dissociation of halide ligand from the catalyst complexes, leading to often poorly controlled polymerizations.44,45 In this aqueous system, water or conjugated monomers and nitrogen-containing polymers may be also able to solvate the halide ligand or coordinate with the copper, which may cause inefficient deactivation and lead to poor control during the polymerization.46,47
However, when SET-LRP of AM was carried out in water using a prior disproportionation strategy the polymerization was rapid and very high, or even full, conversion could be obtained over minutes to hours even at ambient or subambient temperatures used to reduce the possible loss of terminal halogen groups, Table 1. Size exclusion chromatography (SEC) characterization revealed MW dispersities as low as 1.13 for the final poly(AM) albeit with relatively low DP. On increasing the DP from 80 to 320 and 640, the Mw/Mn increased from 1.13 to 1.24 and 1.35 with a slight reduction in the polymerization rate, Table 1, suggesting fine control during the polymerization although termination still occurred during the long-time scales.
| Monomer | [Initiator]0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[monomer]0:[CuBr]0:[Me6TREN]0 :[monomer]0:[CuBr]0:[Me6TREN]0 |
Time (min) | Con.b (%) |
M n, th (g mol−1) |
M
n, SECc (g mol−1) |
M
n, NMRd (g mol−1) |
M
w/Mne |
|---|---|---|---|---|---|---|---|
| a Reaction conditions: T = 0 °C for all polymerizations; solvent, water (2 mL) for the disproportionation of CuBr/Me6TREN, water (2 mL)/i-PrOH (2 mL) mixture for solubilization of monomer/initiator unless otherwise stated. b Conversion determined by 1H NMR spectroscopy. c Number-average molecular weight determined by aqueous size exclusion chromatography (SEC) using PEO-PEG standards for poly(AM) and by DMF SEC using PMMA standards for poly(NIPAM), poly(PEGA480), poly(HEA) and poly(ManA). d Number-average molecular weight (Mn) calculated by 1H NMR. e Dispersity determined by SEC. f Polymerization initiated by 2,3-dihydroxypropyl 2-bromo-2-methylpropanoate and pure water was used to solubilize the monomer/initiator. g Polymerization initiated by the dopamine-functionalized initiator. | |||||||
| AMf | 1:80:0.8:0.4 |
30 | 97 | 5740 | 3800 | — | 1.13 |
| 1:320:0.8:0.4 |
60 | 98 | 22500 |
11400 |
— | 1.24 | |
| 1:640:0.8:0.4 |
180 | 88 | 40200 |
22700 |
— | 1.35 | |
| AMg | 1:80:0.8:0.4 |
60 | 18 | — | — | — | — |
| 1:80:0.4:0.4 |
60 | 99 | 5990 | 3700 | 7400 | 1.22 | |
| NIPAMg | 1:40:0.4:0.4 |
30 | 95 | 4600 | 7800 | 6640 | 1.10 |
| PEGA480g |
1:20:0.4:0.4 |
60 | 99 | 9900 | 12700 |
12540 |
1.12 |
| HEAg | 1:40:0.4:0.4 |
60 | 100 | 4950 | 8300 | 6100 | 1.17 |
| ManAg | 1:10:0.4:0.4 |
90 | 100 | 4030 | 7800 | 5150 | 1.15 |
The experimental MW (Mn) from SEC varied from the theoretically MW which is ascribed to the use of PEO-PEG molecular weight SEC calibration standards rather than narrow dispersed poly(AM) used in the aqueous SEC. However, with the increase of DP from 80 to 320 and 640, the measured Mn increased linearly, which indicated the living nature of the polymerization, Table 1 & Fig. S3.† In order to test the chain extension ability of poly(AM), in situ addition of 2-hydroxyethyl acrylate (HEA) after high conversion polymerization of AM was successfully performed for block copolymerization. SEC revealed a significant shift of elution trace after chain extension indicating MW increase and the Mw/Mn only increased from 1.14 to 1.37, Fig. S4.† The 1H NMR spectrum of the final polymer also confirmed the ratio of AM and HEA, Fig. S5.† It is hypothesized that the Cu0, rather than CuI, acts as the activator and the Me6TREN ligand is able to strongly coordinate with the CuII, which inhibits the competitive coordination of water, AM and poly(AM) to the activators and deactivators, are two key factors for the successful polymerization of AM in water.
We were inspired by the exceptional ability of aqueous SET-LRP for the successful polymerization of monomers usually prone to result in catalyst deactivation and decided to apply this for the synthesis of dopamine terminal-functionalized polymers. In one polymerization of AM from unprotected dopamine-functionalized initiator utilizing the same catalyst ratio and 2,3-dihydroxypropyl 2-bromo-2-methylpropanoate, the polymerization reached 18% after 1 h and SEC did not detect any significant polymer peaks, possibly due to the low MW of obtained polymers. We believe that the competitive coordination of catechol groups to the copper caused the slow polymerization. Thus the ratio of [CuBr]/[Me6TREN] was decreased from 2:1 to 1:1 aiming to increase the amount of Me6TREN to stabilize the deactivator and thus promote disproportionation. In this case, the polymerization proceeded faster with the conversion reaching >99% in 1 h and the Mw/Mn of final polymer was as low as 1.22, Fig. 1A.
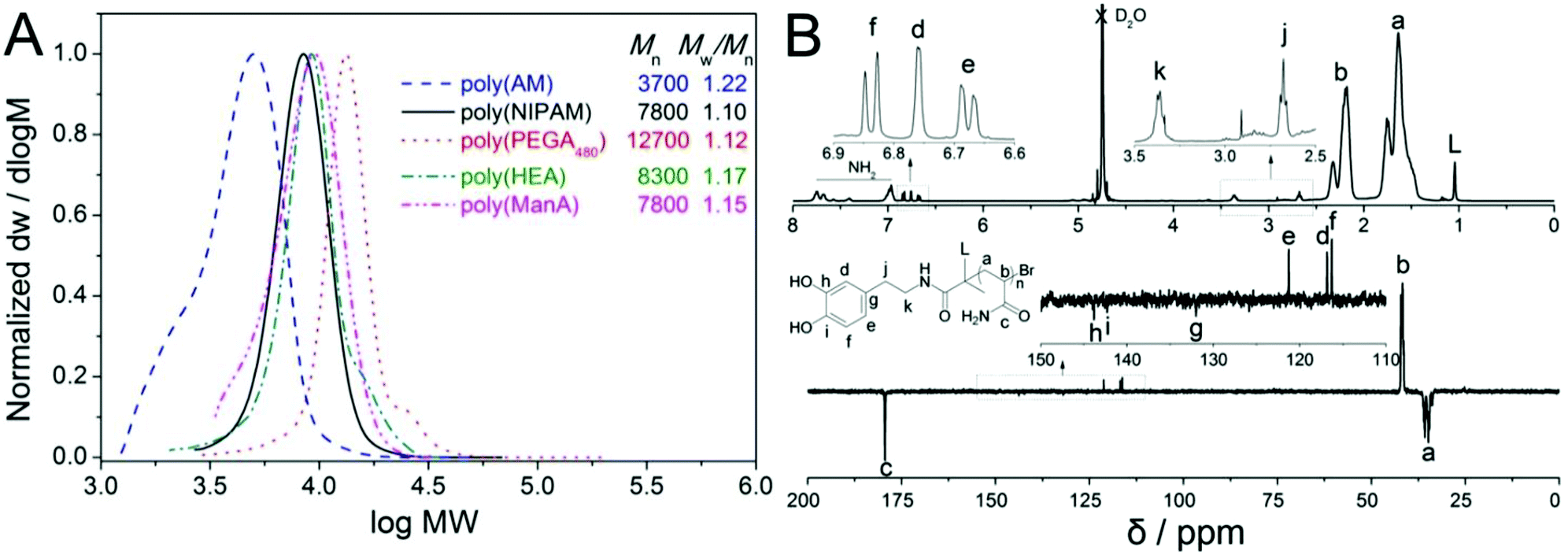 | ||
| Fig. 1 Molecular weight distributions of hydrophilic homo-polymers synthesised via aqueous SET-LRP as measured by SEC (A) and 1H & 13C NMR spectra of dopamine-terminal poly(AM) (B). | ||
1H and 13C NMR spectra of the final product displayed characteristic resonances at 6.6–6.9 ppm (for He, Hd and Hf), 2.5–3.5 ppm (for Hk and Hj, enlarged part) and 115–145 ppm (for Ce, Cd, Cf, Cg, Ch and Ci, enlarged part) respectively, Fig. 1B, which clearly showed the presence of a terminal catechol unit. The average MW calculated by comparing the integration of protons from the catechol groups with that from the polymer backbones was 7400 g mol−1, which was almost 20% higher than the theoretical MW. A considerable difference in experimental and theoretical MW has often been observed in TMM-LRP when using amide functional initiators, which has been attributed to possible termination at low chain length.48
In order to demonstrate the versatility of this strategy, aqueous polymerization of four additional monomers NIPAM, PEGA480, HEA and ManA were carried out using our dopamine-functionalized initiator. Detailed polymerization conditions and results of polymerization of these monomers are summarized in Table 1. Generally low DP ranging from 10 to 40 was chosen in order to confirm the presence of catechol residues in the final polymer products by NMR. All polymerizations went fast and high conversion (>95%) was obtained in 30 to 90 minutes, Table 1.
It is noted that a significant induction period of about one hour was often observed when a dopamine-functionalized chain transfer agent was utilized in RAFT polymerization.16 However, no significant induction period was observed these current polymerizations and often polymerization proceeded so fast that samples could not be taken in time for the first-order kinetic plots to be measured.
SEC characterization confirmed a narrow MW distribution of obtained polymers as low as 1.10 with no significant radical–radical coupling side reactions observed even after full conversion polymerization or when the reaction mixtures were left overnight, except small shoulder peaks for the poly(PEGA480) and poly(HEA) as is often observed, Fig. 1A. 1H and 13C NMR characterization also confirmed the presence of catechol groups (Fig. S9–S16†) and even after dialysis and lypholization no significant oxidation of the catechol groups by oxygen in the air was observed by NMR. The average MW revealed by NMR was ∼20–30% higher than the theoretical values, Table 1, providing further evidence of the possible occurrence of radical coupling or disproportionation termination at the starting stage of polymerization. All these results strongly proved the successful synthesis of well-defined hydrophilic functional polymers entailed with terminal dopamine fragment.
This polymerization retains high chain end fidelity allowing for the synthesis of multi-block copolymerization at low temperatures (0 °C) which in turn significantly reduces the rate of hydrolysis of terminal halogen groups.26,49 In order to check the chain extension ability and also to incorporate a fluorescence tag for dual-model detection, the in situ block copolymerization of a Rhodamine B acrylamide-based monomer (Rhod AM) was performed. After full monomer conversion, as monitored by the disappearance of vinyl groups by 1H NMR spectra (Fig. S17†), a degassed solution of Rhod AM/TEA was directly added to the reaction for further copolymerization, Scheme 1. After chain extension reaction, SEC revealed a significant MW increase and the Mw/Mn remained narrow indicating fine control during the polymerization, Fig. 2A. The 1H NMR spectrum of the pink copolymer product showed typical resonances from both the terminal catechol and side Rhodamine B unit at 6–8 ppm, Fig. 2B. Besides the fluorescent glycopolymer, dopamine-terminal poly(HEA)-b-(Rhod AM) and poly(PEGA480)-b-(Rhod AM) were also successfully synthesized and characterized (Fig. S18–S23†) for further immobilization to magnetic iron oxide particles.
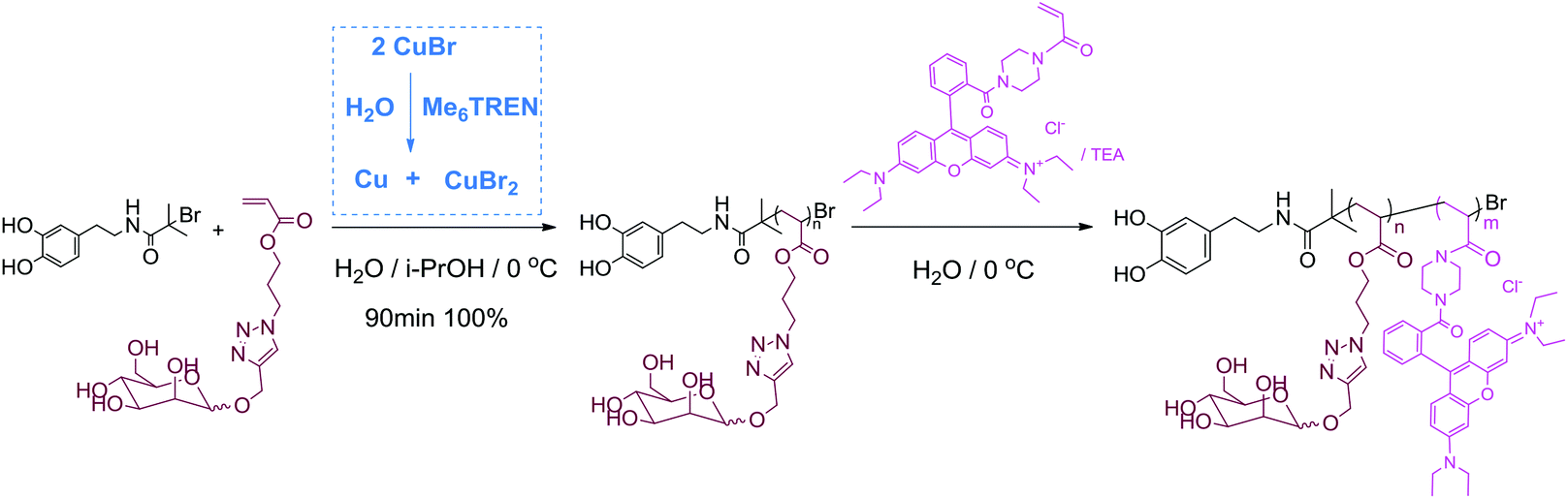 | ||
| Scheme 1 Synthesis of dopamine-terminal fluorescent glycopolymers by aqueous SET-LRP. | ||
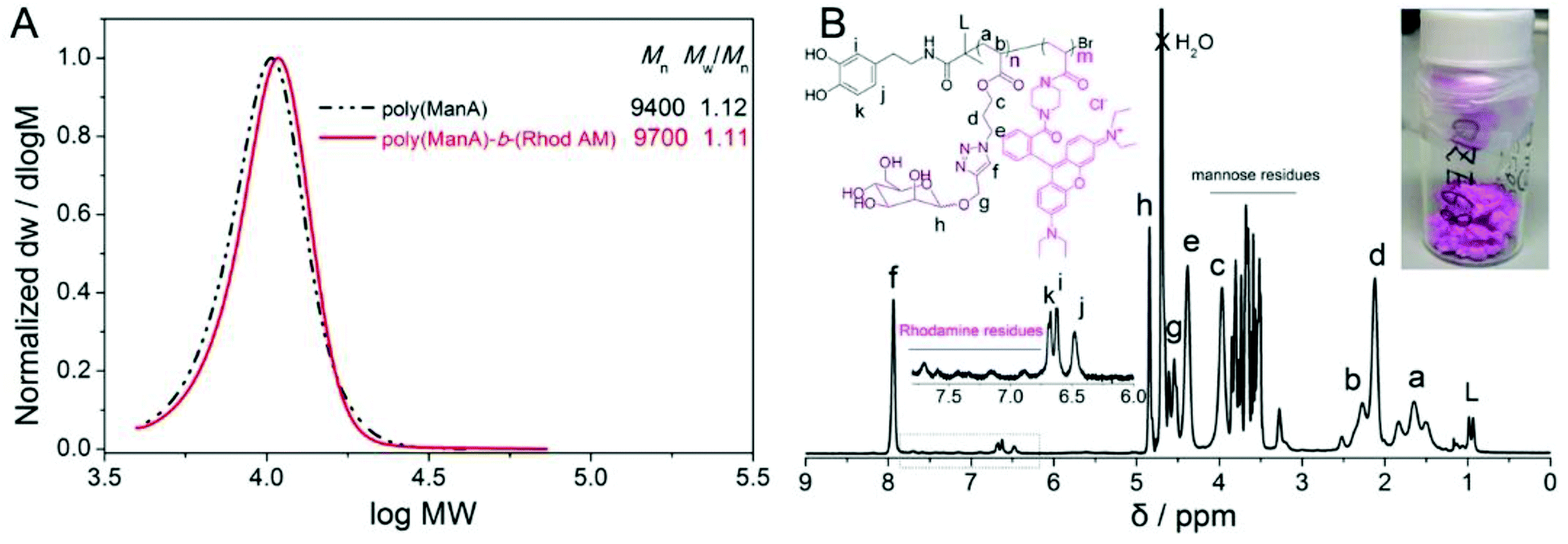 | ||
| Fig. 2 Molecular weight distributions of dopamine-terminal poly(ManA) and poly(ManA)-b-(Rhod AM) by aqueous SET-LRP as measured by DMF SEC (A) and 1H NMR spectrum of poly(ManA)-b-(Rhod AM) (B, the inset showed the pink fluffy product). | ||
In summary, well-defined dopamine-terminal fluorescent copolymers have been successfully prepared by aqueous SET-LRP, which showed dramatic tolerance to the catechol groups and resulted in polymers with various functional groups, controlled MW and narrow MW distribution.
Preparation and characterization of polymer-coated fluorescent magnetic nanoparticles
The dopamine-terminal hydrophilic functional copolymers were used to fabricate water dispersible magnetic iron oxide nanoparticles by precipitation of the Fe(II) and Fe(III) salts by NH4OH in aqueous medium and subsequently coated with oleic acid.50 Nanoparticles prepared by this strategy were stabilized in water and TEM images showed irregularly shaped particles with varied diameter ranging from ∼5 to ∼20 nm, Fig. 3A. Excess amounts of dopamine-terminal copolymers were subsequently used for ligand exchange reaction with the oleic acid coated iron oxide nanoparticles in water under nitrogen protection at 40 °C, Scheme 2.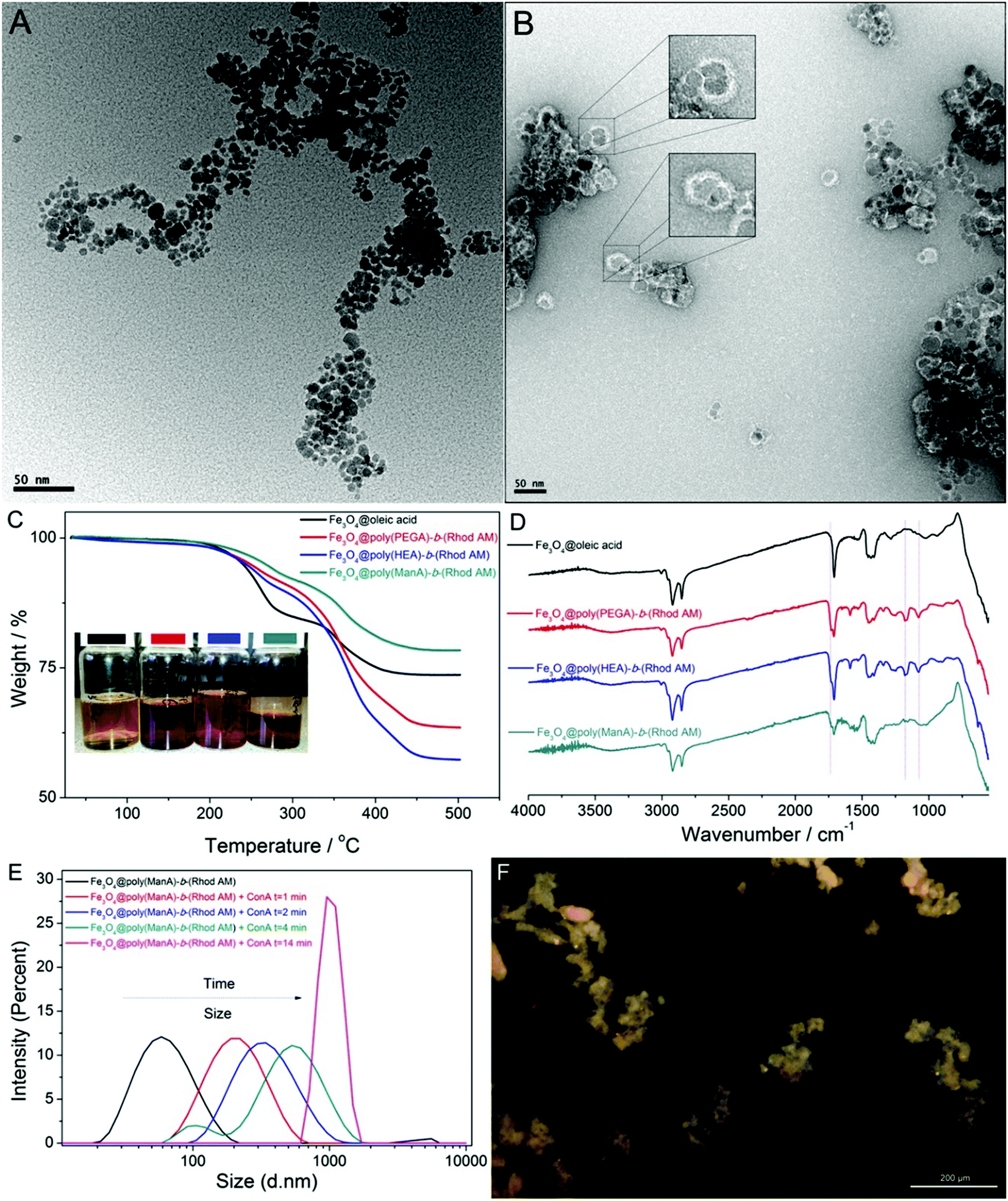 | ||
| Fig. 3 TEM images of Fe3O4@oleic acid (A) and Fe3O4@poly(HEA)-b-(Rhod AM) (B); TGA (C, the inset showed the suspension of Fe3O4 particles) and FTIR (D) of Fe3O4 particles coated with oleic acid or dopamine-terminal fluorescent polymers; DLS (E) results and optical microscopy image (F) of Fe3O4@poly(ManA)-b-(Rhod AM) after addition of ConA. | ||
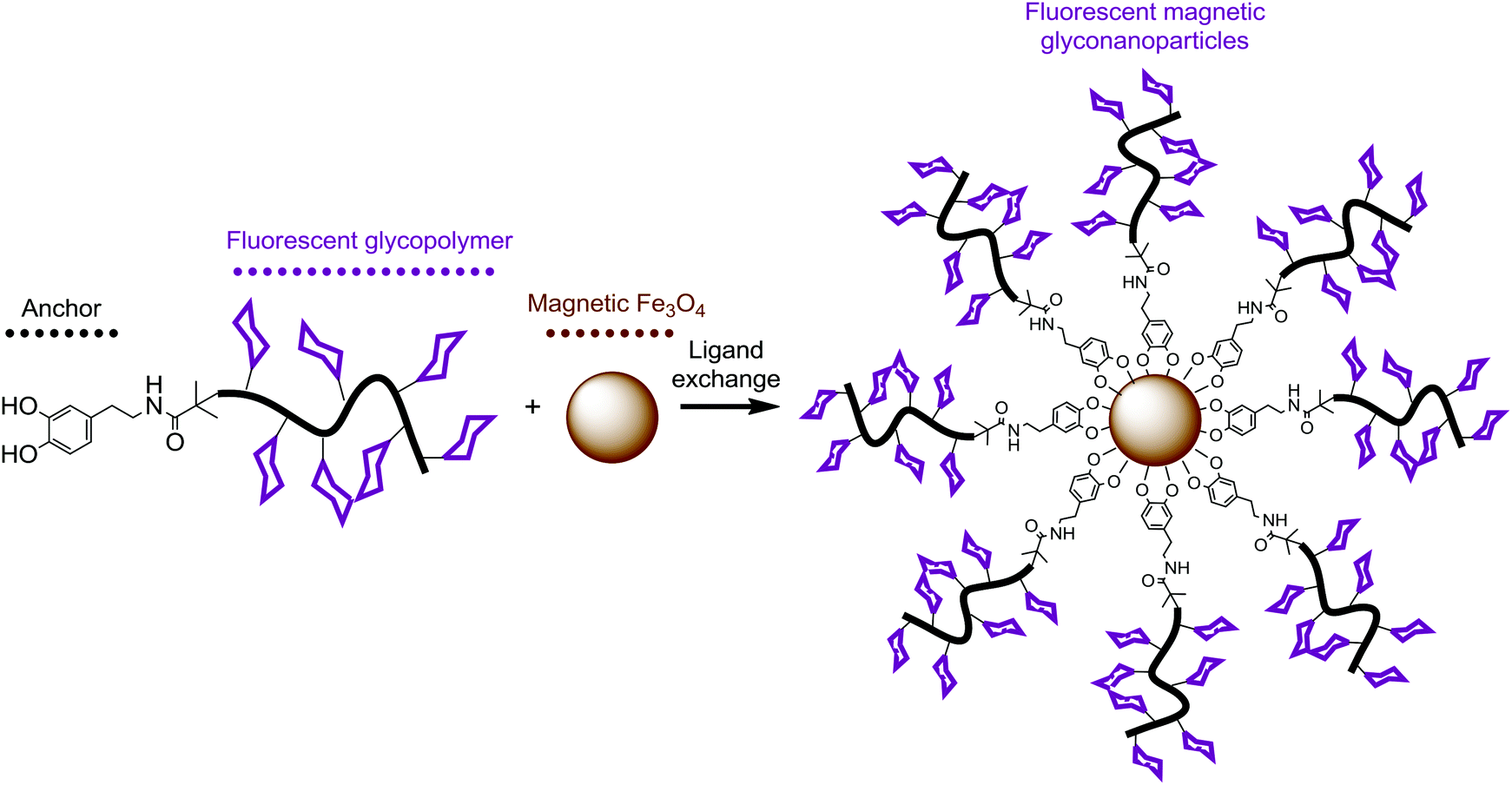 | ||
| Scheme 2 Schematic illustration for the functionalization of magnetic iron oxide particles with dopamine-terminal fluorescent glycopolymers. | ||
After the exchange reaction, the polymer coated nanoparticles were purified from the unbounded polymers via repeated separation and re-dispersion using high speed centrifuge and a strong magnet. Due to the presence of highly water-soluble and pink polymers on the surface of the nanoparticles after exchange with oleic acid, the polymer coated particles are readily dispersible in water without observed precipitation in weeks and the suspension showed a red colour rather than the light yellow colour of original iron oxide particles, Fig. 3C.
The staining technique improved the contrast of TEM images and revealed the presence of a thin and dense polymer layer (5–10 nm) on the surface of particles, which clearly proved that the bio-inspired anchor of dopamine-terminal polymer to transition metal oxide particles was efficient and successful, Fig. 3B. A part of the modified nanoparticles was separated and lyophilized as purple dried powders for further characterization using thermogravimetric analysis (TGA) and FTIR. Compared with the original oleic acid coated particles, TGA analysis of poly(PEGA480)-b-(Rhod AM) and poly(HEA)-b-(Rhod AM) coated particles showed higher weight loss (Fig. 3C), probably due to the presence of much higher MW polymers on the surface. Interestingly, particles coated with relatively low DP and less amount of poly(ManA)-b-(Rhod AM) showed less weight loss during TGA test, which was similarly as one previous report.51 It was considered that the “grafting to” strategy generally had low efficiency in immobilizing polymers to the surface due to steric hindrance effect. Thus theoretically less polymers would be on the surface after ligand exchange reaction and the relatively low MW of poly(ManA)-b-(Rhod AM) further resulted in less weight loss on TGA. FTIR analysis (Fig. 3D) showed clear evidence of the polymer's presence on the particle surface by comparing the spectra of particles coated with oleic acid or block copolymers. The characteristic polymer IR absorbance peaks such as the carbonyl stretching at ∼1750 cm−1 and the –CH2–CH2–O stretching at ∼1000–1250 cm−1 were detected subsequent to the exchange reaction, indicating the success of functionalization.
In order to further prove the presence of carbohydrate on the surface of obtained glycoparticles, we added excess of concanavalin A (ConA) to the particle suspension to check the aggregation behavior caused by carbohydrate–protein interaction. Due to the recognition of lectin with mannose-rich polymers, the particles aggregated fast and the intensity average diameter measured by DLS increased from ∼50 nm to ∼200 nm after 1 minute and ∼600 nm after 4 minute (Fig. 3E). Just after addition of ConA for 14 min, the presence of large irregularly shaped particles are already over the detect limit of our DLS system (shown as a narrow sharp peak, Fig. 3E) and could be even directly observed by optical microscopy (Fig. 3F).
In summary, the dopamine-terminal group anchor the copolymers to the surface of iron oxide is efficient and successful as revealed by varied characterization tools. The presence of these polymers increases the stability of particles in water and also endowed the particles different properties derived from the polymers.
Cytotoxicity and cell uptake of fluorescent magnetic nanoparticles
The cellular uptake of polymer coated nanoparticles by A549 cells (Tsinghua University, PRC) was investigated using confocal laser scanning microscope (CLSM) and the cell morphology was also observed to evaluate the cytotoxicity of nanoparticles. A549 cells were incubated in the presence of different concentrations of nanoparticles for up to 24 hours. After washing with PBS buffer to remove excess particles, the cells were first observed under bright field and the images clearly demonstrated the maintained cell morphology under different concentrations of nanoparticles ranging from 10 to 120 μg ml−1, Fig. 4, which suggested that these particles have good biocompatibility. To further define the viability of A549 cells after being treated with different magnetic nanoparticles, a cell counting kit-8 (CCK-8) assay was utilized.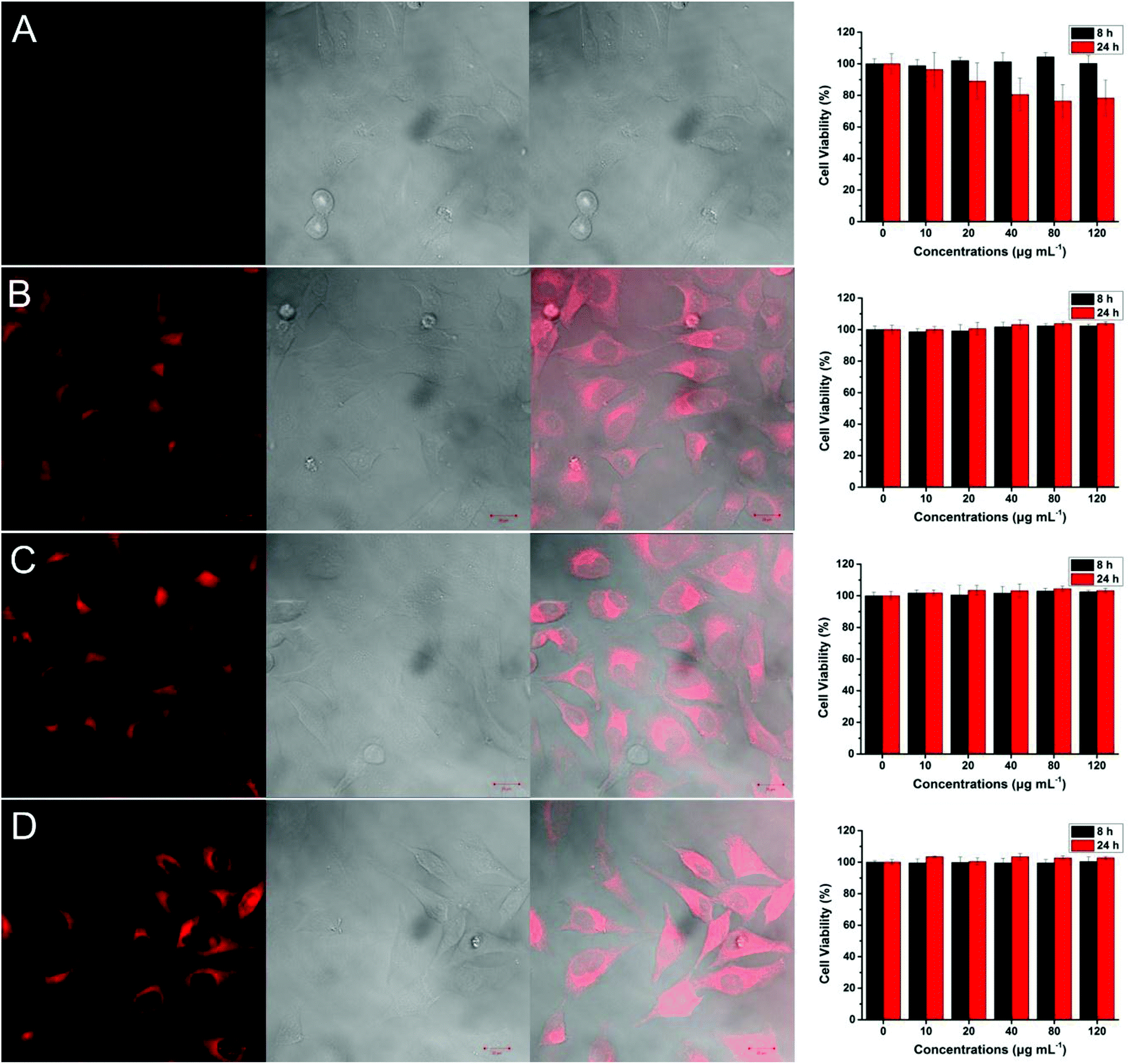 | ||
| Fig. 4 Confocal laser scanning microscopy (CLSM) images and cell viability of A549 cells with Fe3O4@oleic acid (A); Fe3O4@poly(HEA)-b-(Rhod AM) (B); Fe3O4@poly(PEGA480)-b-(Rhod AM) (C); Fe3O4@poly(ManA)-b-(Rhod AM) (D). In each image the left: excited with 405 nm laser; the middle: bright field; the right: merged image of bright field and fluorescent field. The cell viability was determined by CCK-8 assay. The concentration of Fe3O4 nanocomposites for CLSM is 100 μg mL−1. | ||
The cell viability decreased to around 80% when the concentration of Fe3O4@oleic acid was increased to 120 μg ml−1, which highlighted the adverse effect of Fe3O4 nanoparticles especially under relatively high concentrations, Fig. 4A. However, we did not observe any significant viability decrease when Fe3O4@poly(HEA)-b-(Rhod AM), Fe3O4@poly(PEGA480)-b-(Rhod AM) and Fe3O4@poly(ManA)-b-(Rhod AM) were present during the incubation, Fig. 4B, C and D. Compared with the oleic acid, it is hypothesized that the presence of dense hydrophilic polymer brushes on the surface of Fe3O4 nanoparticles, which obviously had better water solubility, higher molecular weight, different functional groups and charge state, would theoretically lead to better coverage of the inorganic metal oxide core and better biocompatibility.
Previous research reported that the cell uptake of particles coated with mannose functionalized polymer tended to be mediated by the mannose receptors on the A549 cell surface and was faster and more significant than particle coated with PEG or glucose functionalized polymer.52 When excited with a 405 nm laser, the cells incubated with Fe3O4@poly(HEA)-b-(Rhod AM), Fe3O4@poly(PEGA480)-b-(Rhod AM) and Fe3O4@poly(ManA)-b-(Rhod AM) particles showed very strong fluorescence in the cytoplasm area (Fig. 4), while the cell nucleus area was much darker and the cells incubated with Fe3O4@oleic acid were completely dark under same excitation. These results further proved the immobilization of fluorescent polymers on the particle surface and the cells could take up these nanoparticles mainly into cytoplasm area. Although the cells were incubated with particles in same concentration and incubation time, the cells with Fe3O4@poly(ManA)-b-(Rhod AM) proved to be no less or even more lighter than under CLSM images under visual observation (Fig. 4D). It needs to be emphasized that the Fe3O4@poly(ManA)-b-(Rhod AM) showed least weight loss during TGA test (Fig. 3C) as least glycopolymer was used for surface modification compared with poly(HEA)-b-(Rhod AM) and poly(PEGA480)-b-(Rhod AM) (ESI†). These indicated that the cells may take more Fe3O4@poly(ManA)-b-(Rhod AM) than Fe3O4@poly(HEA)-b-(Rhod AM) and Fe3O4@poly(PEGA480)-b-(Rhod AM) after same time, which also proved that the cellular uptake of mannose functionalized particles was faster than the others. Considering the excellent fluorescence and different cellular uptake behaviour, these particles may have potential application in biomedical imaging and targeting therapy.
Conclusions
In summary, well-defined hydrophilic polymers with terminal catechol groups have been successfully prepared by aqueous SET-LRP directly from an unprotected dopamine-functionalized initiator. The polymerizations showed excellent compatibility with the catechol groups as well as different functional monomers leading to final polymers with controlled MW and narrow MW distributions. Furthermore, the grafting of several fluorescent block copolymers to iron oxide particles led to dual-modal fluorescent nano materials as characterized by TEM, FTIR, TGA and DLS etc. Investigation of the polymer-coated nanoparticles showed no significant cytotoxic effect, indicating its significant biocompatibility. Cellular-uptake of these nanoparticles by A549 cells was very efficient and suggested potential application in bioimaging, cell–glycopolymer interaction and RME process studies. The presented synthetic route opens up a novel approach for synthesis of well-defined catechol polymers as well as surface functionalization of various materials.Acknowledgements
Q. Z. acknowledges the financial support from University of Warwick and the China Scholarship Council. D. M. H. was a Royal Society/Wolfson Fellow during this work.Notes and references
- X. Li and G. Chen, Polym. Chem., 2015, 6, 1417–1430 RSC.
- W. Jiang, Y. S. KimBetty, J. T. Rutka and C. W. ChanWarren, Nat. Nanotechnol., 2008, 3, 145–150 CrossRef CAS PubMed.
- X. Chen, O. Ramström and M. Yan, Nano Res., 2014, 7, 1381–1403 CrossRef CAS PubMed.
- X. Li, M. Bao, Y. Weng, K. Yang, W. Zhang and G. Chen, J. Mater. Chem. B, 2014, 2, 5569–5575 RSC.
- A. S. Zahr, C. A. Davis and M. V. Pishko, Langmuir, 2006, 22, 8178–8185 CrossRef CAS PubMed.
- Y. Goto, R. Matsuno, T. Konno, M. Takai and K. Ishihara, Biomacromolecules, 2008, 9, 3252–3257 CrossRef CAS PubMed.
- K. El-Boubbou, C. Gruden and X. Huang, J. Am. Chem. Soc., 2007, 129, 13392–13393 CrossRef CAS PubMed.
- C.-H. Lai, J. Hütter, C.-W. Hsu, H. Tanaka, S. Varela-Aramburu, L. De Cola, B. Lepenies and P. H. Seeberger, Nano Lett., 2016, 16, 807–811 CrossRef CAS PubMed.
- C. Xu, K. Xu, H. Gu, R. Zheng, H. Liu, X. Zhang, Z. Guo and B. Xu, J. Am. Chem. Soc., 2004, 126, 9938–9939 CrossRef CAS PubMed.
- P. J. Endres, T. Paunesku, S. Vogt, T. J. Meade and G. E. Woloschak, J. Am. Chem. Soc., 2007, 129, 15760–15761 CrossRef CAS PubMed.
- W. O. Yah, H. Xu, H. Soejima, W. Ma, Y. Lvov and A. Takahara, J. Am. Chem. Soc., 2012, 134, 12134–12137 CrossRef CAS PubMed.
- K. Cheng, S. Peng, C. Xu and S. Sun, J. Am. Chem. Soc., 2009, 131, 10637–10644 CrossRef CAS PubMed.
- M. Mazur, A. Barras, V. Kuncser, A. Galatanu, V. Zaitzev, K. V. Turcheniuk, P. Woisel, J. Lyskawa, W. Laure, A. Siriwardena, R. Boukherroub and S. Szunerits, Nanoscale, 2013, 5, 2692–2702 RSC.
- J. Su, F. Chen, V. L. Cryns and P. B. Messersmith, J. Am. Chem. Soc., 2011, 133, 11850–11853 CrossRef CAS PubMed.
- J. Liu, W. Yang, H. M. Zareie, J. J. Gooding and T. P. Davis, Macromolecules, 2009, 42, 2931–2939 CrossRef CAS.
- C. Zobrist, J. Sobocinski, J. Lyskawa, D. Fournier, V. Miri, M. Traisnel, M. Jimenez and P. Woisel, Macromolecules, 2011, 44, 5883–5892 CrossRef CAS.
- W. Scarano, H. Lu and M. H. Stenzel, Chem. Commun., 2014, 50, 6390–6393 RSC.
- Z. Jia, V. A. Bobrin, N. P. Truong, M. Gillard and M. J. Monteiro, J. Am. Chem. Soc., 2014, 136, 5824–5827 CrossRef CAS PubMed.
- N. Holten-Andersen, M. J. Harrington, H. Birkedal, B. P. Lee, P. B. Messersmith, K. Y. C. Lee and J. H. Waite, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2651–2655 CrossRef CAS PubMed.
- A. Neves, L. M. Rossi, A. J. Bortoluzzi, B. Szpoganicz, C. Wiezbicki, E. Schwingel, W. Haase and S. Ostrovsky, Inorg. Chem., 2002, 41, 1788–1794 CrossRef CAS PubMed.
- G. Li, G. Cheng, H. Xue, S. Chen, F. Zhang and S. Jiang, Biomaterials, 2008, 29, 4592–4597 CrossRef CAS PubMed.
- C. Gao, G. Li, H. Xue, W. Yang, F. Zhang and S. Jiang, Biomaterials, 2010, 31, 1486–1492 CrossRef CAS PubMed.
- G. Li, H. Xue, C. Gao, F. Zhang and S. Jiang, Macromolecules, 2010, 43, 14–16 CrossRef CAS PubMed.
- M. Chanana, S. Jahn, R. Georgieva, J.-F. Lutz, H. Bäumler and D. Wang, Chem. Mater., 2009, 21, 1906–1914 CrossRef CAS.
- G. Rayner, University of Warwick, 2012.
- Q. Zhang, P. Wilson, Z. Li, R. McHale, J. Godfrey, A. Anastasaki, C. Waldron and D. M. Haddleton, J. Am. Chem. Soc., 2013, 135, 7355–7363 CrossRef CAS PubMed.
- G. R. Jones, Z. Li, A. Anastasaki, D. J. Lloyd, P. Wilson, Q. Zhang and D. M. Haddleton, Macromolecules, 2016, 49, 483–489 CrossRef CAS.
- S. Son and B. A. Lewis, J. Agric. Food Chem., 2002, 50, 468–472 CrossRef CAS PubMed.
- C. Zhang, K. Li and J. Simonsen, J. Appl. Polym. Sci., 2003, 89, 1078–1084 CrossRef CAS.
- J. Yang, J. Keijsers, M. van Heek, A. Stuiver, M. A. Cohen Stuart and M. Kamperman, Polym. Chem., 2015, 6, 3121–3130 RSC.
- H. O. Ham, Z. Liu, K. H. A. Lau, H. Lee and P. B. Messersmith, Angew. Chem., Int. Ed., 2011, 50, 732–736 CrossRef CAS PubMed.
- M. Álvarez-Paino, G. Marcelo, A. Muñoz-Bonilla and M. Fernández-García, Macromolecules, 2013, 46, 2951–2962 CrossRef.
- V. Thavasi, R. P. A. Bettens and L. P. Leong, J. Phys. Chem. A, 2009, 113, 3068–3077 CrossRef CAS PubMed.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS PubMed.
- V. Percec, A. V. Popov, E. Ramirez-Castillo, M. Monteiro, B. Barboiu, O. Weichold, A. D. Asandei and C. M. Mitchell, J. Am. Chem. Soc., 2002, 124, 4940–4941 CrossRef CAS PubMed.
- B. M. Rosen and V. Percec, Chem. Rev., 2009, 109, 5069–5119 CrossRef CAS PubMed.
- S. K. Jewrajka and B. M. Mandal, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 2483–2494 CrossRef CAS.
- S. K. Jewrajka and B. M. Mandal, Macromolecules, 2003, 36, 311–317 CrossRef CAS.
- D. A. Z. Wever, P. Raffa, F. Picchioni and A. A. Broekhuis, Macromolecules, 2012, 45, 4040–4045 CrossRef CAS.
- F. Alsubaie, A. Anastasaki, V. Nikolaou, A. Simula, G. Nurumbetov, P. Wilson, K. Kempe and D. M. Haddleton, Macromolecules, 2015, 48, 6421–6432 CrossRef CAS.
- A. Anastasaki, V. Nikolaou, G. Nurumbetov, P. Wilson, K. Kempe, J. F. Quinn, T. P. Davis, M. R. Whittaker and D. M. Haddleton, Chem. Rev., 2016, 116, 835–877 CrossRef CAS PubMed.
- V. Nikolaou, A. Simula, M. Droesbeke, N. Risangud, A. Anastasaki, K. Kempe, P. Wilson and D. M. Haddleton, Polym. Chem., 2016, 7, 2452–2456 RSC.
- A. Anastasaki, V. Nikolaou and D. M. Haddleton, Polym. Chem., 2016, 7, 1002–1026 RSC.
- N. V. Tsarevsky, T. Pintauer and K. Matyjaszewski, Macromolecules, 2004, 37, 9768–9778 CrossRef CAS.
- N. V. Tsarevsky and K. Matyjaszewski, Chem. Rev., 2007, 107, 2270–2299 CrossRef CAS PubMed.
- P. Y. Zavalii, M. G. Mys'kiv and V. S. Fundamenskii, Sov. Phys. Crystallogr., 1984, 29, 34–37 Search PubMed.
- K. B. Girma, V. Lorenz, S. Blaurock and F. T. Edelmann, Z. Anorg. Allg. Chem., 2008, 634, 267–273 CrossRef CAS.
- A. Limer and D. M. Haddleton, Macromolecules, 2006, 39, 1353–1358 CrossRef CAS.
- A. H. Soeriyadi, C. Boyer, F. Nyström, P. B. Zetterlund and M. R. Whittaker, J. Am. Chem. Soc., 2011, 133, 11128–11131 CrossRef CAS PubMed.
- D. Maity and D. C. Agrawal, J. Magn. Magn. Mater., 2007, 308, 46–55 CrossRef CAS.
- M. Arslan, T. N. Gevrek, J. Lyskawa, S. Szunerits, R. Boukherroub, R. Sanyal, P. Woisel and A. Sanyal, Macromolecules, 2014, 47, 5124–5134 CrossRef CAS.
- J. S. Basuki, L. Esser, H. T. T. Duong, Q. Zhang, P. Wilson, M. R. Whittaker, D. M. Haddleton, C. Boyer and T. P. Davis, Chem. Sci., 2014, 5, 715–726 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6py01709f |
| This journal is © The Royal Society of Chemistry 2016 |