
Synthesis of a polymethyl(methacrylate)-polystyrene-based diblock copolymer containing biotin for selective protein nanopatterning†
A.
Lagunas
*ab,
B.
Sasso
c,
N.
Tesson
c,
C.
Cantos
c,
E.
Martínez
dae and
J.
Samitier
bae
aNetworking Biomedical Research Center (CIBER) in Bioengineering, Biomaterials and Nanomedicine (CIBER-BBN), Monforte de Lemos 3-5, Pabellón 11, 28029 Madrid, Spain
bNanobioengineering group, Institute for Bioengineering of Catalonia (IBEC), Baldiri Reixac 15-21, 08028 Barcelona, Spain. E-mail: alagunas@ibecbarcelona.eu
cEnantia, S. L., Baldiri Reixac 10, 08028 Barcelona, Spain
dBiomimetic Systems for Cell Engineering group, Institute for Bioengineering of Catalonia (IBEC), Baldiri Reixac 15-21, 08028 Barcelona, Spain
eElectronics Department, University of Barcelona (UB), Martí i Franquès 1-11, 08028 Barcelona, Spain
First published on 22nd October 2015
Abstract
Protein patterning is of interest in high-throughput screening. Due to an increase in demand for further miniaturization of protein assays, block copolymers (BCPs) that can undergo large-area phase separation into nanometer-size domains have attracted great attention as substrates for protein nanopatterning. Here we report the synthesis of a polymethyl(methacrylate)-polystyrene-based diblock copolymer which, once spin-coated, is capable of self-segregating into cylindrical polystyrene (PS) domains. In this copolymer, the PS block was modified to introduce biotin below 10% molar in order to achieve molecular recognition of streptavidin. The PMMA matrix used to introduce poly(ethylene glycol) enabled us to obtain an antifouling environment that prevents unspecific protein adsorption outside the domains. The use of the biotin–streptavidin pair in this BCP makes it suitable for nanopatterning of other biotinylated proteins of interest for the purposes of cell biology, biosensors, and tissue engineering.
Introduction
Protein patterning is of interest in high-throughput screening. Protein microarrays offer significant advantages for sensing applications when compared to the alternative well-plate format assays. In this regard, the former allow short diffusion times and parallel detection of multiple targets and, in addition, require only tiny amounts of the sample.1 The lack of protein amplification methods analogous to PCR for nucleic acid analysis together with the small amounts in which many proteins exert their biological functions call for further miniaturization of protein assays. Moreover, miniaturization through confinement to nanoscale dimensions allows for interrogation of protein interactions at the molecular level.2Several methods have been developed to produce protein nanoarrays, including electron-beam lithography,3 AFM-based patterning,4 and colloidal lithography.5 Of these, only colloidal lithography is suitable to produce large-scale arrays and it can be easily implemented in laboratories.
Block copolymers (BCPs), in which the integrating polymer blocks are immiscible enough to undergo phase separation and self-segregation into large areas, thus generating nanometer-sized domains, are attracting attention as substrates for protein nanopatterning. The use of BCP thin film nanopatterns has been explored for the selective adsorption of proteins.6 In these studies, non-specific protein adsorption in the non-patterned regions was poorly controlled, and proteins were retained in a completely hydrophobic environment, which may be detrimental to protein activity. It is generally recognized that the optimal activity for immobilized protein molecules often requires a hydrophilic and non-fouling surface environment to preserve the protein structure and function.7 Other examples describe the covalent immobilization of proteins onto the functional group-bearing nanodomains in BCPs. Cooper White and co-workers reported the covalent immobilization of His-tagged GFP on maleimide-functionalized nanodomains modified with a zinc-chelating peptide by a thiol–ene reaction,8 and Shen et al. described the covalent immobilization of azide-tagged proteins onto alkyne-functionalized nanostructured BCPs.9 These chemical modifications after copolymer synthesis also have several drawbacks. Among them, the set of reactions required to achieve the final material may not take place in quantitative yields because of the intrinsic heterogeneity of the medium. This limitation makes it difficult to control the final amount of protein in the nanodomains. Moreover, chemical immobilization may not show sufficient specificity, thereby leading to several protein orientations that affect the function of the final protein, and similar to adsorption processes in hydrophobic environments, proteins directly chemisorbed onto surfaces are prone to denature. Cornelissen and co-workers reported the immobilization of proteins by molecular recognition on BCPs. Molecular recognition is more sensitive to the structure of the protein and leads to a unique orientation. The authors included biotin in the OH termination of poly(ethylene glycol) (PEG) in a poly(ethylene glycol)-polystyrene block copolymer (PEG-b-PS) for selective molecular recognition of streptavidin. Nevertheless, the final copolymer resulted in an excess of biotin groups, which caused the segregation of streptavidin. These authors then turned their attention to different mixtures of biotin- and non-biotin-containing copolymers to obtain the final nanopatterned surfaces.10
Here we report the synthesis of a new polymethyl(methacrylate) (PMMA)-PS-based diblock copolymer, capable of segregating into cylindrical PS domains once spin-coated onto flat silicon surfaces. The PS block is modified during polymer synthesis to introduce biotin below 10% molar in order to minimize the effects of steric hindrance on the final biotinylated domains and to ensure correct molecular recognition of streptavidin. Moreover, the PMMA matrix is used to introduce PEG, thus achieving an anti-fouling environment that prevents unspecific protein adsorption outside the domains. Using this approach, we synthesized BCPs with a polydispersity index (PDI) below 1.5 and these allow the selective immobilization of streptavidin. The use of the biotin–streptavidin pair makes the presented platform suitable for nanopatterning of other biotinylated proteins.
Experimental
General
The monomers, poly(ethylene glycol) methyl ether methacrylate (PEGMA OMe; Mn = 300 g mol−1), styrene (≥99%) and 4-vinylbenzyl chloride (VBC; 90%) were obtained from Aldrich. After passing through inhibitor-removing columns (Sigma-Aldrich), they were stored at −20 °C. Biotin (99%) was obtained from Pure Bulk and used as received. The initiator benzoyl peroxide (BPO; 75%) was supplied by Aldrich and recrystallized from methanol. The tetrahydrofuran (THF) used in the synthesis was freshly distilled from sodium benzophenone. Toluene (analytical grade) was obtained from Aldrich and used as received. N,N-Dimethylformamide (DMF) was purchased from Panreac and dried over 3 Å molecular sieves (5% w/v) before use. All other reagent chemicals were obtained from Aldrich and/or Panreac and used without purification unless otherwise stated. The solvents used for column chromatography were of AR grade.Analysis of the various polymers was performed by gel permeation chromatography/size-exclusion chromatography (GPC/SEC) in THF (Scharlau, 99.9%, stabilized with BHT) using Varian columns (2 × PLgel 5 μ MIXED-C 300 × 7.5 mm + 1 × PLgel 5 μ GUARD 50 × 7.5 mm). The flow rate was maintained at 1 mL min−1 using an Agilent 1260 Infinity isocratic HPLC pump. Analyses were performed by injection of 50 μL of polymer solution (1 mg mL−1) in THF. Detection was performed using an Agilent 1260 Infinity Refractive Index Detector (RID). The molecular weight and polydispersity data were determined using the Agilent Chemstation software package, according to poly(methyl methacrylate) calibration. Proton nuclear magnetic resonance (1H-NMR) was also performed using a Varian Mercury 400 MHz apparatus.
Absolute toluene (99.7%) over molecular sieves used for thin film preparation was from Sigma-Aldrich. Four-inch silicon wafers were obtained from D+T MICROELECTRONICA, AIE at CNM (UAB-Bellaterra), and cleaned for 10 min in piranha solution before use: H2SO4 95–98% (Panreac Química S.A.U.) and 33% w/v H2O2 (BASF) at a 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 volume ratio. Caution: piranha acid is a strong oxidizer and a strong acid. It should be handled with extreme care, as it reacts violently with most organic materials. Millex® (33 mm) filters were from Millipore. Polymer solutions were spin-coated at room temperature using a Laurell Model WS-400A-6TFM/LITE, and analyzed by atomic force microscopy (AFM) using a Dimension 3100 AFM instrument (Veeco Instruments) equipped with a silicon AFM tip (Budgetsensors, spring constant 40 N m−1) and operated in tapping mode at room temperature in air. The topographic images obtained were processed with WSxM software (Nanotec Electronica).11
:3 volume ratio. Caution: piranha acid is a strong oxidizer and a strong acid. It should be handled with extreme care, as it reacts violently with most organic materials. Millex® (33 mm) filters were from Millipore. Polymer solutions were spin-coated at room temperature using a Laurell Model WS-400A-6TFM/LITE, and analyzed by atomic force microscopy (AFM) using a Dimension 3100 AFM instrument (Veeco Instruments) equipped with a silicon AFM tip (Budgetsensors, spring constant 40 N m−1) and operated in tapping mode at room temperature in air. The topographic images obtained were processed with WSxM software (Nanotec Electronica).11
Image thresholds were obtained manually from AFM height images and processed with Image J 1.44p freeware (http://imagej.nih.gov/ij). Nanodomain positions were used to obtain minimum interdomain distances using a custom-generated MATLAB code (The MATHWORKS, Inc.; ESI†). The interdomain distances were analyzed with OriginPro 8.5.0 SR1 (OriginLab Corp.). At least three images were computed per sample in two independent experiments.
Streptavidin conjugated to 10 nm colloidal gold from Streptomyces avidinii ∼2.5 A520 units per mL (streptavidin–AuNPs) was from Sigma-Aldrich. Deionized water (18 MΩ cm Milli-Q, Millipore) was used for rinsing the samples.
Synthesis of the reversible addition fragmentation chain transfer polymerization (RAFT) agent cumyl dithiobenzoate (CDB)
CDB was synthesized by adapting a previously described procedure.12 All glassware were dried at 120 °C overnight before use. Bromobenzene (0.42 mL, 0.63 g, 4 mmol) in dry THF (5 mL) was added to warm magnesium turnings (0.996 g, 41 mmol) activated with a catalytic amount of iodine in a three-necked 1 L round-bottom flask under a nitrogen atmosphere. The sudden appearance of brownish iodine color indicated the start of the reaction. The solution of bromobenzene (3.79 mL, 5.65 g, 36 mmol) in dry THF (15 mL) was added dropwise at such a rate as to maintain the reaction and the temperature around 60 °C. Upon completion of the addition, the mixture was left to stir and refluxed for 15 min. The empty dropping funnel was recharged with carbon disulfide (2.42 mL, 3.05 g, 40 mmol). An ice bath was applied to maintain the temperature at 0 °C while carbon disulfide was added. The mixture was stirred for about 30 min at 0 °C and 1 h at room temperature. The Grignard product was hydrolyzed with cold water, and 1 N HCL was added to dissolve the salts formed. Excess Mg turnings were removed by filtration, and after removal of THF under reduced pressure the reaction mixture was acidified to pH 1 by adding fuming HCl. The product was extracted with diethyl ether (3 × 150 mL). After drying the organic phase with MgSO4 and evaporating it to dryness under vacuum, the crude dithiobenzoic acid was obtained as dark-red brown oil. Dithiobenzoic acid, α-methylstyrene (6.44 mL, 5.85 g, 49.5 mmol) and carbon tetrachloride (23.4 mL) were mixed under nitrogen and heated at 70 °C overnight. After evaporation of the solvent and excess monomer using a rotary evaporator, the residue (10.7296 g) was purified by column chromatography on alumina activity III with n-hexane to obtain CDB as dark-purple oil (5.0370 g). The Rf value of TLC was 0.6 in n-hexane. Yield = 46%. Purity = 96% (HPLC). 1H-NMR (DMSO-d6, δ in ppm): 7.82–7.77 (m, 2H), 7.62–7.53 (m, 3H), 7.45–7.38 (m, 2H), 7.36–7.29 (m, 2H), 7.25–7.19 (m, 1H), 1.96 (s, 6H).RAFT polymerization of PEGMA OMe using CDB as the RAFT agent
12.38 mL (43.35 mmol) of PEGMA OMe, 33.7 mg (0.1 mmol) of CDB, and 3.0 mg (0.01 mmol) of BPO were placed in a 100 mL dry Schlenk tube and dissolved in 43.5 mL of toluene. The deep purple solution was degassed by five freeze–evacuate–thaw cycles. The reactor was sealed under vacuum and placed in a thermostatic oil bath at 80 °C to initiate the polymerization. At the end of the reaction (15 h), the glass tube was quenched in ice-cold water and opened, and the crude solution was diluted with dichloromethane (DCM), and precipitated in a large amount of hexane. The polymer was recovered by decanting off the organics. It was then redissolved in DCM and reprecipitated in hexane four more times. The PEGMA OMe homopolymer, P(PEGMA) 1, was dissolved in toluene before drying under reduced pressure at room temperature for at least 24 h until a constant weight was obtained (17.8747 g). GPC: total molecular weight (Mn) = 43000 Da; PDI = 1.33. 1H-NMR (acetone-d6, δ in ppm): 7.96–7.89 (m, Raft end-group), 7.53–7.46 (m, Raft end-group), 7.42–7.34 (m, Raft end-group), 7.34–7.26 (m, Raft end-group), 4.14 (s, 2H, CO–OC![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 2), 3.75 (s, 2H, CO–O–C–C2–O), 3.70–3.56 (m, 10H, –O–CH2–C), 3.52 (s, 2H, –C2–O–CH3), 3.34 (s, 3H, O–C3), 2.17–1.79 (m, 2H, C2), 1.24–0.83 (m, 3H, C3).
2), 3.75 (s, 2H, CO–O–C–C2–O), 3.70–3.56 (m, 10H, –O–CH2–C), 3.52 (s, 2H, –C2–O–CH3), 3.34 (s, 3H, O–C3), 2.17–1.79 (m, 2H, C2), 1.24–0.83 (m, 3H, C3).
RAFT polymerization of styrene using P(PEGMA) as the macro-RAFT agent
The preparation of the brush-type amphiphilic diblock copolymer poly(PEGMA)-b-polystyrene (P(PEGMA)-b-PS) involved two consecutive RAFT polymerizations: (1) synthesis of P(PEGMA) 1via RAFT polymerization of PEGMA OMe using CDB as the RAFT agent and BPO as the free-radical initiator (as described above), and (2) synthesis of a diblock copolymer, P(PEGMA)-b-PS, via RAFT polymerization of styrene using 1 as the macroRAFT agent and BPO as the free-radical initiator.3.5210 g of P(PEGMA) 1 (Mn = 40000 Da, PDI 1.41, 0.0826 mmol), 3.22 mL of styrene (28.10 mmol), and 2.0 mg (0.01 mmol) of BPO were placed in a 100 mL dry Schlenk tube and dissolved in 15.4 mL of toluene. The deep purple solution was degassed by five freeze–evacuate–thaw cycles. The reactor was sealed under vacuum and placed in a thermostatic oil bath at 80 °C to initiate the polymerization. At the end of the reaction (24 h), the glass tube was placed in ice-cold water, opened and the reaction crude was diluted with dichloromethane (DCM) and precipitated in a large amount of hexane. The polymer was recovered by decanting off the organics. It was then redissolved in DCM and reprecipitated in hexane. The polymer, P(PEGMA)-b-PS was dissolved in DCM and evaporated and dried under reduced pressure at room temperature for at least 24 h until a constant weight was obtained (1.0600 g). GPC: Mn = 52300 Da; PDI = 1.45. P(PEGMA):PS = 77:23. 1H-NMR (acetone-d6, δ in ppm): 7.96–7.81 (m, Raft end-group), 7.64–7.52 (m, Raft end-group), 7.47–6.37 (m, 1.5 H, H arom. S), 4.15 (s, 2H, CO–OC2), 3.74 (s, 2H, CO–O–C–C2–O), 3.70–3.57 (m, 12H, –O–CH2–C), 3.52 (s, 2H, –C2–O–CH3), 3.34 (s, 3H, O–C3), 2.08–1.82 (m, 2H, C2), 1.75–1.22 (m, 0.5H, C2), 1.19–0.78 (m, 3H, C3).
P(PEGMA)-b-PS thin film preparation
10.0 mg of P(PEGMA)-b-PS (Mn = 52300 Da, %PS = 23, PDI = 1.45) was dissolved in 2 mL of toluene for 2 h at room temperature under magnetic stirring. The resulting 5 mg mL−1 solution was filtered and spin-coated at 3000 rpm for 40 s onto 2 × 2 cm silicon wafers previously cleaned with piranha solution. The thin films obtained were analyzed by AFM without any further treatment.
RAFT polymerization of styrene and VBC using P(PEGMA) as the macro-RAFT agent
The procedures used for the block copolymerization of styrene and VBC were similar to those used for the RAFT polymerization of PEGMA OMe. 5.75 mL of styrene (50.18 mmol), 0.37 mL of VBC (2.63 mmol), 2.0 mg of BPO (0.01 mmol) and 3.5395 g of P(PEGMA) 1 (Mn = 43000 Da, 0.08260 mmol) were dissolved in 41.8 mL of toluene in a 100 mL dry Schlenk tube under stirring. The homogeneous solution was degassed by five freeze–evacuate–thaw cycles. The glass tube was then sealed under vacuum. Polymerization was carried out at 80 °C for 24 h. At the end of the polymerization reaction, the glass tube was placed in ice-cold water and opened, and the reaction crude was diluted with DCM and precipitated in a large amount of hexane. The polymer was recovered by decanting off the organics. It was then redissolved in DCM and reprecipitated in hexane four times. The polymer, poly(PEGMA)-b-poly(styrene-co-VBC) (P(PEGMA)-b-P(S-co-VBC)) 2 was dried under reduced pressure at room temperature for at least 24 h until a constant weight was obtained (3.6160 g). GPC: Mn = 56000 Da; PDI = 1.41. P(PEGMA):P(S-co-VBC) = 69:31 and styrene:VBC = 94:6. 1H-NMR (acetone-d6, δ in ppm): 7.94–7.82 (m, Raft end-group), 7.62–7.50 (m, Raft end-group), 7.44–6.39 (m, 2.3 H, H arom. S and VBC), 4.75–4.51 (m, 0.06 H, C2–Cl) −4.14 (s, 2H, CO–OC2), 3.75 (s, 2H, CO–O–C–C2–O), 3.70–3.57 (m, 12H, –O–CH2–C), 3.52 (s, 2H, –C2–O–CH3), 3.34 (s, 3H, O–C3), 2.13–1.82 (m, 2H, C2), 1.76–1.22 (m, 0.7H, C2), 1.24–0.87 (m, 3H, C3).
Derivatization of P(PEGMA)-b-P(S-co-VBC) 2 with biotin
Into a 25 mL round-bottom flask, P(PEGMA)-b-P(S-co-VBC) 2 (1.3842 g, 0.02470 mmol), biotin (243.8 mg, 1.0 mmol), and K2CO3 (221.2 mg, 1.6 mmol) were introduced. The flask was purged by means of 3 volumes per N2 cycle. Anhydrous DMF was added (15 mL), and the solution was stirred at 50 °C overnight. After cooling to room temperature, it was concentrated under vacuum. The crude product was dissolved in THF (25 mL), and the solution was filtered over celite and evaporated to dryness under vacuum (T < 40 °C). Filtration was repeated, affording 452.6 mg of the desired product poly(PEGMA)-b-poly(styrene-co-biotin styrene) (P(PEGMA)-b-P(S-co-bioS)) 3 as a yellowish polymer. GPC: Mn = 40000 Da; PDI = 1.33, with a P(PEGMA):P(S-co-bioS) = 70:30 and styrene:BioS = 95:5. 1H-NMR (acetone-d6, δ in ppm): 7.53–7.36 (m, 2.3H arom.), 5.21–4.92 (m, 0.06 H, CH2–O–CO–), 4.51–4.38 (m, 0.03H, CH–NH–CO), 4.14 (s, 2H, CO–OCH2), 3.75 (s, 2H, CO–O–C–CH2–O), 3.70–3.57 (m, 12H, –O–CH2–C), 3.52 (s, 2H, –CH2–O–CH3), 3.34 (s, 3H, O–CH3), 2.74–2.62 (m, 0.03H, S–CH2–), 2.46–2.33 (m, 0.06H, –CH2–O–CO–CH2–), 2.06–1.77 (m, 2H, CH2), 1.77–1.24 (m, 0.8H, CH2), 1.22–0.75 (m, 3H, CH3) (Raft group not detected by 1H NMR).
Selective streptavidin immobilization onto biotin-containing domains of P(PEGMA)-b-P(S-co-bioS) 3
10 mg of 3 (Mn = 40000 Da, PDI = 1.33) were dissolved in 2 mL of toluene for 2 h at room temperature under magnetic stirring. The resulting 5 mg mL−1 solution was filtered and spin-coated at 1500 rpm for 40 s onto 2 × 2 cm silicon wafers previously cleaned with piranha solution. The thin films obtained were analyzed by AFM without any further treatment and then incubated overnight at room temperature with streptavidin–AuNPs. The incubated films were washed in Milli-Q water and dried with compressed air. The resulting functionalized films were analyzed by AFM.
As a control of non-specific protein adsorption, P(PEGMA)-b-PS nanostructured films were also incubated with streptavidin–AuNPs.
Results and discussion
RAFT homopolymerization of PEGMA OMe
RAFT was selected for the homopolymerization of PEGMA OMe. RAFT is an extremely versatile, controlled, free-radical polymerization technique for the synthesis of well-defined polymer architectures of predictable molecular weight and narrow polydispersity.13 Thiocarbonylthio compounds such as CDB show effectiveness in the control of radical polymerization systems, in particular for methacrylate derivatives.14 PEGMA OMe was polymerized in the presence of CDB and BPO as the free-radical initiator in toluene (Scheme 1).15 In contrast with previous reports, where polar solvents or mixtures of them were used to perform RAFT reactions with methacrylic acid derivatives,16 toluene was our solvent of choice during diblock copolymer synthesis, in order to ensure the solubility of the final product in a solvent which facilitates thin film formation and phase separation.17 PEGMA OMe homopolymer (P(PEGMA)) 1 was obtained with a molecular weight of 43000 Da and a PDI of 1.33, achieving 62% of conversion. The product was characterized by GPC and NMR.
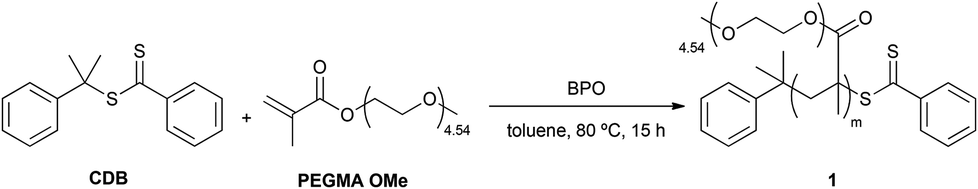 | ||
| Scheme 1 Synthesis of P(PEGMA) 1; reaction conditions: PEGMA OMe 300 (1.00 mol L−1), [CDB]0/[BPO]0 = 10:1, [PEGMA OMe 300]0/[CDB]0 = 350:1. | ||
An analogous procedure was developed using PEGMA OH instead of PEGMA OMe as the initial monomer. Although we were able to obtain oligomers with low PDI (PDI = 1.10), attempts to synthesize high molecular weight polymers failed due to gelation. The gelation products resulted were insoluble in the most common solvents (dichloromethane, THF, methanol, dioxane, dimethylsulphoxide, ethyl acetate, water, diethyl ether, and chloroform) and even in solvents whose molecular structure resembles the lateral monomer unit of PEGMA (1,2-dimethoxyethane and dyglime). Workup, purification and characterization could not be performed for these products.
Synthesis of the diblock/random copolymer P(PEGMA)-b-P(S-co-bioS)
Theoretical studies indicate that for the simplest case of a non-crystalline flexible coil AB diblock copolymer, the composition of the AB block (i.e. the volume fraction f of block A) controls the geometry of the macrodomain structure, while the size of the domains, typically in the range of tens of nanometers, is mostly influenced by the length of the blocks. The desired morphology (cylinders) can be obtained with high compositional asymmetry: when the volume fraction f of the A block (here a PS-based block) is 21–33%, A can form hexagonally packed cylinders within the B block matrix (P(PEGMA)).18Therefore, a diblock copolymer with a Mn of ∼50000 Da and a PDI ∼1 was designed, with a PS-based block Mn ∼ 10000 Da and a P(PEGMA) block Mn ∼ 40000 Da. For such a copolymer, it is possible to calculate that PS f = 20%, f being the volume fraction determined by GPC. Given that GPC is a chromatographic method in which polymer molecules in solution are separated on the basis of size and not weight, the f value can provide an approximation to the true volume fraction.
In an initial approach, a brush-type amphiphilic diblock copolymer P(PEGMA)-b-PS was synthesized by RAFT polymerization of styrene using 1 as the macroRAFT agent and BPO as the free-radical initiator (Scheme 2).
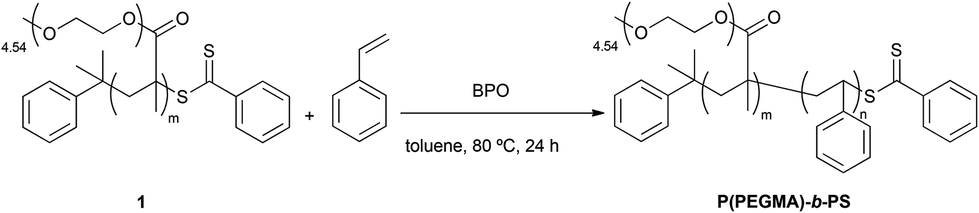 | ||
| Scheme 2 Synthesis of P(PEGMA)-b-PS; reaction conditions: styrene (1.50 mol L−1), [styrene]0/[P(PEGMA)]0 = 320:1, [P(PEGMA)]0/[BPO]0 = 11:1. | ||
Both 1H NMR and GPC confirmed that P(PEGMA)-b-PS was obtained with a molecular weight of 52300 Da and a PDI of 1.45, with polystyrene in 23% molar percentage. These characteristics are suitable to allow phase separation showing the cylindrical patterning of PS within the P(PEGMA) matrix. Therefore, a 5 mg mL−1 solution of P(PEGMA)-b-PS in toluene was filtered and spin-coated at 3000 rpm for 40 s onto silicon wafers previously treated with piranha solution. AFM was used to analyze the nanostructured film (Fig. 1).
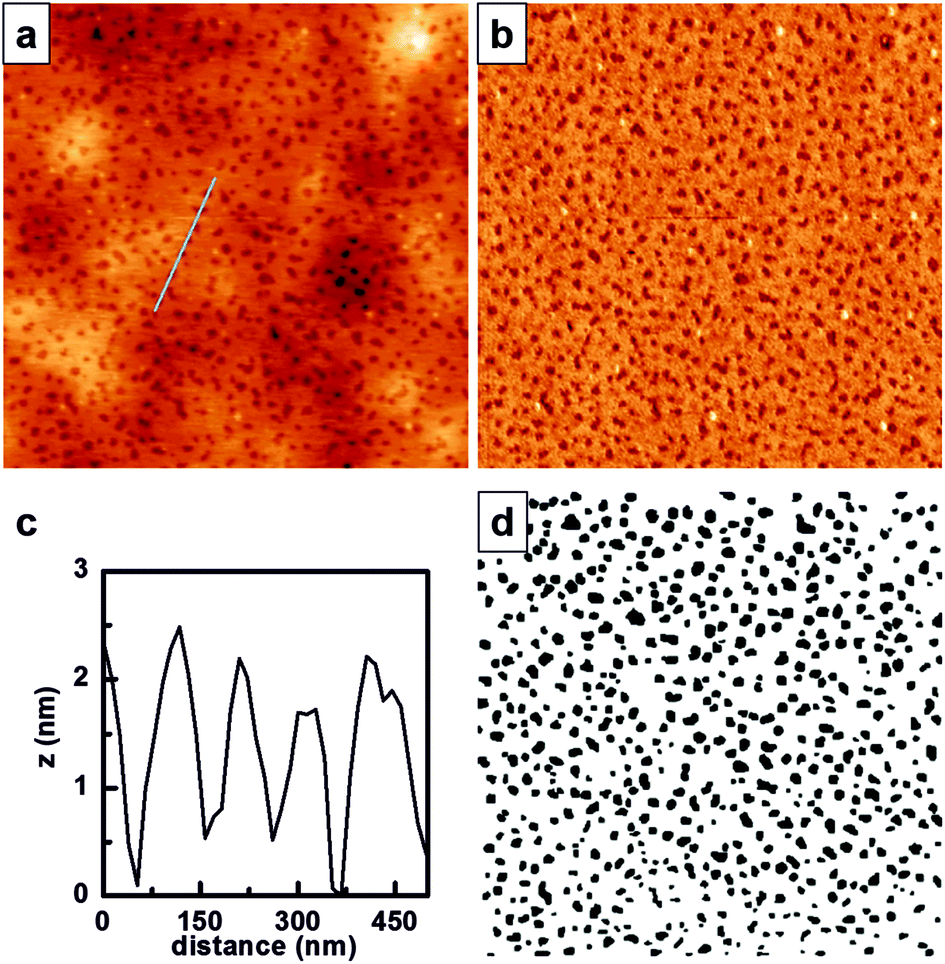 | ||
| Fig. 1 Micro-phase separation of P(PEGMA)-b-PS films on silicon surfaces, analyzed with AFM in air. (a) 3 × 3 μm representative AFM topographical image, and (b) the corresponding AFM phase image. (c) Cross-sectional profile indicated in (a), and (d) threshold image obtained from (a). | ||
P(PEGMA)-b-PS showed phase separation when spin-coated onto flat silicon surfaces (Fig. 1a), with PS domains appearing lower in topography and darker in the phase image (Fig. 1b). The cross-sectional profile indicated in Fig. 1a and shown in Fig. 1c reveals that the PS domains are buried ∼1.5 nm in the P(PEGMA) matrix. This is an inverted topography when compared to what is obtained for polystyrene-b-poly(2-hydroxyethyl methacrylate) (PS-b-PHEMA),10 and could be explained by the increased extension of the ethylene glycol chain in the polymethacrylate block. The threshold image (Fig. 1c) was obtained from AFM height images and further processed (Fig. S1, ESI†) to obtain the Feret diameter of each PS domain, which was estimated to be 74 ± 21 nm, and the minimum interdomain distance, which was 89 ± 20 nm.
In order to introduce biotin into the PS domains, the RAFT polymerization reaction described in Scheme 2 was modified using biotin-styrene (Scheme S2 and Table S1, ESI†). Although the synthesis of the biotin-styrene monomer was successful, the solubility of this molecule compromised its use as a co-monomer for preparing the final block/random copolymer. Therefore, an alternative synthetic approach was designed in which the diblock/random precursor copolymer P(PEGMA)-b-P(S-co-VBC) 2 was synthesized via random polymerization of VBC, using P(PEGMA) as the macro-RAFT agent (Scheme 3).
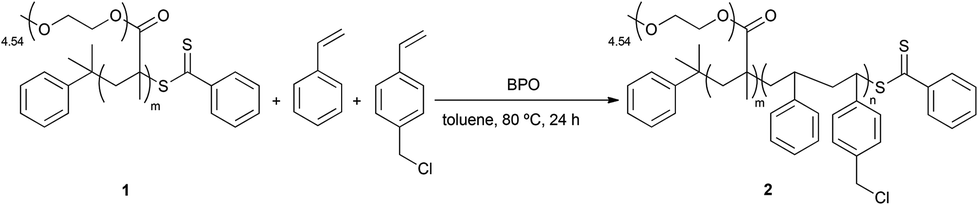 | ||
| Scheme 3 Synthesis of P(PEGMA)-b-P(S-co-VBC) 2; reaction conditions: styrene (1.20 mol L−1), [styrene + VBC]0/[P(PEGMA)]0 = 640:1, [P(PEGMA)]0/[BPO]0 = 10:1, VBC 5%, in toluene, 80 °C, 24 h. | ||
Both 1H NMR and GPC confirmed that 2 was obtained with a molecular weight of 56000 Da and a PDI of 1.41 and with P(PEGMA):P(S-co-VBC) = 69:31 and styrene:VBC = 94:6. The molar concentration of VBC was maintained below 10% in order to minimize the effects of steric hindrance on the final biotinylated domains.10
As shown in Scheme 4, the derivatization of 2 with biotin led to the formation of P(PEGMA)-b-P(S-co-bioS) 3 with a molecular weight of 40000 Da and a PDI of 1.33 with P(PEGMA):P(S-co-bioS) = 70:30 and styrene:BioS = 95:5. The presence of biotin was assessed by 1H NMR. The signal corresponding to CH2–Cl from 2 at 4.75–4.51 ppm disappeared and a new signal appeared, located at 5.21–4.92 and with a similar integration (0.05 H). This signal can be attributed to the formation of an ester between 2 and biotin (Fig. S12, ESI†).
 | ||
| Scheme 4 Synthesis of P(PEGMA)-b-P(S-co-bioS) 3; reaction conditions: biotin (0.07 mol L−1), [biotin]0/[P(PEGMA)-b-P(S-co-VBC)]0 = 40:1, [K2CO3]0/[biotin]0 = 1.6:1, anhydrous DMF, 50 °C, o/n. | ||
Selective streptavidin immobilization onto biotin-containing domains of P(PEGMA)-b-P(S-co-bioS)
The biotin–streptavidin complex with a dissociation constant of about 10−14 M is the strongest non-covalent interaction reported. It ensures rapid recognition in a highly sensitivity binding assay, thus minimizing the side reaction effects such as unspecific adsorption.19With the aim to selectively immobilize streptavidin onto the biotin-containing domains of 3, we first spin-coated a 5 mg mL−1 filtered solution of 3 in toluene at 1500 rpm for 40 s onto silicon wafers previously treated with piranha solution. Higher spin rates lead to non-homogeneous coating. The resulting surfaces were analyzed by AFM (Fig. 2a). Phase separation was observed with an estimated Feret diameter of 61 ± 12 nm for the biotin-containing PS domains, and a minimum inter-domain distance 117 ± 32 nm. Therefore, taking into account streptavidin dimensions (4.5 × 4.5 × 5.3 nm3),20 it is expected that streptavidin molecules can fit well into the PS domains.
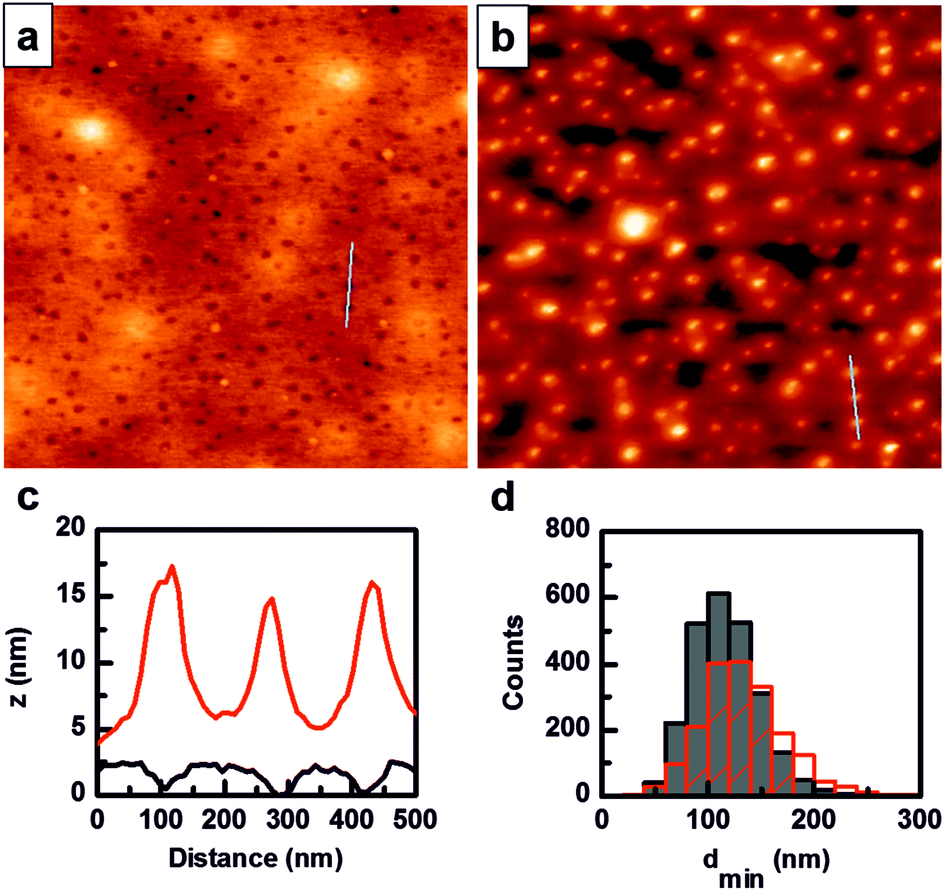 | ||
| Fig. 2 Selective immobilization of streptavidin–AuNPs on biotin-containing domains of P(PEGMA)-b-P(S-co-bioS) 3. 3 × 3 μm representative AFM topographical images of: (a) 3 spin-coated onto flat silicon surfaces; (b) 3 spin-coated onto flat silicon surfaces, after incubation with streptavidin–AuNPs; (c) cross-sectional profiles indicated in (a) (black line) and in (b) (red line); and (d) minimum inter-domain distance (dmin) superposed histograms of 3 before (solid graph) and after (dashed graph) incubation with streptavidin–AuNPs. | ||
The resulting nanostructured films were incubated with a suspension of streptavidin, labeled with streptavidin–AuNPs and imaged in AFM after washing with Milli-Q water. Fig. 2b shows uniformly distributed features that correlate with the original biotin-containing PS domains and scale with the size of streptavidin, partially buried in the PS domains, plus the 10 nm gold particle (cross-sectional profile in Fig. 2c). The superimposed minimum inter-domain distance histograms obtained before and after streptavidin–AuNP incubation confirmed the good correlation between the distribution of the biotin-containing PS domains and the localization of streptavidin. As a control, P(PEGMA)-b-PS without biotin nanostructured films were also incubated with streptavidin–AuNPs. After washing these films with Milli-Q water, the AFM images showed that the pattern was no longer visible. P(PEGMA)-b-PS without biotin nanostructured films proved to be unstable when exposed to protein solution. This observation could be attributed to the different water affinities of the PS and P(PEGMA) blocks, and the extensive swelling of the PEG block.21
Conclusions
In summary, here we describe the complete synthesis of P(PEGMA)-b-P(S-co-bioS) diblock copolymer 3. The modular approach followed allows for facile modification of the characteristics of the final copolymer. In this particular case, 3 is capable of forming thin films that self-segregate into cylindrical nano-domains of PS, which contain less than a 10% molar of biotin, within a non-fouling matrix of P(PEGMA).Protein nanopatterning has been achieved on thin films of 3 by selective immobilization of streptavidin into the biotin-containing domains through molecular recognition, with no unspecific adsorption within the P(PEGMA) matrix. We believe that the synthetic approach reported herein is suitable for the production of large-scale protein nanoarrays based on the self-assembly of BCPs and the biotin–streptavidin molecular recognition. The biotin–streptavidin pair provides more sensitive protein immobilization regarding the protein structure and function and fixes the protein orientation, thus making our platform suitable for extension to a range of applications that involve the nanopatterning of other biotinylated proteins.
Acknowledgements
This work was supported by the Networking Biomedical Research Center in Bioengineering, Biomaterials and Nanomedicine (CIBER-BBN), Spain. CIBER-BBN is an initiative funded by the VI National R&D&i Plan 2008–2011, Iniciativa Ingenio 2010, Consolider Program, CIBER Actions, and the Instituto de Salud Carlos III, with the support of the European Regional Development Fund. This work has been financially supported by the Commission for Universities and Research of the Department of Innovation, Universities, and Enterprise of the Generalitat de Catalunya (2014 SGR 1442). This work was funded by the projects OLIGOCODES (MAT2012-38573-C02) and MINAHE5 (TEC2014-51940-C2-2-R), awarded by the Spanish Ministry of Economy and Competitiveness. Financial support from the European Union through the GLAM project (Grant Agreement-634928) under the Horizon 2020 Program is gratefully acknowledged.Notes and references
- K. B. Lee, E. Y. Kim, C. A. Mirkin and S. M. Wolinski, Nano Lett., 2004, 4, 1869 CrossRef CAS; J. Vörös, T. Blättler and M. Textor, MRS Bull., 2005, 30, 202 CrossRef.
- V. Vogel and M. Sheetz, Nat. Rev. Mol. Cell Biol., 2006, 7, 265 CrossRef CAS PubMed; V. C. Hirschfeld-Warneken, M. Arnold, A. Cavalcanti-Adam, M. Lopez-García, H. Kessler and J. P. Spatz, Eur. J. Cell Biol., 2008, 87, 743 CrossRef PubMed; M. Théry, J. Cell Sci., 2010, 123, 4201 CrossRef PubMed.
- A. Biebricher, A. Paul, P. Tinnefeld, A. Golzhauser and M. Sauer, J. Biotechnol., 2004, 112, 97 CrossRef CAS PubMed.
- A. Tinazli, J. Piehler, M. Beuttler, R. Guckenberger and R. Tampé, Nat. Nanotechnol., 2007, 2, 220 CrossRef CAS PubMed; S. Oberhansl, M. Hirtz, A. Lagunas, R. Eritja, E. Martínez, H. Fuchs and J. Samitier, Small, 2012, 8, 541 CrossRef PubMed; S. Oberhansl, A. G. Castaño, A. Lagunas, E. Prats-Alfonso, M. Hirtz, F. Albericio, H. Fuchs, J. Samitier and E. Martinez, RSC Adv., 2014, 4, 56809 RSC.
- M. Arnold, E. A. Cavalcanti-Adam, R. Glass, J. Blummel, W. Eck, M. Kantlehner, H. Kessler and J. P. Spatz, ChemPhysChem, 2004, 5, 383 CrossRef CAS PubMed.
- K. H. A. Lau, J. Bang, D. H. Kim and W. Knoll, Adv. Funct. Mater., 2008, 18, 3148 CrossRef CAS; J. Zemla, M. Lekka, J. Raczkowska, A. Bernasik, J. Rysz and A. Budkowski, Biomacromolecules, 2009, 10, 2101 CrossRef PubMed; D. Liu, C. A. C. Abdullah, R. P. Sear and J. L. Keddie, Soft Matter, 2010, 6, 5408 RSC; J. Racine, H. Cheradame, Y. S. Chu, J. P. Thiery and I. Rodriguez, Biotechnol. Bioeng., 2011, 108, 983 CrossRef PubMed.
- B. T. Houseman, J. H. Huh, S. J. Kron and M. Mrksich, Nat. Biotechnol., 2002, 20, 270 CrossRef CAS PubMed; J. Groll, E. V. Amirgoulova, T. Ameringer, C. D. Heyes, C. Rocker, G. U. Nienhaus and M. Möllers, J. Am. Chem. Soc., 2004, 126, 4234 CrossRef PubMed.
- P. A. George, M. R. Doran, T. I. Croll, T. P. Munro and J. J. Cooper White, Biomaterials, 2009, 30, 4732 CrossRef CAS PubMed.
- L. Shen, A. Garland, Y. Wang, Z. Li, C. W. Bielawski, A. Guo and X.-Y. Zhu, Small, 2012, 20, 3169 CrossRef PubMed.
- I. C. Reynhout, G. Delaittre, H.-C. Him, R. J. M. Nolte and J. J. L. M. Cornelissen, J. Mater. Chem. B, 2013, 1, 3026 RSC.
- I. Horcas, R. Fernández, J. M. Gómez-Rodríguez, J. Colchero, J. Gómez-Herrero and A. M. Baro, Rev. Sci. Instrum., 2007, 78, 013705 CrossRef CAS PubMed.
- T. P. Le, G. Moad, E. Rizzardo and S. H. Thang, PCT Int. Appl, WO9801478, 1998 Search PubMed.
- D. J. Keddie, Chem. Soc. Rev., 2014, 43, 496 RSC.
- J. Chiefari, Y. K. (Bill) Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Lee, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559 CrossRef CAS.
- Z. Cheng, X. Zhu, E. T. Kang and K. G. Neoh, Langmuir, 2005, 21, 7180 CrossRef CAS PubMed; D. Fournier, R. Hoogenboom, H. M. L. Thijs, R. M. Paulus and U. S. Schubert, Macromolecules, 2007, 40, 915 CrossRef; N. A. A. Rossi, Y. Zou, M. D. Scott and J. N. Kizhakkedathu, Macromolecules, 2008, 41, 5272 CrossRef.
- D. Rinaldi, T. Hamaide, C. Graillat, F. D'Agosto, R. Spitz, S. Georges, M. Mosquet and P. Maitrasse, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3045 CrossRef CAS; W. Zhao, G. Gody, S. Dong, P. B. Zetterlund and S. Perrier, Polym. Chem., 2014, 5, 6990 RSC.
- K. E. Strawhecker and S. K. Kumar, Macromolecules, 2001, 34, 4669 CrossRef CAS; I. W. Hamley, Prog. Polym. Sci., 2009, 34, 1161 CrossRef.
- I. W. Hamley, Prog. Polym. Sci., 2009, 34, 1161 CrossRef CAS.
- A. Lagunas, J. Comelles, E. Martínez and J. Samitier, Langmuir, 2010, 26, 14154 CrossRef CAS PubMed.
- A. Lagunas, J. Comelles, S. Oberhansl, V. Hortigüela, E. Martínez and J. Samitier, Nanomedicine, 2013, 9, 694 CrossRef CAS PubMed.
- Z. Lin, D. H. Kim, X. Wu, L. Boosahda, D. Stone, L. LaRose and T. P. Russell, Adv. Mater., 2002, 14, 1373 CrossRef CAS; L. Shen and X.-Y. Zhu, Langmuir, 2011, 27, 7059 CrossRef PubMed; L. Shen, J. Zhu and H. Liang, J. Chem. Phys., 2015, 142, 101908 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental data, calculation of nanodomain characteristics from AFM images and 1H NMR and GPC spectra of the different products synthesized. See DOI: 10.1039/c5py01601k |
| This journal is © The Royal Society of Chemistry 2016 |