DOI:
10.1039/C5PY01407G
(Paper)
Polym. Chem., 2016,
7, 198-211
Electrochemical synthesis of electrochromic polycarbazole films from N-phenyl-3,6-bis(N-carbazolyl)carbazoles†
Received
2nd September 2015
, Accepted 20th October 2015
First published on 22nd October 2015
Abstract
Two compounds with a tri-carbazole (3Cz) structure, namely 3,6-di(carbazol-9-yl)-N-(4-nitrophenyl)carbazole (NO2-3Cz) and 3,6-di(carbazol-9-yl)-N-(4-aminophenyl)carbazole (NH2-3Cz), were synthesized and electropolymerized into robust polymer films on the electrode surface in an electrolyte solution via the oxidative coupling reactions. The electro-generated polymer films exhibited reversible electrochemical oxidation processes, with significant electrochromic behaviour. The colour of P(NO2-3Cz) films changed from the pale yellow neutral state to yellow-green as a radical cation and then to blue when fully oxidized. Upon oxidation, the colour of P(NH2-3Cz) films changed from colourless to pale green and finally to blue. Furthermore, we also synthesized and characterized some N-phenylcarbazoles with different substituents on the phenyl group. Based on a comparative study, the possible electropolymerization mechanisms of the NO2-3Cz and NH2-3Cz monomers are suggested; that of the former is through carbazole–carbazole coupling, and the latter is through both carbazole–carbazole and NH2–carbazole coupling.
Introduction
Electrochromism (EC) refers to the reversible electromagnetic absorbance/transmittance and color change resulting from the oxidation or the reduction of the material in response to an externally applied potential by electrochemical means.1 This functionality is of great interest for a wide range of applications, including smart windows,2 memory elements,3 electrochromic displays,4 and camouflage materials.5 Recent high-profile commercialization of electrochromic materials includes the Boeing 787 Dreamliner windows manufactured by Gentex.6 The most studied electrochromic materials include transition metal oxides,7 inorganic coordination complexes,8 small organic molecules,9 and conjugated polymers.10 Among the available electrochromic materials, π-conjugated polymers have attracted tremendous attention because of combining several advantages such as mechanical flexibility, high optical contrast ratios, long-term redox stability and easy color tuning through structural control.11 In addition, triarylamine-containing condensation-type high performance polymers such as aromatic polyamides and polyimides have been developed as a new family of electrochromic materials.12–14
Polymers containing carbazole moieties in the main chain or side chain have attracted much attention because of their unique properties, which allow various optoelectronic applications such as photoconductive, electroluminescent, electrochromic, and photorefractive materials.15 Carbazole-based derivatives simultaneously possess carrier-transport properties and sufficiently high triplet energy levels, and therefore, oligocarbazoles or carbazole dendrons via 3(6), 9-linkages have been used as effective host materials for phosphorescent metal complexes.16 Carbazole can be substituted or polymerized either at the 3- and 6-positions or 2- and 7-positions and a wide variety of alkyl and aryl chains can be added to the nitrogen atom without altering the planar conformation of the resulting polymers. Using different synthetic strategies and substitution patterns, the physico-chemical properties of poly(3,6-carbazole)s and poly(2,7-carbazole)s can be fine-tuned, leading to high performance materials for a number of electronic applications.17 On the other hand, the electrochemical oxidation of carbazole and N-substituted derivatives was first studied by Ambrose and Nelson.18a 76 ring-substituted carbazoles were further studied systematically for their substituent effects by using electrochemical and spectroscopic techniques.18b For the N-phenylcarbazoles (NPCs) with both the 3 and 6 carbazole ring positions unprotected, these compounds underwent an initial one-electron oxidation to generate a very reactive cation radical; two of these were then coupled at the 3 positions to yield an N,N′-diphenyl-3,3′-bicarbazyl. This carbazole oxidative dimerization reaction has been employed efficiently to fabricate electroactive polymeric films for potential applications in electronic and optoelectronic devices.19 Compared with the chemical routes, electrochemical polymerization has several advantages in the synthesis of conducting polymer films, such as one-step polymer film formation with good mechanical properties on the electrode.20 This not only enlarges the scope of candidate polymers, but also omits the procedure of film casting.
In this work, we have synthesized a series of NPCs (Scheme 1) in which the para position of the N-phenyl group has been substituted with various groups such as cyano, carboxyl, nitro, and amino groups. Their electrochemistry and electropolymerization and the substituent effect on the redox properties of carbazoles were investigated. We found that the oxidized forms of NPC, CN-NPC, COOH-NPC, and NO2-NPC underwent dimerization to the corresponding biscarbazoles, whereas NH2-NPC could undergo electrochemical polymerization to form polymer thin films on the electrode surface via a electropolymerization mechanism likely similar to that of polyaniline. Then two N-phenyl-3,6-bis(N-carbazolyl)carbazoles, i.e., NO2-3Cz and NH2-3Cz as shown in Scheme 2, were prepared and the P(NO2-3Cz) and P(NH2-3Cz) films were prepared directly on the electrode surface via electrochemical oxidative coupling of carbazole–carbazole and amine–carbazole. The electrochromic properties of the resulting polycarbazole films were evaluated by spectroelectrochemical and electrochromic switching studies.
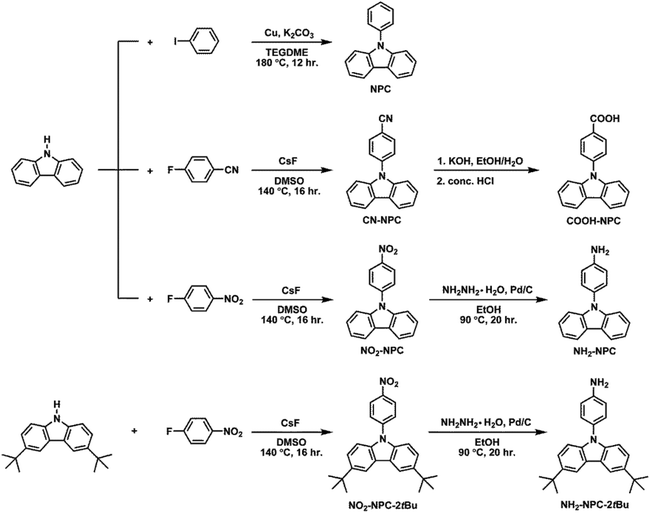 |
| | Scheme 1 Synthetic routes to N-phenylcarbazoles. | |
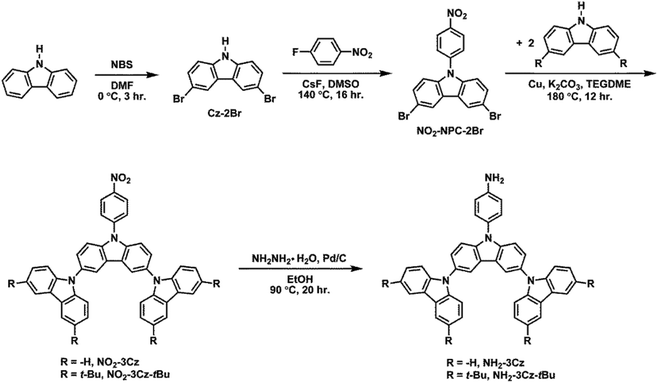 |
| | Scheme 2 Synthetic routes to 3,6-bis(N-carbazolyl)-N-phenylcarbazoles. | |
Experimental section
Materials and instrumentation
N-Phenylcarbazole (NPC), N-(4-nitrophenyl)carbazole (NO2-NPC), N-(4-aminophenyl)carbazole (NH2-NPC), 3,6-di-tert-butyl-9-(4-nitrophenyl)carbazole (NO2-NPC-2tBu) and 3,6-di-tert-butyl-9-(4-aminophenyl)carbazole (NH2-NPC-2tBu) were synthesized according to literature methods.21N-(4-Carboxyphenyl)carbazole (COOH-NPC) was synthesized by the cesium fluoride (CsF)-mediated N-arylation reaction of carbazole with p-fluorobenzonitrile, followed by the alkaline hydrolysis of the intermediate nitrile compound N-(4-cyanophenyl)carbazole (CN-NPC). The synthetic route and chemical structures of the N-phenylcarbazole derivatives are illustrated in Scheme 1. The functionalities of all the synthesized compounds were confirmed by the IR analysis shown in Fig. S1.† The synthetic details and electrochemical properties (Fig. S2†) of model compounds 3,6-di(3,6-di-tert-butylcarbazole-9-yl)-N-(4-nitrophenyl)carbazole (NO2-3Cz-tBu) and 3,6-Di(3,6-di-tert-butylcarbazole-9-yl)-N-(4-aminophenyl)carbazole (NH2-3Cz-tBu) are included in the ESI.† All other reagents and solvents were used as received from commercial sources.
Infrared (IR) spectra were recorded on a Horiba FT-720 FT-IR spectrometer. 1H and 13C NMR spectra were recorded on a Bruker Avance III HD-600 MHz NMR spectrometer with tetramethylsilane (TMS) as an internal standard. Electrochemical measurements were performed with a CH Instruments 750A electrochemical analyzer. The polymers were electropolymerized from 5 × 10−4 M monomers in a 0.1 M Bu4NClO4/dichloromethane solution via repetitive cycling at a scan rate of 50 mV s−1. Thin films on ITO-glass obtained by two and five cycles between 0 V and 1.5 V at a scan rate of 50 mV s−1 were used for UV-visible absorption measurements and spectroelectrochemistry studies, respectively. Voltammograms are presented with the positive potential pointing to the left and with increasing anodic currents pointing downwards. Cyclic voltammetry (CV) was conducted with the use of a three-electrode cell in which ITO (polymer films area about 0.8 cm × 1.25 cm) was used as a working electrode. A platinum wire was used as an auxiliary electrode. All cell potentials were obtained with the use of a home-made Ag/AgCl, KCl (sat.) reference electrode. Ferrocene was used as an external reference for calibration (+0.48 V vs. Ag/AgCl). Spectroelectrochemistry analyses were carried out with an electrolytic cell, which was composed of a 1 cm cuvette, ITO as a working electrode, a platinum wire as an auxiliary electrode, and Ag/AgCl as a reference electrode. Absorption spectra in the spectroelectrochemical experiments were recorded with an Agilent 8453 UV-visible diode array spectrophotometer. Colorimetric data of the electrochromic films were obtained on an Admesy Brontes colorimeter.
Monomer synthesis
3,6-Di(carbazol-9-yl)-N-(4-nitrophenyl)carbazole (NO2-3Cz).
A mixture of 3,6-dibromo-N-(4-nitrophenyl)carbazole (0.03 mol, 13.38 g), carbazole (0.063 mol, 10.53 g), copper powder (0.063 mol, 4 g), potassium carbonate (0.065 mol, 8.98 g) and triethyleneglycol dimethyl ether (TEGDME) (35 mL) was stirred under a nitrogen atmosphere at 180 °C for 12 h. The reaction mixture was poured into an excess of methanol to precipitate the product. The yellow precipitate was collected by filtration and washed thoroughly with methanol. The crude product was filtered and re-precipitated three times from the DMF/methanol solvent pair to afford 11.69 g of yellow powder. FT-IR (KBr) (Fig. S3†): 1500, 1322 cm−1 (–NO2 stretch). 1H NMR (600 MHz, CDCl3, δ, ppm) (Fig. S4†): 7.27 (t, J = 6.0 Hz, 4H, Hb), 7.38 (m, 8H, Hc + Hd), 7.64 (d, J = 8.4 Hz, 2H, He), 7.70 (d, J = 8.4 Hz, 2H, Hf), 7.93 (d, J = 8.4 Hz, 2H, Hh), 8.14 (d, J = 7.8 Hz, 4H, Ha), 8.27 (s, 2H, Hg), 8.56 (d, J = 8.4 Hz, 2H, Hi). 13C NMR (150 MHz, CDCl3, δ, ppm) (Fig. S5†): 109.56 (C5), 111.15 (C9), 119.91 (C3), 119.96 (C12), 120.38 (C2), 123.28 (C1), 124.83 (C11), 125.84 (C15), 125.98 (C4), 126.70 (C8), 127.06 (C14), 130.52 (C13), 131.57 (C7), 139.63 (C10), 141.60 (C6), 146.49 (C16).
3,6-Di(carbazol-9-yl)-N-(4-aminophenyl)carbazole (NH2-3Cz).
In the 1000 mL three-neck round-bottomed flask equipped with a stirring bar, 6.19 g (0.01 mol) of the nitro compound NO2-3Cz and 0.05 g of 10% Pd/C were dissolved/suspended in 600 mL ethanol. The suspension solution was heated to reflux under a nitrogen atmosphere, and 1.3 mL of hydrazine monohydrate was added to the reaction mixture. After a further 20 h of reflux, the solution was filtered to remove Pd/C, and the filtrate was cooled under a nitrogen flow to precipitate colorless powder. The product was collected by filtration and dried in vacuo at 80 °C to give 5.06 g of NH2-3Cz as white fine powder in 86% yield. FT-IR (KBr) (Fig. S3†): 3472, 3373 cm−1 (–NH2 stretch). 1H NMR (600 MHz, CDCl3, δ, ppm) (Fig. S6†): 3.83 (s, 2H, –NH2), 6.85 (d, J = 8.4 Hz, 2H, Hi), 7.26 (m, 4H, Hb), 7.37 (m, 8H, Hc + Hd), 7.39 (d, J = 8.4 Hz, 2H, Hh), 7.54 (m, 4H, Hf + He), 8.13 (d, J = 7.2 Hz, 2H, Ha), 8.23 (s, 2H, Hg). 13C NMR (150 MHz, CDCl3, δ, ppm) (Fig. S7†): 109.74 (C5), 111.32 (C9), 116.01 (C15), 119.63 (C3 + C12), 120.26 (C2), 123.12 (C1), 123.53 (C11), 125.86 (C4), 126.07 (C8), 127.31 (C12), 128.51 (C14), 129.91 (C7), 141.35 (C10), 141.86 (C6), 146.60 (C16).
Electrochemical polymerization
Electrochemical polymerization was performed with a CH Instruments 750A electrochemical analyzer. The polymers were synthesized from 5 × 10−4 M monomers in 0.1 M Bu4NClO4/dichloromethane (CH2Cl2) or acetonitrile (MeCN) solution via cyclic voltammetry repetitive cycling at a scan rate of 50 mV s−1 for ten cycles. The polymer was deposited onto the surface of the working electrode (ITO/glass surface, polymer film area about 0.8 cm × 1.25 cm), and the film was rinsed with plenty of acetone for the removal of unreacted monomer, inorganic salts and other organic impurities formed during the process.
Fabrication of electrochromic devices
Electrochromic polymer films were electrodeposited on the ITO-coated glass substrate by the electropolymerization method described above. A gel electrolyte based on poly(methyl methacrylate) (PMMA) (Mw: 120![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) and LiClO4 was plasticized with propylene carbonate to form a highly transparent and conductive gel. PMMA (1 g) was dissolved in dry MeCN (4 mL), and LiClO4 (0.1 g) was added to the polymer solution as the supporting electrolyte. Then propylene carbonate (1.5 g) was added as a plasticizer. The mixture was then gently heated until gelation. The gel electrolyte was spread on the polymer-coated side of the electrode, and the electrodes were sandwiched. Finally, an epoxy resin was used to seal the device.
000) and LiClO4 was plasticized with propylene carbonate to form a highly transparent and conductive gel. PMMA (1 g) was dissolved in dry MeCN (4 mL), and LiClO4 (0.1 g) was added to the polymer solution as the supporting electrolyte. Then propylene carbonate (1.5 g) was added as a plasticizer. The mixture was then gently heated until gelation. The gel electrolyte was spread on the polymer-coated side of the electrode, and the electrodes were sandwiched. Finally, an epoxy resin was used to seal the device.
Results and discussion
Monomer synthesis
3,6-Bis(N-carbazolyl)-N-phenylcarbazoles were synthesized starting from carbazole by a four-step reaction sequence as shown in Scheme 2. Cz-2Br was synthesized by bromination of carbazole with N-bromosuccinimide (NBS). Nucleophilic fluoro-displacement reaction of p-fluoronitrobenzene with Cz-2Br in the presence of cesium fluoride (CsF) resulted in NO2-NPC-2Br,22 which was subsequently condensed with carbazole and 3,6-di-tert-butylcarbazole, respectively, to NO2-3Cz and NO2-3Cz-tBu via the Ullmann reaction. The amino compounds NH2-3Cz and NH2-3Cz-tBu were prepared by the hydrazine Pd/C-catalyzed reduction of their precursor nitro compounds. FT-IR and NMR spectroscopic techniques were used to identify the structures of the synthesized compounds. The FT-IR spectra of NO2-3Cz and NO2-3Cz-tBu gave two characteristic bands at around 1500 and 1322–1337 cm−1 ascribed to the nitro groups (Fig. S3†). The N–H stretching absorptions of NH2-3Cz and NH2-3Cz-tBu occur at about 3472 and 3373 cm−1. NO2-3Cz-tBu and NH2-3Cz-tBu showed an additional aliphatic C–H stretching absorption at around 2955–2863 cm−1 due to the presence of tert-butyl groups. The 1H, 13C and two-dimensional (2-D) NMR spectra of NO2-3Cz and NH2-3Cz are compiled in Fig. S4–S7.† The 1H NMR spectra confirm that the nitro group has been completely transformed into amino groups by the high field shift of aromatic protons Hh and Hi and the resonance signals at around 3.84 ppm corresponding to the amino protons. Assignments of each carbon and proton assisted by the 2-D NMR spectra are also indicated in these spectra, and they are in good agreement with their proposed molecular structures.
Electrochemical properties
N-Phenylcarbazoles.
Fig. 1(a) shows the repetitive cyclic voltammetry (CV) diagrams of 5 × 10−4 M of NO2-NPC in 0.1 M Bu4NClO4/MeCN solution at a potential scan rate of 50 mV s−1. These four NPC derivatives exhibit similar CV behavior. For the first positive scan, a sharp oxidation peak at Epa = 1.37–1.53 V is observed. In the reverse scan, two reduction peaks are observed. In the second scan, a new oxidation wave at Epa = 1.09–1.20 V appears, which indicates that the carbazole radical cations are involved in very fast electrochemical reactions that produce a carbazole dimer via the ring–ring coupling reaction between the carbazole units [Scheme 3(a)].18 In the continuous scans, the new oxidation wave gradually grows; however, no significant increase in redox wave current density is observed. The biscarbazoles seem to be the major products during the oxidation processes of these NPCs, indicating that they are stable in the oxidized states. No polymer films grew up on the electrode surface. Similar results were observed for other NPCs such as NPC, CN-NPC, and COOH-NPC (see Fig. S8†). The results are similar to those found for other NPC analogs reported in the literature.23 The CV result of NH2-NPC is completely different [Fig. 1(b)]. In the first scan, two oxidation peaks at 1.00 V and 1.36 V are observed, which may be attributed to the oxidation of the aromatic primary amino group (aniline-like) and the carbazole unit, respectively. As the CV scan continued, two new oxidation peaks at Epa = +0.84 V and +1.18 V gradually grew and the redox wave current intensities increased. We also found that a polymer film was deposited on the electrode surface (as can be seen in the inset of Fig. 1(b)). We propose that the NH2–carbazole and carbazole–carbazole undergo oxidative coupling reactions simultaneously during the CV scanning. A possible reaction path is suggested in Scheme 3(b). The polymer films are insoluble in polar organic solvents like N-methyl-2-pyrrolidine (NMP) and even in concentrated sulfuric acid, but the film can be removed from the ITO-glass surface after immersing in NMP or concentrated sulfuric acid for a certain period of time. This insolubility of the polymer film may be caused by the fact that crosslinking reactions or tight packing of the polymer chains occurred during the electropolymerization process.
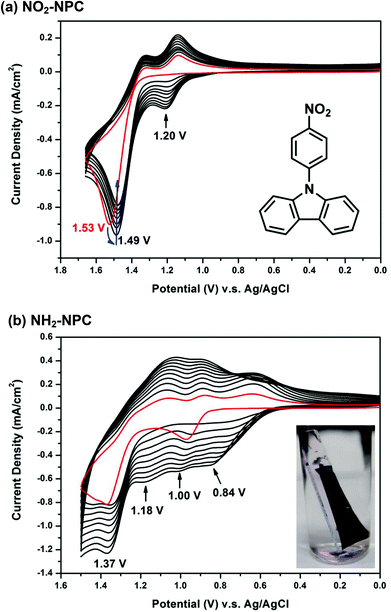 |
| | Fig. 1 Repetitive cyclic voltammograms of 5 × 10−4 M (a) NO2-NPC and (b) NH2-NPC in 0.1 M Bu4NClO4/MeCN solution at a scan rate of 50 mV s−1. The inset in (b) displays the escaped polymer film from the ITO glass surface by immersing the film-coated ITO glass in conc. H2SO4. | |
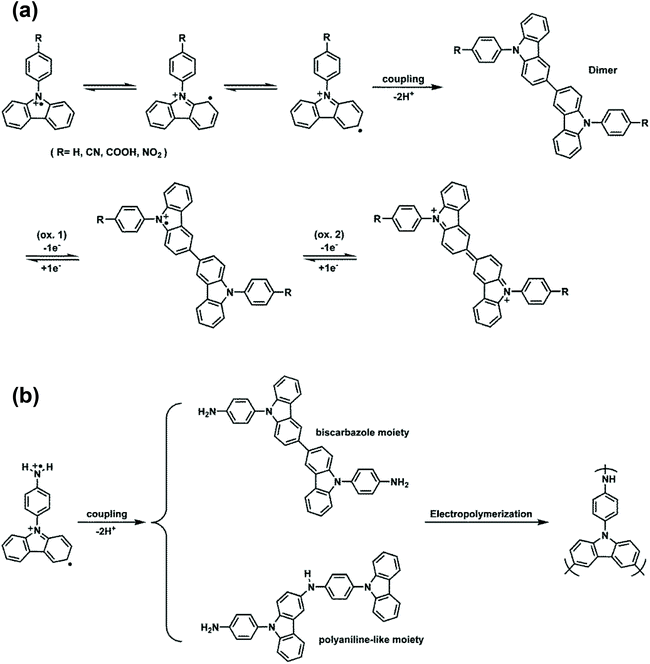 |
| | Scheme 3 (a) Electrochemical dimerization of NPCs (R = –H, –CN, –COOH, –NO2) and subsequent oxidation of the resulting biscarbazoles. (b) Electro-coupling and polymerization of NH2-NPC. | |
N-Phenyl-3,6-bis(N-carbazolyl)carbazoles.
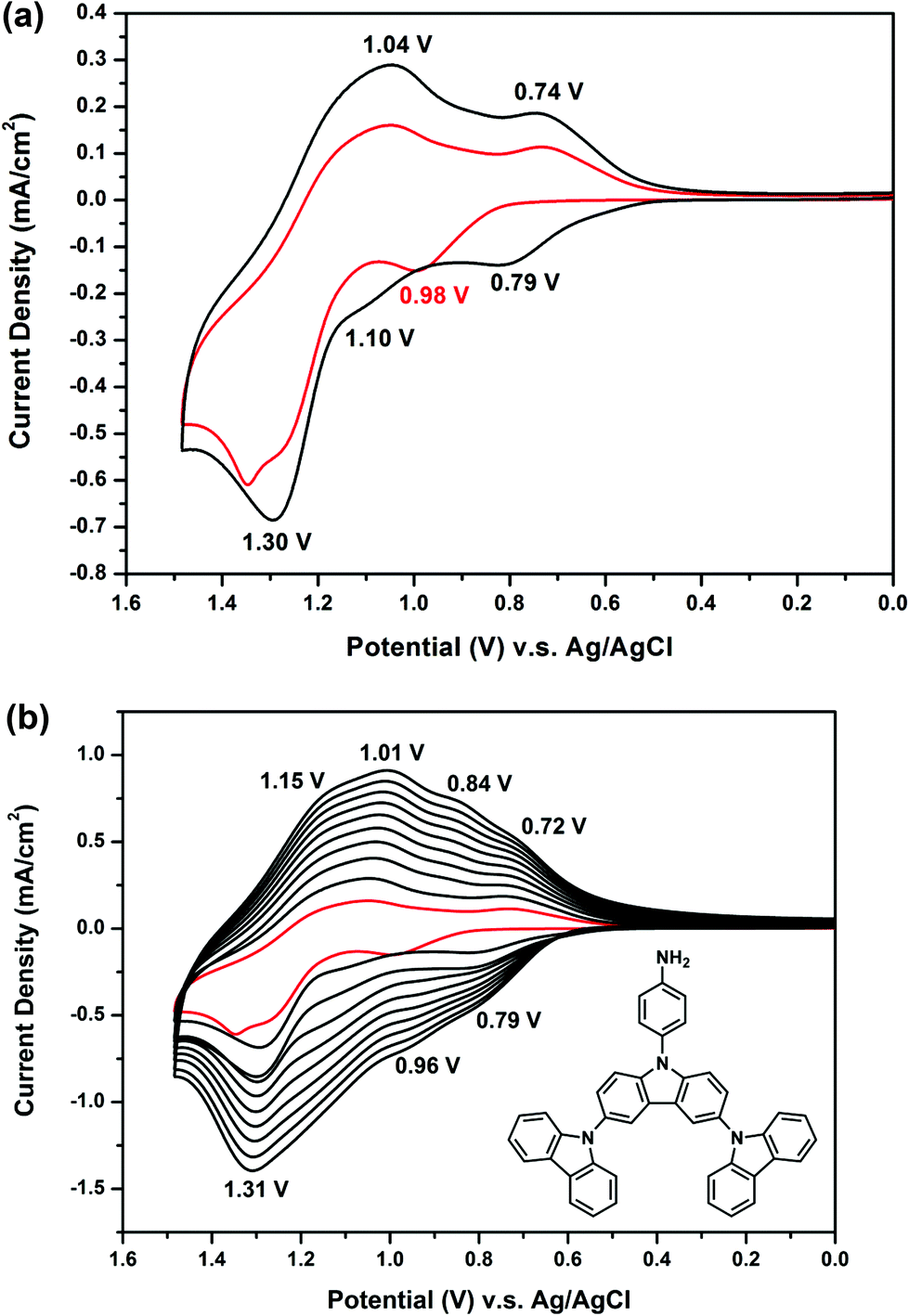
Fig. 2(a) presents the first and second CV scans of 5 × 10−4 M NO2-3Cz in 0.1 M Bu4NClO4/CH2Cl2via repetitive cycling at a potential scan rate of 50 mV s−1. In the first scan, only one oxidation peak at 1.46 V is observed, which can be attributed to the oxidation of carbazole. A new oxidation peak appears at about 1.10 V during the second scan, similar to that observed in most NPCs discussed earlier. The carbazole coupling reactions occurring in NO2-3Cz leads to polymerization. A possible polycarbazole structure may develop as that shown in Scheme 4(a). Thus, as the CV scan continued, the redox wave current densities increased and an obvious polymer film formed on the electrode surface [Fig. 2(b)]. The CV result of NH2-3Cz is similar to that of NH2-NPC. Fig. 3(a) displays the first two consecutive CV scans of NH2-3Cz. In the first scan, this compound displayed two oxidation peaks at 0.98 V and 1.30 V, which is attributed to the oxidation of the aniline-like structure and the carbazole unit, respectively. In the second scan, two new oxidation peaks appeared at 0.79 V and 1.10 V. It is known that the biscarbazole unit oxidized at a lower potential than the parent carbazole unit. Thus, we attribute the new oxidation wave at 1.10 V to the biscarbazole unit oxidation and that at 0.79 V to polyaniline-like unit oxidation [Scheme 4(b)]. After several CV cycles, individual oxidation peaks emerged to one broad peak, and the increase in the redox wave current densities implied that the amount of conducting polymers deposited on the electrode was increasing [Fig. 3(b)]. The polymer with polyaniline-like and biscarbazole units was successfully electrodeposited onto the ITO-glass electrode surface. The lower oxidation potential of NH2-3Cz compared to NO2-3Cz can be explained by the electron-donating effect of the amino group.
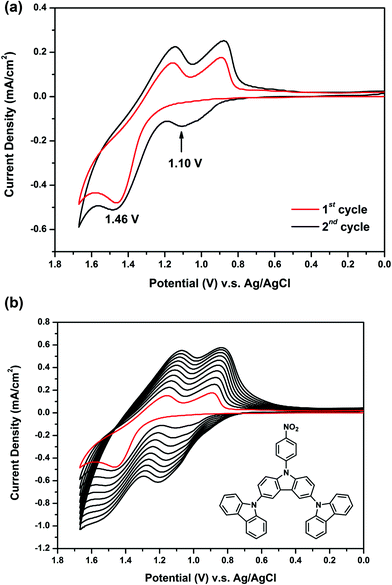 |
| | Fig. 2 Cyclic voltammograms and repeated potential scanning of 5 × 10−4 M NO2-3Cz in 0.1 M Bu4NClO4/CH2Cl2 at a scan rate of 50 mV s−1. | |
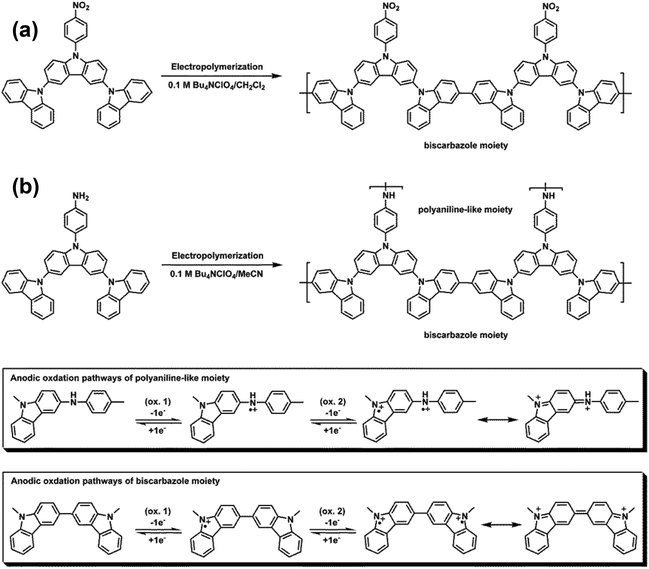 |
| | Scheme 4 Electropolymerization of NO2-3Cz and NH2-3Cz, together with proposed anodic oxidation pathways of the polyaniline-like moiety and biscarbazole moiety. | |
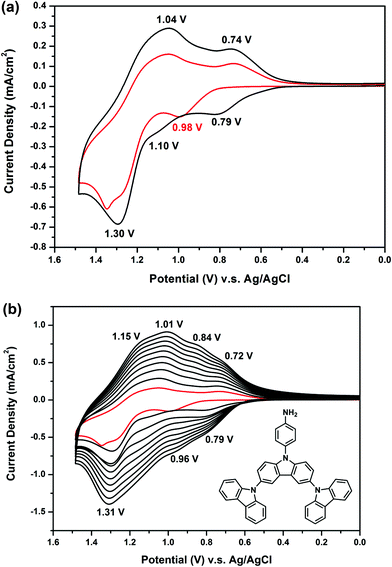 |
| | Fig. 3 Cyclic voltammograms and repeated potential scanning of 5 × 10−4 M NH2-3Cz in 0.1 M Bu4NClO4/MeCN at a scan rate of 50 mV s−1. | |
Optical and electrochemical properties of the polymer films
The representative UV-vis absorption spectra of monomers NO2-3Cz and NH2-3Cz in CH2Cl2 and the polymer films of P(NO2-3Cz) and P(NH2-3Cz) in the solid state on an ITO electrode are shown in Fig. 4. The spectra of the monomers show absorption bands with maximum peaks at 293 nm and absorption onsets at 458 and 365 nm, respectively. The polymer films of P(NO2-3Cz) and P(NH2-3Cz) show absorption maxima at 305 and 304 nm, respectively, and the wavelength at the absorption edge of the former is much longer than that of the latter possibly due to the charge-transfer complexing effect. The red-shift of absorption maximum and onset of the P(NH2-3Cz) film compared to the NH2-3Cz monomer implies an extended π-conjugation length.
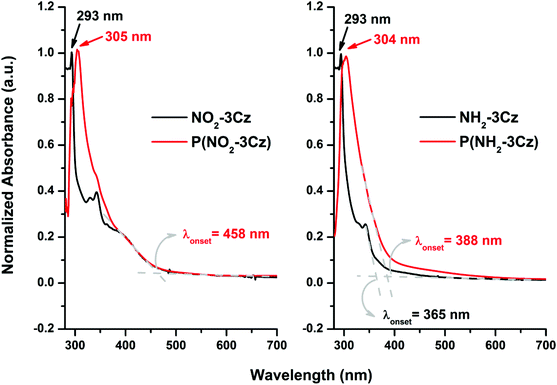 |
| | Fig. 4 UV-vis absorption spectra of NO2-3Cz and NH2-3Cz in CH2Cl2 and the P(NO2-3Cz) and P(NH2-3Cz) films on ITO-glass. | |
The electrochemical behavior of the electrodeposited polymer films was investigated by CV in a monomer-free 0.1 M Bu4NClO4/MeCN solution. The quantitative details are summarized in Table 1. The CV diagrams of the polymer films of P(NO2-3Cz) and P(NH2-3Cz) are shown in Fig. 5. There are two reversible oxidation redox couples at the half-wave potentials (E1/2) of 1.06 and 1.28 V for P(NO2-3Cz), and half-wave potentials of 1.04 and 1.24 V for P(NH2-3Cz). The oxidation half-wave potential of the P(NO2-3Cz) film started to oxidize at a higher potential (Eonset = 0.88 V) than P(NH2-3Cz) (Eonset = 0.71 V), attributed to the electronic effects arising from the electron-withdrawing properties of the nitro substituents. The P(NH2-3Cz) film showed only two redox waves, although three oxidation processes of the polyaniline-like and biscarbazole moieties were expected (Scheme 4). This might be attributed to the fact that the oxidation wave of the polyaniline-like structure and the first oxidation wave of the biscarbazole unit merged and became undistinguishable in the CV diagram. For comparative studies, the CV diagrams of the (NO2-NPC) dimer and the P(NH2-NPC) film in a monomer-free 0.1 M Bu4NClO4/MeCN solution were also recorded and illustrated in Fig. S9.† In general, the CV behaviour of the P(NH2-3Cz) film is similar to that of P(NH2-NPC), with a lower oxidation onset potential. However, the CV diagram of the P(NO2-3Cz) film is totally different from that of the (NO2-NPC) dimer. The latter showed a relatively higher oxidation potential and an irreversible CV behaviour. Fig. S10† displays the CV curves of the P(NO2-3Cz) and P(NH2-3Cz) at different scanning rates between 50 and 350 mV s−1 in 0.1 M Bu4NClO4/MeCN solution. The polymer films exhibited two reversible redox couples, attributed to the radical cation and dication states of biscarbazole, respectively (Scheme 4). In the scan rate dependence experiments for P(NO2-3Cz) and P(NH2-3Cz) films, both anodic and cathodic peak current values increase linearly with increasing scan rate, indicating that the electrochemical processes are reversible and not diffusion limited, and the electroactive polymer is well adhered to the working electrode (ITO glass) surface.
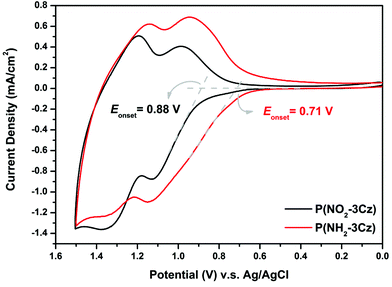 |
| | Fig. 5 Cyclic voltammograms of P(NO2-3Cz) and P(NH2-3Cz) films on the ITO-coated glass slide in monomer-free 0.1 M Bu4NClO4/MeCN solution at a scan rate of 50 mV s−1. | |
Table 1 Optical and electrochemical properties of the electrosynthesized polymers
| Polymers |
UV-vis absorption wavelengtha (nm) |
Oxidation potentialb (V) |
E
gc (eV) |
HOMOd (eV) |
LUMOd (eV) |
|
λ
max
|
λ
onset
|
E
onset
|
E
Ox11/2
|
E
Ox21/2
|
|
UV-vis absorption maximum and onset wavelengths for the polymer films.
Read from the first CV scans, in acetonitrile at a scan rate of 50 mV s−1 (versus Ag/AgCl).
Optical bandgap calculated from absorption edge of the polymer film: Eg = 1240/λonset.
The HOMO and LUMO energy levels were calculated from EOx11/2 values of CV diagrams and were referenced to ferrocene (4.8 eV relative to vacuum energy level; Eonset = 0.37 V; E1/2 = 0.44 V in acetonitrile). EHOMO = EOx11/2 + 4.8–0.44 (eV); ELUMO = EHOMO − Eg.
|
| P(NO2-3Cz) |
305 |
458 |
0.88 |
1.06 |
1.28 |
2.71 |
5.42 |
2.71 |
| P(NH2-3Cz) |
304 |
388 |
0.71 |
1.04 |
1.24 |
3.20 |
5.40 |
2.20 |
The energy levels of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) of the corresponding polymers were estimated from the Eox1/2 values. Assuming that the HOMO energy level for the ferrocene/ferrocenium (Fc/Fc+) standard is 4.80 eV with respect to the zero vacuum level, the HOMO levels for P(NO2-3Cz) and P(NH2-3Cz) were calculated to be 5.42 and 5.40 eV (relative to the vacuum energy level), respectively. Their LUMO energy levels deduced from the band gap calculated from the optical absorption edge were 2.71 and 2.20 eV, respectively (Table 1). According to the HOMO and LUMO energy levels obtained, the polymers in this study might be used as hole injection and transport materials.
Spectroelectrochemical properties
Spectroelectrochemistry was performed on the electro-generated polymeric films on ITO glass to clarify its electronic structure and optical behavior upon oxidation. The result of the polymer P(NO2-3Cz) film is presented in Fig. 6(a) as the UV-vis-NIR absorbance curves correlated to electrode potentials. In the neutral form, the P(NO2-3Cz) film exhibited strong absorption at wavelengths around 304 nm characteristic of π–π* transitions, and it was pale yellow due to some absorption in the region of 400–500 nm. Upon oxidation (increasing the applied voltage from 0 to 1.13 V), the absorption of the π–π* transition at 304 nm gradually decreased while a new absorption peak at 432 nm and a broadband from about 600 nm extending to the near-IR (NIR) region grew up, which we assigned to the formation of biscarbazole radical cations. The NIR absorption of the polymer can be attributed to the intervalence charge transfer (IVCT) transitions of mixed-valent biscarbazole units or charge-resonance bands in the case of Robin-Day class III systems.24 When the applied potential was increased to 1.35 V, the absorption at 432 nm decreased gradually accompanied by a broad absorption band at around 710 nm. Meanwhile, they were associated with significant color changes upon oxidation [from pale yellow (L*: 70; a*: 2; b*: 11) to a deeper yellow (L*: 60; a*: 2; b*: 17) and to blue (L*: 49; a*: −5; b*: 2)] that were homogeneous across the ITO glass and easy to detect with the naked eye [Fig. 6(a), inset].
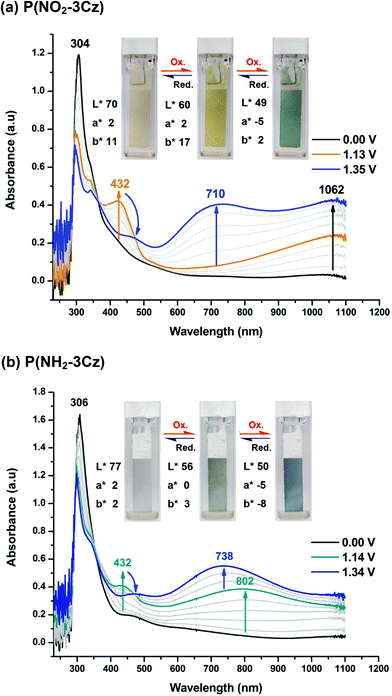 |
| | Fig. 6 Spectroelectrograms and color changes of (a) P(NO2-3Cz) and (b) P(NH2-3Cz) thin films on ITO-coated glass in 0.1 M Bu4NClO4/MeCN at various applied voltages. | |
Fig. 6(b) shows the spectral changes of the electro-deposited P(NH2-3Cz) film upon incremental oxidative scans from 0 to 1.34 V. In the neutral form, the P(NH2-3Cz) film exhibited strong absorption at a λmax of 306 nm and was almost colorless. As the applied voltage was stepped from 0 to 1.14 V, the absorbance at 306 nm decreased, and a new peak at 432 nm and a broad absorption band centered at 802 nm gradually increased in intensity. In the same time, the film turned green (L*: 56; a*: 0; b*: 3). We attribute these spectral changes to the formation of a stable cation radical of biscarbazole units and polyaniline-like units. At a higher applied voltage of 1.34 V, the absorbance at 432 nm decreased and a new broad absorption band at 738 nm, which indicates the formation of dication states. Meanwhile, the film changed color from green to blue (L*: 50; a*: −5; b*: −8). The inset of Fig. 6(b) shows images of the P(NH2-3Cz) film in uncharged (neutral, colorless), semi-oxidized (green) and fully oxidized states (blue). For comparison, the spectroelectrochemical diagrams of the electrodeposited films of P(NH2-NPC) and (NO2-NPC) dimer are included in Fig. S11.† Similar spectral and color changes of the P(NH2-3Cz) and P(NH2-NPC) films imply the simultaneous formation of cation radicals of biscarbazole units and polyaniline-like units.
Electrochromic switching
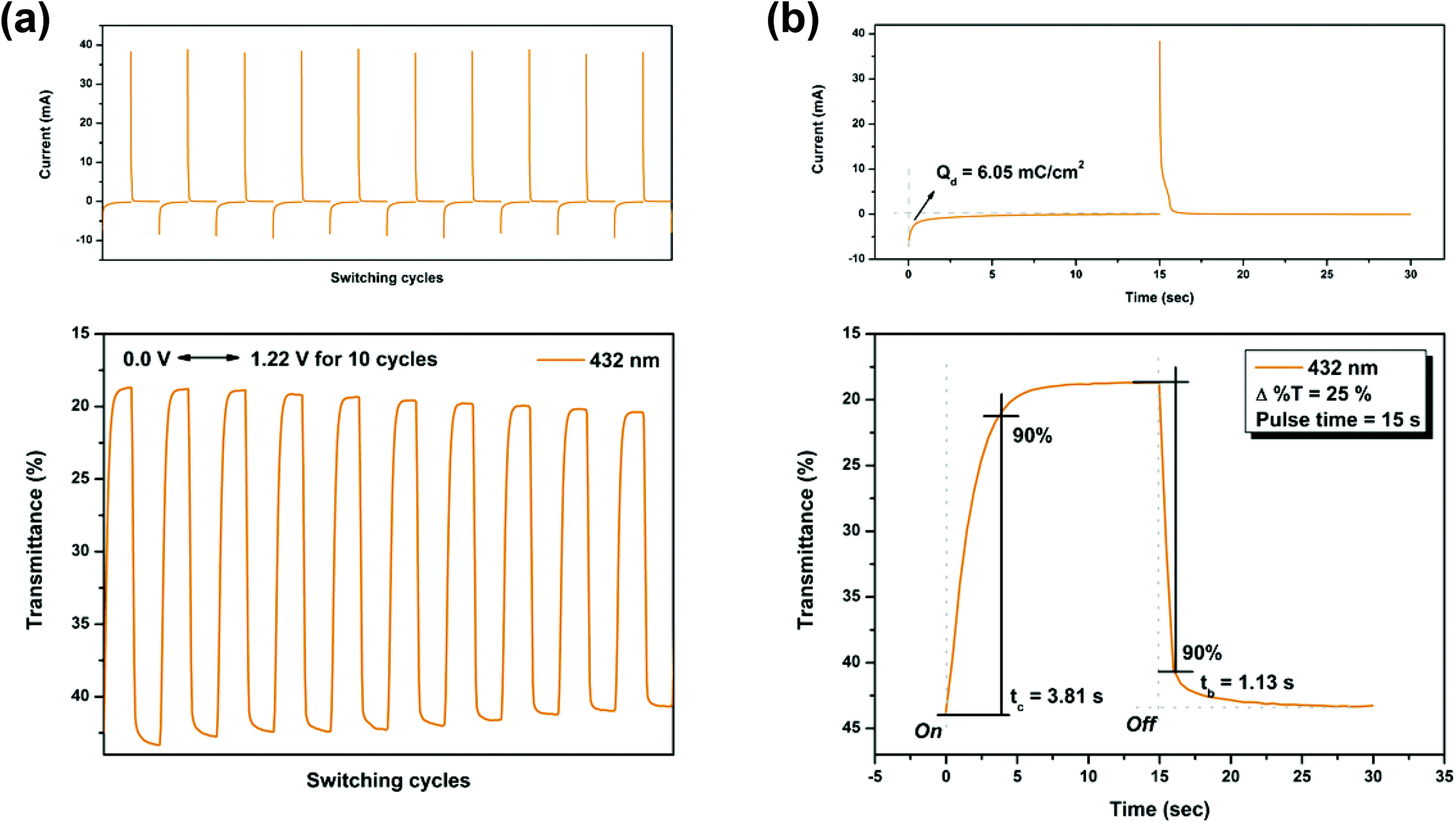
Electrochromic switching studies for the P(NO2-3Cz) and P(NH2-3Cz) were performed to monitor the percent transmittance changes (Δ%T) as a function of time at their absorption maximum (λmax) and to determine the response time by stepping potential repeatedly between the neutral and oxidized states. The active area of the polymer film on ITO-glass is approximately 1 cm2. Fig. 7 and 8 depict the optical transmittance of P(NO2-3Cz) films at 432 and 710 nm as a function of time by applying square-wave potential steps between 0 and 1.22 V and between 0 and 1.38 V with a pulse width of 15 s. The response time was calculated at 90% of the full transmittance change, because it is difficult to perceive any further color change with the naked eye beyond this point. As shown in Fig. 7(b), P(NO2-3Cz) attained 90% of a complete coloring and bleaching in 3.81 and 1.13 s, respectively. The optical contrast measured as Δ%T of P(NO2-3Cz) between neutral pale yellow and oxidized deeper yellow states was found to be 25% at 432 nm. The polymer film exhibited a moderate stability in the first ten switching cycles between the neutral and first oxidation states. As the applied voltage was stepped between 0 and 1.38 V, the P(NO2-3Cz) exhibited an initial Δ%T up to 52% at 710 nm for the oxidized blue state and required 4.23 s for the coloring step and 1.06 s for the bleaching step. However, the polymer film displayed a larger loss of optical contrast after ten switching cycles between the neutral and the second oxidized states (from 52% to 38%). The electrochromic coloration efficiency (CE; η) can be calculated via optical density using the following equation:
where ΔOD is the optical absorbance change, and Qd (mC cm−2) is the inject/ejected charge during a redox step. On the basis of this equation, the CE values of P(NO2-3Cz) were calculated as 62 cm2 C−1 (at 432 nm) and 35 cm2 C−1 (at 710 nm).
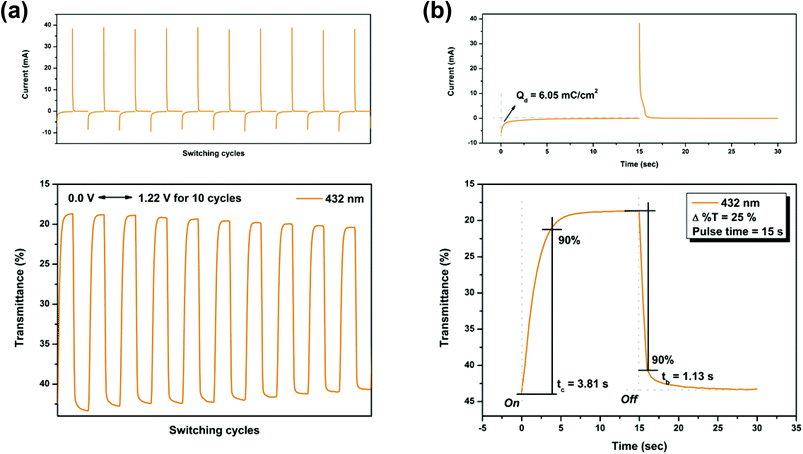 |
| | Fig. 7 Potential step absorptiometry of the cast films of P(NO2-3Cz) on the ITO-glass slide (coated area ∼1 cm2) (in MeCN with 0.1 M Bu4NClO4 as the supporting electrolyte) by applying a potential step: (a) optical switching at potential 0.00 V ⇔ 1.22 V (10 cycles) with a pulse width of 15 s, monitored at λmax = 432 nm; (b) the 1st cycle transmittance change for the P(NO2-3Cz) thin film. | |
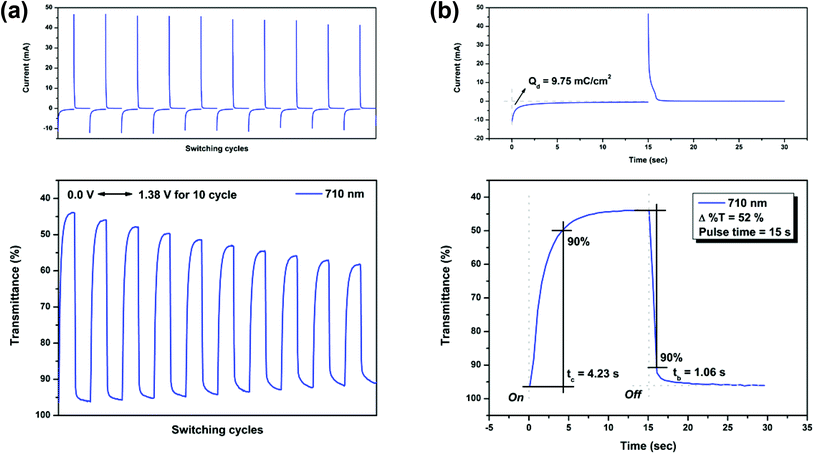 |
| | Fig. 8 Potential step absorptiometry of the cast films of P(NO2-3Cz) on the ITO-glass slide (coated area ∼1 cm2) (in MeCN with 0.1 M Bu4NClO4 as the supporting electrolyte) by applying a potential step: (a) optical switching at potential 0.00 V ⇔ 1.38 V (10 cycles) with a pulse width of 15 s, monitored at λmax = 710 nm; (b) the 1st cycle transmittance change for the P(NO2-3Cz) thin film. | |
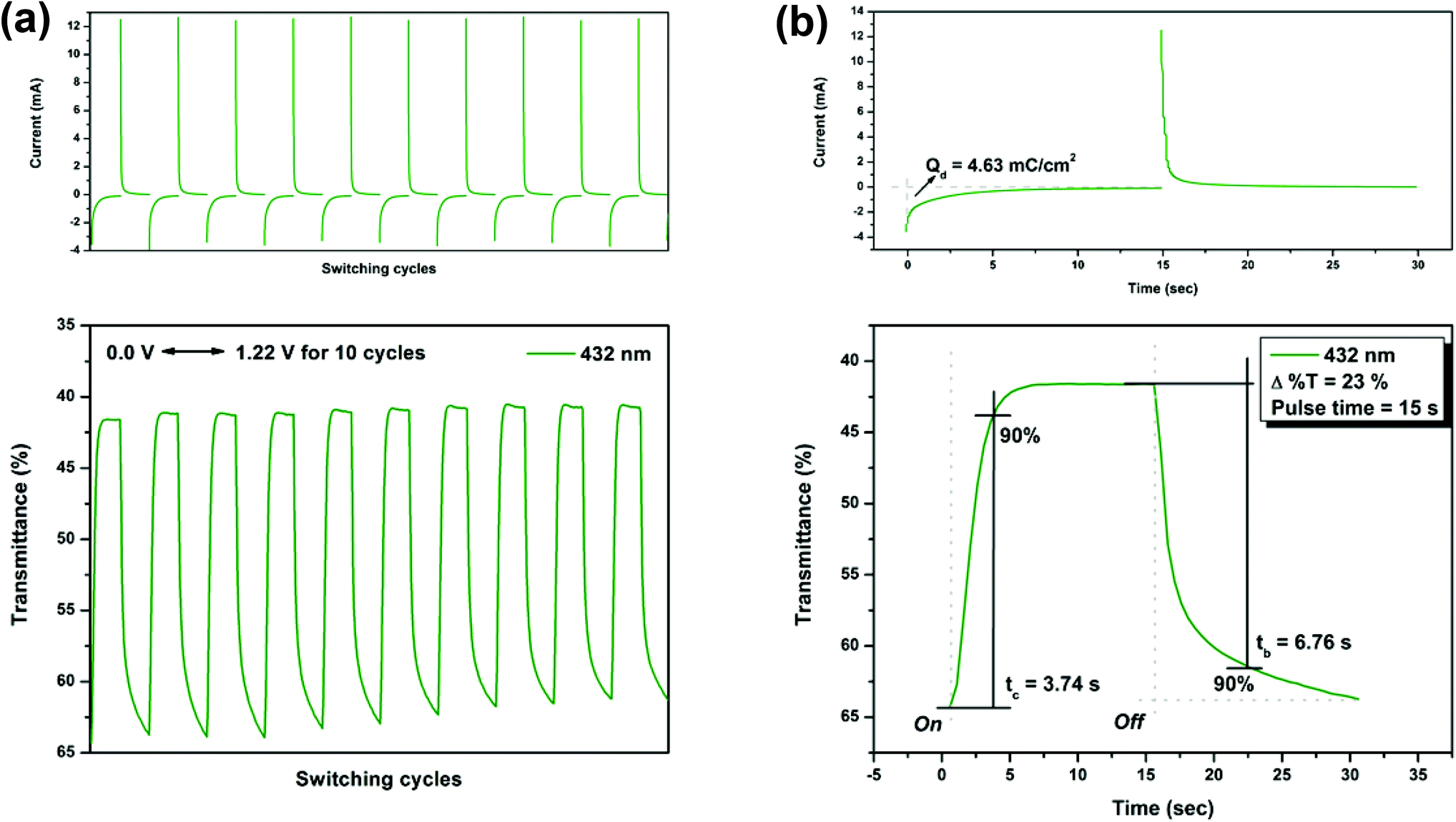
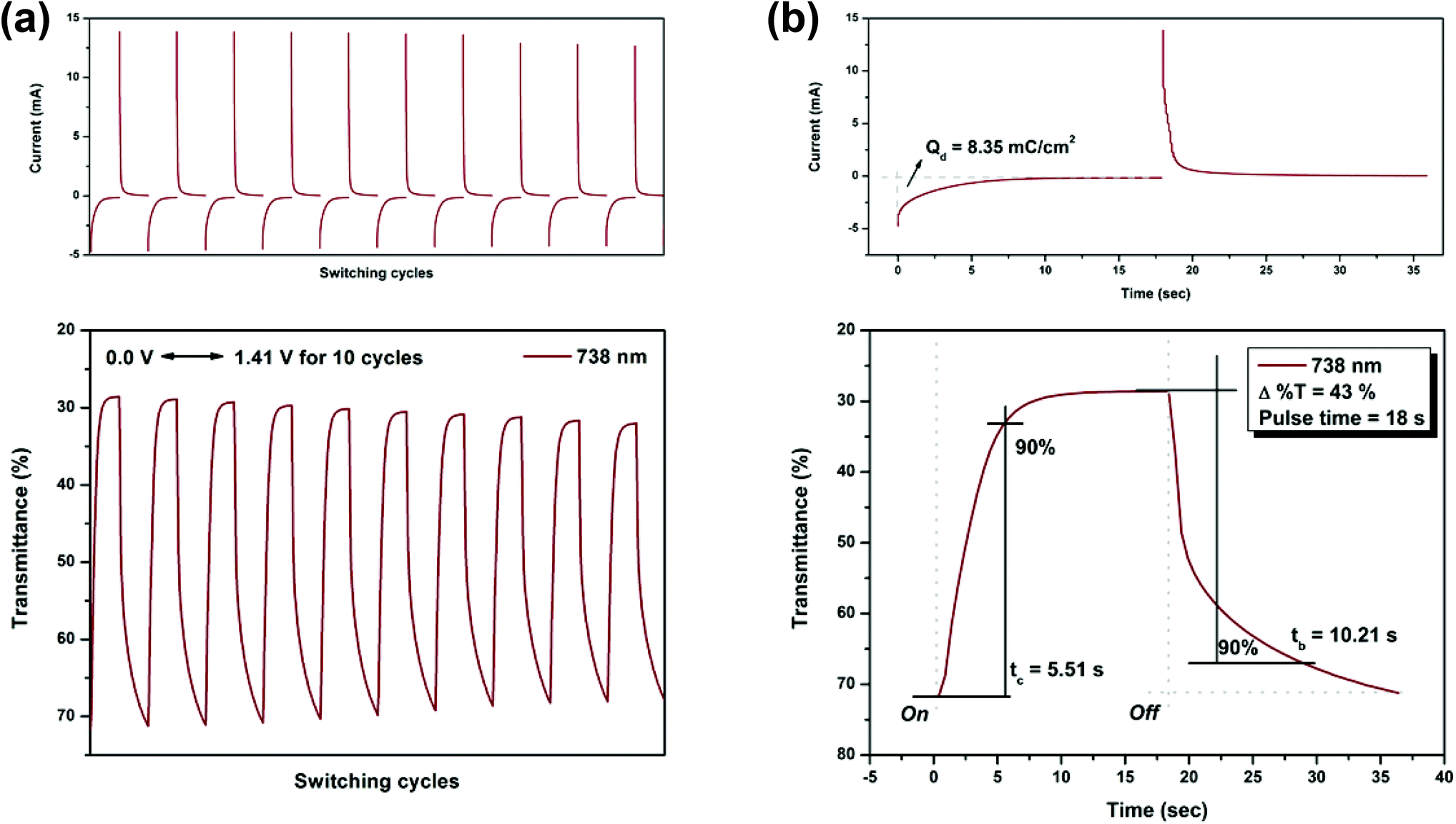
On the other hand, the switching stability of P(NH2-3Cz) at λmax = 432 and λmax = 738 was investigated by monitoring the optical contrast (Δ%T) of the thin film upon repeated square-wave potential steps between 0 and 1.22 V with a pulse width of 15 s, and between 0 and 1.41 V with a pulse width of 18 s (see Fig. 9 and 10). In this case, a response time was required for 90% full-transmittance change of 3.74 s for the coloration step and 6.76 s for the bleaching step at 432 nm and 5.51 s for the coloration step and 10.21 s for the bleaching step at 738 nm. In addition, the optical contrast measured as Δ%T recorded in neutral and oxidized forms was found to be 23% at 432 nm and 43% at 738 nm. As compared to P(NO2-3Cz), the P(NH2-3Cz) film revealed a better electrochromic switching stability possibly due to the increased electrochemical stability associated with the aniline-like main chain. As shown in Table 2, the CE values of P(NH2-3Cz) were calculated to be 41 cm2 C−1 at 432 nm and 48 cm2 C−1 at 738 nm by chronoamperometry.
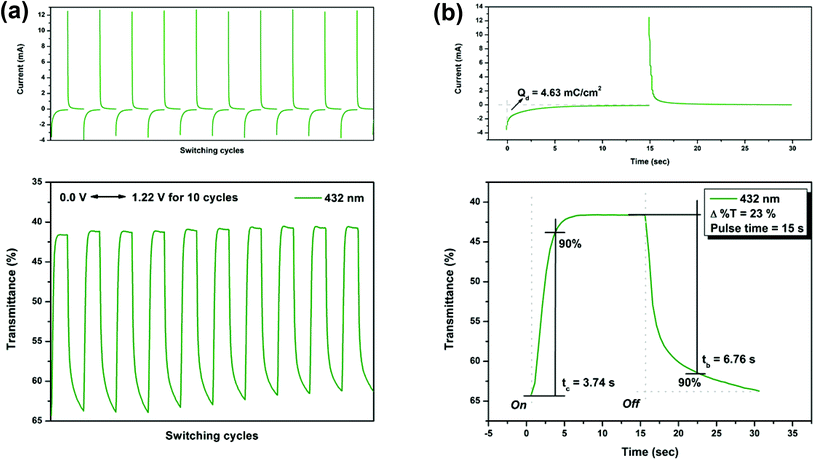 |
| | Fig. 9 Potential step absorptiometry of the cast films of P(NH2-3Cz) on the ITO-glass slide (coated area ∼1 cm2) (in MeCN with 0.1 M Bu4NClO4 as the supporting electrolyte) by applying a potential step: (a) optical switching at potential 0.00 V ⇔ 1.22 V (10 cycles) with a pulse width of 15 s, monitored at λmax = 432 nm; (b) the 1st cycle transmittance change for the P(NH2-3Cz) thin film. | |
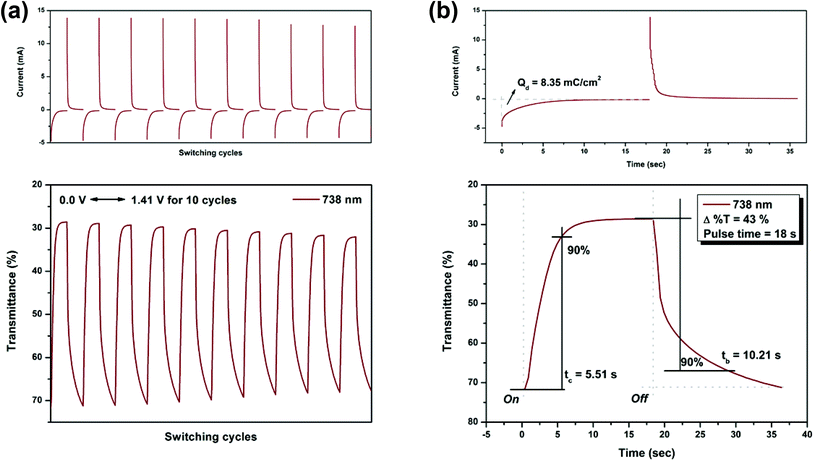 |
| | Fig. 10 Potential step absorptiometry of the cast films of P(NH2-3Cz) on the ITO-glass slide (coated area ∼1 cm2) (in MeCN with 0.1 M Bu4NClO4 as the supporting electrolyte) by applying a potential step: (a) optical switching at potential 0.00 V ⇔ 1.41 V (10 cycles) with a pulse width of 18 s, monitored at λmax = 738 nm; (b) the 1st cycle transmittance change for the P(NH2-3Cz) thin film. | |
Table 2 Electrochromic properties of the polymer films
| Polymer |
λ
maxa (nm) |
Δ%T |
Response timeb |
ΔODc |
Q
dd (mC cm−2) |
CEe (cm2 C−1) |
|
t
c (s) |
t
b (s) |
|
Wavelength of absorption maximum.
Time for 90% of the full-transmittance change.
Optical density change (ΔOD) = log[Tbleached/Tcolored], where Tcolored and Tbleached are the maximum transmittance in the oxidized and neutral states, respectively.
Q
d is ejected charge, determined from the in situ experiments.
Coloration efficiency (CE) = ΔOD/Qd.
|
| P(NO2-3Cz) |
432 |
25 |
3.81 |
1.13 |
0.375 |
6.05 |
62 |
| 710 |
52 |
4.23 |
1.06 |
0.343 |
9.75 |
35 |
| P(NH2-3Cz) |
432 |
23 |
3.74 |
6.67 |
0.189 |
4.63 |
41 |
| 738 |
43 |
5.51 |
10.21 |
0.404 |
8.35 |
48 |
Electrochromic devices
Based on the foregoing results, it can be concluded that these polymers can be used in the construction of electrochromic devices and optical displays. Therefore, single layer electrochromic cells were fabricated as preliminary investigations for the electrochromic applications of these electro-generated polycarbazole films. A schematic illustration of the electrochromic cell is shown in Fig. 11(a). The polymer films were electrodeposited onto ITO-coated glass, thoroughly rinsed, and then dried. Afterwards, the gel electrolyte was spread on the polymer-coated side of the electrode and the electrodes were sandwiched. To prevent leakage, an epoxy resin was applied to seal the device. An electrochromic cell based on P(NO2-3Cz) was fabricated which is shown in Fig. 11(b). The P(NO2-3Cz) film is pale yellow in the neutral form. When the applied voltage was increased to 2.3 V, the color changed to a deeper yellow. Upon further oxidation at an applied potential of 3.0 V, the color changed to blue. When the potential was subsequently set back at 0.0 V, the polymer film turned back to the original color. In the other case, the P(NH2-3Cz) film is colorless in the neutral form. When voltage was applied (from 0.0 to 2.2 V and 2.4 V), the color changed to yellow green and blue (Fig. 11(c)).
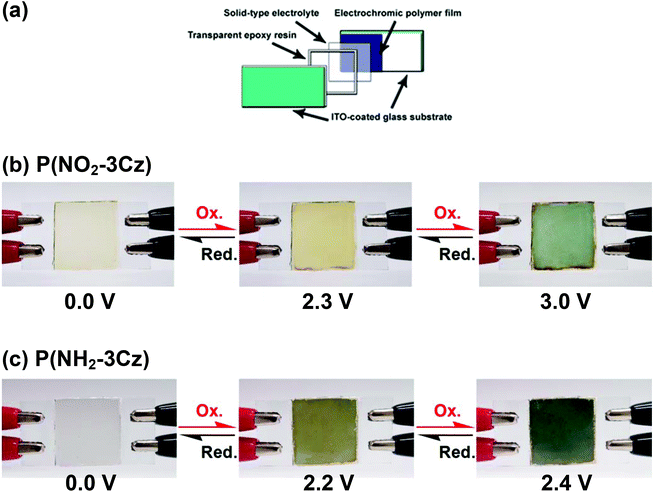 |
| | Fig. 11 (a) Schematic illustration of the structure of the electrochromic devices. (b) and (c) Photos of the sandwich-type ITO-coated glass electrochromic cell, using P(NO2-3Cz) and P(NH2-3Cz) as the active layer. | |
Conclusions
A series of N-phenylcarbazole (NPC) derivatives with different substituents on the para position of the N-phenyl group have been synthesized and characterized for their electrochemical properties. The oxidized forms of NPC, CN-NPC, COOH-NPC, and NO2-NPC underwent dimerization to the corresponding biscarbazoles, whereas NH2-NPC could undergo electrochemical polymerization to form polymer thin films on the electrode surface via the electropolymerization mechanism likely similar to that of polyaniline. 3,6-Bis(N-carbazolyl)-substituted NPCs (NO2-3Cz and NH2-3Cz) could be readily electropolymerized into polymer films on the electrode surface in an electrolyte solution via the oxidative coupling reactions. The resulting polymer film of P(NO2-3Cz) exhibited two reversible oxidation redox couples due to oxidations of the biscarbazole unit, and the P(NH2-3Cz) film showed multistep redox processes attributed to oxidations of the biscarbazole and amino-carbazole moieties. Both of the electro-generated polymer films exhibited obvious color changes during the oxidation processes. The P(NH2-3Cz) film showed a slightly lower onset oxidation potential and higher electrochromic stability than the P(NO2-3Cz) film due to the presence of polyaniline-like structure. The electro-generated polymer films could be promising materials for electrochromic applications.
Acknowledgements
The authors acknowledge the financial support from the Ministry of Science and Technology of Taiwan, ROC.
References
-
(a) D. R. Rosseinsky and R. J. Mortimer, Adv. Mater., 2001, 13, 783–793 CrossRef CAS;
(b)
P. M. S. Monk, R. J. Mortimer and D. R. Rosseinsky, Electrochromism and Electrochromic Devices, Cambridge University Press, Cambridge, UK, 2007 Search PubMed.
-
(a) R. D. Rauh, Electrochim. Acta, 1999, 44, 3165–3176 CrossRef CAS;
(b) R. Baetens, B. P. Jelle and A. Gustavsen, Sol. Energy Mater. Sol. Cells, 2010, 94, 87–105 CrossRef CAS.
- G. Sonmez and H. B. Sonmez, J. Mater. Chem., 2006, 16, 2473–2477 RSC.
-
(a) R. J. Mortimer, A. L. Dyer and J. R. Reynolds, Displays, 2006, 27, 2–18 CrossRef CAS;
(b) P. M. Beaujuge, S. Ellinger and J. R. Reynolds, Nat. Mater., 2008, 7, 795–799 CrossRef CAS PubMed.
- S. Beaupre, A.-C. Breton, J. Dumas and M. Leclerc, Chem. Mater., 2009, 21, 1504–1513 CrossRef CAS.
-
http://www.gentex.com
.
-
(a) W. C. Dautremont-Smith, Displays, 1982, 3, 3–20 CrossRef CAS;
(b) W. C. Dautremont-Smith, Displays, 1982, 3, 67–80 CrossRef CAS;
(c) C. G. Granqvist, E. Avendano and A. Azens, Thin Solid Films, 2003, 442, 201–211 CrossRef CAS;
(d) S. Y. Park, J. M. Lee, C. H. Noh and S. U. Son, J. Mater. Chem., 2009, 19, 7959–7964 RSC;
(e) D. T. Gillapie, R. C. Tenent and A. C. Dillon, J. Mater. Chem., 2010, 20, 9585–9592 RSC.
-
(a) S. Bernhard, J. I. Goldsmith, K. Takada and H. D. Abruna, Inorg. Chem., 2003, 42, 4389–4393 CrossRef CAS PubMed;
(b) F. S. Han, M. Higuchi and D. G. Kurth, Adv. Mater., 2007, 19, 3928–3931 CrossRef CAS;
(c) F. S. Ham, M. Higuchi and D. G. Kurth, J. Am. Chem. Soc., 2008, 130, 2073–2081 CrossRef PubMed;
(d) A. Maier, A. R. Rabindranath and B. Tieke, Adv. Mater., 2009, 21, 959–963 CrossRef CAS;
(e) A. Maier, H. Fakhrnabavi, A. R. Rabindranath and B. Tieke, J. Mater. Chem., 2011, 21, 5795–5804 RSC.
-
(a) R. J. Mortimer, Electrochim. Acta, 1999, 44, 2971–2981 CrossRef CAS;
(b) P. R. Somani and S. Radhakrishnan, Mater. Chem. Phys., 2002, 77, 117–133 CrossRef;
(c) Y. Shirota, J. Mater. Chem., 2000, 10, 1–25 RSC.
-
(a) A. A. Argun, P.-H. Aubert, B. C. Thompson, I. Schwendeman, C. L. Gaupp, J. Hwang, N. J. Pinto, D. B. Tanner, A. G. MacDiarmid and J. R. Reynolds, Chem. Mater., 2004, 16, 4401–4412 CrossRef CAS;
(b) J. Roncali, P. Blanchard and P. Frere, J. Mater. Chem., 2005, 15, 1589–1610 RSC;
(c) R. M. Walczak and J. R. Reynolds, Adv. Mater., 2006, 18, 1121–1131 CrossRef CAS;
(d) R. M. Walczak, J.-H. Jung, J. S. Cowart, Jr. and J. R. Reynolds, Macromolecules, 2007, 40, 7777–7785 CrossRef CAS;
(e) A. Patra, Y. H. Wijsboom, S. S. Zade, M. Li, Y. Sheynin, G. Leitus and M. Bendikov, J. Am. Chem.
Soc., 2008, 130, 6734–6736 CAS;
(f) A. Patra and M. Bendikov, J. Mater. Chem., 2010, 20, 422–433 RSC.
-
(a) P. M. Beaujuge and J. R. Reynolds, Chem. Rev., 2010, 110, 268–320 CrossRef CAS PubMed;
(b) A. Patra, Y. H. Wijsboom, G. Leitus and M. Bendikov, Chem. Mater., 2011, 23, 896–906 CrossRef CAS;
(c) A. Balan, D. Baran and L. Toppare, Polym. Chem., 2011, 2, 1029–1043 RSC;
(d) G. Gunbas and L. Toppare, Chem. Commun., 2012, 48, 1083–1101 RSC.
-
(a) H.-J. Yen, H.-Y. Lin and G.-S. Liou, Chem. Mater., 2011, 23, 1874–1882 CrossRef CAS;
(b) H.-J. Yen, H.-Y. Lin and G.-S. Liou, J. Mater. Chem., 2011, 21, 6230–6237 RSC;
(c) H.-J. Yen and G.-S. Liou, Polym. Chem., 2012, 3, 255–264 RSC;
(d) H.-J. Yen, C.-J. Chen and G.-S. Liou, Adv. Funct. Mater., 2013, 23, 5307–5316 CrossRef CAS;
(e) H.-J. Yen and G.-S. Liou, Chem. Commun., 2013, 49, 9797–9799 RSC.
-
(a) Y.-C. Kung and S.-H. Hsiao, J. Mater. Chem., 2010, 20, 5481–5492 RSC;
(b) Y.-C. Kung and S.-H. Hsiao, J. Mater. Chem., 2011, 21, 1746–1754 RSC;
(c) S.-H. Hsiao, H.-M. Wang, P.-C. Chang, Y.-R. Kung and T.-M. Lee, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2925–2938 CrossRef CAS;
(d) H.-M. Wang and S.-H. Hsiao, J. Mater. Chem. C, 2014, 2, 1553–1564 RSC;
(e) S.-H. Hsiao, H.-M. Wang and S.-H. Liao, Polym. Chem., 2014, 5, 2473–2483 RSC.
-
(a) H. Niu, H. Kang, J. Cai, C. Wang, X. Bai and W. Wang, Polym. Chem., 2011, 2, 2804–2817 RSC;
(b) L. Ma, H. Niu, J. Cai, P. Zhao, C. Wang, Y. Lian, X. Bai and W. Wang, J. Mater. Chem. C, 2014, 2, 2272–2282 RSC;
(c) Y. Wang, Y. Liang, J. Zhu, X. Bai, X. Jiang, Q. Zhang and H. Niu, RSC Adv., 2015, 5, 11071–11076 RSC.
-
(a) J. V. Grazulevicius, P. Strohriegl, J. Pielichowski and K. Pielichowski, Prog. Polym. Sci., 2003, 28, 1297–1353 CrossRef CAS;
(b) A. Iraqi and I. Wataru, Chem. Mater., 2004, 16, 442–448 CrossRef CAS;
(c) A. Iraqi and I. Wataru, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 6041–6051 CrossRef CAS;
(d) H. Yi, A. Iraqi, M. Stevenson, C. J. Elliott and D. G. Lidzey, Macromol. Rapid Commun., 2007, 28, 1155–1160 CrossRef CAS.
-
(a) J. Ding, B. Zhang, J. Lu, Z. Xie, L. Wang, X. Jing and F. Wang, Adv. Mater., 2009, 21, 4983–4986 CrossRef CAS PubMed;
(b) C.-C. Lee, M.-k. Leung, P.-Y. Lee, T.-L. Chiu, J.-H. Lee, C. Liu and P.-T. Chou, Macromolecules, 2012, 45, 751–765 CrossRef CAS;
(c) D. Xia, B. Wang, C. Chen, S. Wang, B. Zhang, J. Ding, L. Wang, X. Jing and F. Wang, Angew. Chem., Int. Ed., 2014, 53, 1048–1052 CrossRef CAS PubMed;
(d) Y. Wang, S. Wang, N. Zhao, B. Gao, S. Shao, J. Ding, L. Wang, X. Jing and F. Wang, Polym. Chem., 2015, 6, 1180–1191 RSC;
(e) F. Dumur, Org. Electron., 2015, 25, 345–361 CrossRef CAS.
-
(a) Y. Pelous, G. Froyer, D. Ades, C. Chevrot and A. Siove, Polym. Commun., 1990, 31, 341 CAS;
(b) J.-F. Morin, M. Leclerc, D. Ades and A. Siove, Macromol. Rapid Commun., 2005, 26, 761–778 CrossRef;
(c) N. Bloudin and M. Leclerc, Acc. Chem. Res., 2008, 41, 1110–1119 CrossRef PubMed;
(d) P.-L. T. Boudreault, S. Beaupre and M. Leclerc, Polym. Chem., 2010, 1, 127–136 RSC.
-
(a) J. F. Ambrose and R. F. Nelson, J. Electrochem. Soc., 1968, 115, 1159–1164 CrossRef CAS;
(b) J. F. Ambrose, L. L. Carpenter and R. F. Nelson, J. Electrochem. Soc., 1975, 122, 876–894 CrossRef CAS.
-
(a) S. Koyuncu, B. Gultekin, C. Zafer, H. Bilgili, M. Can, S. Demic, I. Kaya and S. Icli, Electrochim. Acta, 2009, 54, 5694–5702 CrossRef CAS;
(b) B. Wang, J. Zhao, R. Liu, J. Liu and Q. He, Sol. Energy Mater. Sol. Cells, 2011, 95, 1867–1874 CrossRef CAS;
(c) O. Usluer, S. Koyuncu, S. Demic and R. A. J. Janssen, J. Polym. Sci., Part B: Polym. Phys., 2011, 49, 333–341 CrossRef CAS;
(d) C. Xu, J. Zhao, M. Wang, Z. Wang, C. Cui, Y. Kong and X. Zhang, Electrochim. Acta, 2012, 75, 28–34 CrossRef CAS.
-
(a) Z. Xu, M. Wang, J. Zhao, C. Cui, W. Fan and J. Liu, Electrochim. Acta, 2014, 125, 241–249 CrossRef CAS;
(b) Z. Xu, M. Wang, W. Fan, J. Zhao and H. Wang, Electrochim. Acta, 2015, 160, 271–280 CrossRef CAS.
-
(a) G.-S. Liou, S.-H. Hsiao and H.-W. Chen, J. Mater. Chem., 2006, 16, 1831–1842 RSC;
(b) H.-M. Wang, S.-H. Hsiao, G.-S. Liou and C.-H. Sun, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 4775–4789 CrossRef CAS.
-
(a) A. Kulasi, H. Yi and A. Iraqi, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 5957–5967 CrossRef CAS;
(b) I. Fabre-Francke, M. Zagorska, G. Louarn, P. Hapiot, A. Pron and S. Sadki, Electrochim. Acta, 2008, 53, 6469–6476 CrossRef CAS.
- S.-K. Chiu, Y.-C. Chung, G.-S. Liou and Y. O. Su, J. Chin. Chem. Soc., 2012, 59, 331–337 CrossRef CAS.
-
(a) M. B. Robin and P. Day, Adv. Inorg. Chem. Radiochem., 1968, 10, 247–422 CrossRef;
(b) C. Lambert and G. Noll, J. Am. Chem. Soc., 1999, 121, 8434–8442 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5py01407g |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.