
Enhancement of the crosslink density, glass transition temperature, and strength of epoxy resin by using functionalized graphene oxide co-curing agents†
Jin Won
Yu‡
ab,
Jin
Jung‡
a,
Yong-Mun
Choi
a,
Jae Hun
Choi
a,
Jaesang
Yu
a,
Jae Kwan
Lee
b,
Nam-Ho
You
a and
Munju
Goh
*a
aCarbon Composite Materials Research Center, Institute of Advanced Composites Materials, Korea Institute of Science and Technology (KIST), Chudong-ro 92, Bondong-eup, Wanju-gun, Jeollabuk-do 565-905, Korea. E-mail: goh@kist.re.kr; Tel: +82 63 2198141
bDepartment of Carbon Materials, Chosun University, Gwangju, 561-756, Korea
First published on 3rd November 2015
Abstract
We synthesized diamine-functionalized graphene oxide, DDS–GO and HMDA–GO, by introducing 4,4′-diaminodiphenyl sulfone (DDS) or hexamethylenediamine (HMDA) into the carboxylic acid groups on graphene oxide (GO) via amide bonds. The introduction of diamines was confirmed by analytical methods such as FT-IR, TG-DTA, XPS, AFM, and optical microscopy. Then, we applied DDS–GO and HMDA–GO as co-curing agents for epoxy (EP) nanocomposites that were prepared by mixing bisphenol-A type EP and DDS curing agent (ca. 21 wt%). Interestingly, when 1.0 wt% of DDS–GO was added to the EP/DDS mixture, the crosslink density (CD) increased from 0.028 to 0.069 mol cm−3. Due to the higher CD, both the glass transition temperature and tensile strength of the EP/DDS/DDS–GO nanocomposite effectively improved from 160.7 °C to 183.4 °C and from 87.4 MPa to 110.3 MPa, respectively.
Introduction
Epoxy resin (EP), the most common thermosetting resin, forms a three-dimensional cross-linked network structure when subjected to exothermic curing. Epoxy resin has been used in various industrial applications because of its good adhesion, chemical resistance, thermal resistance, and mechanical properties. However, despite its good properties, there has been a persistent demand for more functional EP that can be used in potential applications such as in automotive, aerospace, construction, and electronic industries. For example, various structure-modified EPs such as novolac,1 phenoxy,2 crystalline,3 and rubber-modified EPs4 have been developed. Another attempt to improve the properties of cured EP has been to change curing agents and/or co-curing agents, which dramatically affect the thermal and mechanical properties of cured EP.The use of carbon nanomaterials like carbon black,5 carbon nanotubes,6,7 carbon nanofibers,8 and graphene9–12 as fillers to prepare EP nanocomposites has been attracting attention owing to their unique properties. In addition to these nanomaterials, graphene oxide (GO) has been considered a promising nanomaterial for the improvement of the mechanical and thermal properties of polymer nanocomposites such as poly(methyl methacrylate), polystyrene, polyurethane, and poly(vinylidene fluoride) nanocomposites.13,14 Various neat and functionalized GOs have also been adapted for enhancing the glass transition temperature (Tg) and/or mechanical strength of EP nanocomposites.15–20 The crosslink density (CD) is generally recognized as a critical factor influencing the thermal21,22 and mechanical23,24 properties of EP nanocomposites. If the interfacial strength between the GO and EP is low, the CD of nanocomposites will be low, and because of the low CD value, improvements in the thermal and mechanical properties of EP nanocomposites cannot be sufficient and, in some cases, these properties can be worse. In order to improve the interfacial strength between GO and EP, some studies focused on the preparation of functionalized GOs with epoxide terminal groups15–17 and with amine terminal groups.18–20 In these studies, slight improvements in the Tg and tensile strength of cured EP nanocomposites compared to those of neat cured EPs were realized. Further, it has been mentioned that incorporation of nanomaterials into EP could slightly increase the CD of EP nanocomposites,25–30 but there have been few studies that calculated the CD of EP nanocomposites, made an effort to increase the CD of EP nanocomposites, and discussed the effect of the nanomaterials on the CD of EP nanocomposites.
In this work, we report a novel technique to improve the CD of GO-incorporated EP nanocomposites. We synthesized two diamine-functionalized GO, DDS–GO and HMDA–GO, by introducing 4,4′-diaminodiphenyl sulfone (DDS) or hexamethylenediamine (HMDA) into the carboxylic acid groups on GO via amide bonds. We prepared two kinds of mixtures, EP/DDS/DDS–GO and EP/DDS/HMDA–GO, by adding each diamine-functionalized GO as the co-curing agent into the mixture of diglycidyl ether of bisphenol-A (DGEBA)-type EP and 4,4′-diaminodiphenyl sulfone (DDS) curing agent (ca. 21 wt%). By increasing the amounts of DDS–GO from 0 to 1 wt%, the CD of EP/DDS/DDS–GO nanocomposites increased, and thus, we could effectively increase the Tg and tensile strength of EP nanocomposites.
Experimental
Materials
Graphite powder (average particle size <30 μm), potassium permanganate (KMnO4), hexamethylenediamine (HMDA), and N,N′-dicyclohexylcarbodiimide (DCC) were purchased from Sigma-Aldrich Co. (USA). Sulfuric acid (H2SO4; 98%), hydrogen peroxide (H2O2; 30% in water), hydrochloric acid (HCl; 35% in water), and dichloromethane (DCM; 99.5%) were purchased from Daejung Chemical Co. (Korea). 4,4′-Diaminodiphenyl sulfone (DDS) and 4-dimethylaminopyridine (DMAP) were obtained from TCI Co. (Japan). Diglycidyl ether of bisphenol-A (DGEBA)-type EP resin (YD-128) was purchased from Kukdo Chemical Co. (Korea).General procedure for synthesis of graphene oxide (GO)
GO was synthesized using the modified Hummer's method.31 A round-bottom flask containing 98% H2SO4 (80 mL) was cooled to 0 °C. Graphite powder (2 g) was added into the flask and stirred until mixed. KMnO4 (10 g) was slowly added to the chilled graphite solution, and the resulting mixture was kept at 0 °C. The mixture was then taken from the ice bath and stirred at 35 °C for 6 h. Deionized water (DI-water, 0.5 L) was slowly added, and the mixture was further stirred for 2 h. Subsequently, additional DI-water (1.5 L) was added followed by dropwise addition of 30% H2O2 (30 mL). The mixture was left undisturbed for 3 days for the precipitation to complete. The clear supernatant was decanted. The precipitate was repeatedly washed with DI-water followed by centrifugation with 20 ml of HCl solution (1 N), at least five times to remove the residual metal oxides. The resulting GO dispersion was subjected to ultrasonication for 30 min, with occasional agitation at regular intervals of 10 min for monolayer exfoliation. Dialysis was performed with a sealed membrane tube (Spectra/Por 1 Dialysis Tubing, MWCO 6–8000) in DI-water with constant stirring for 7 days. The DI-water was continuously exchanged every 3 h. After dialysis, the clear supernatant was removed by centrifugation at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 30 min. After freeze drying, deep-brown GO powder was obtained.
000 rpm for 30 min. After freeze drying, deep-brown GO powder was obtained.
General procedure for synthesis of diamine-functionalized GO (diamine-GO)
A schematic representation of this process is shown in Fig. 1. All experiments were performed under an argon atmosphere. DCM was distilled prior to use. GO (0.1 g) and the diamine derivative (0.1 g) were added to DCM (200 mL) in a 500 mL flask, followed by ultrasonication for 60 min. After that, DCC and DMAP were added into the flask and ultrasonication was performed for 60 min. The number of moles of DCC and DMAP was the same as that of the diamine derivative. The reaction mixture was taken from the ultrasonicator and stirred at room temperature for 24 h. After the reaction, the mixture was washed with DCM, DI-water, and acetone, and filtered to remove excess diamine. The product was finally dried at 60 °C in a vacuum for 24 h.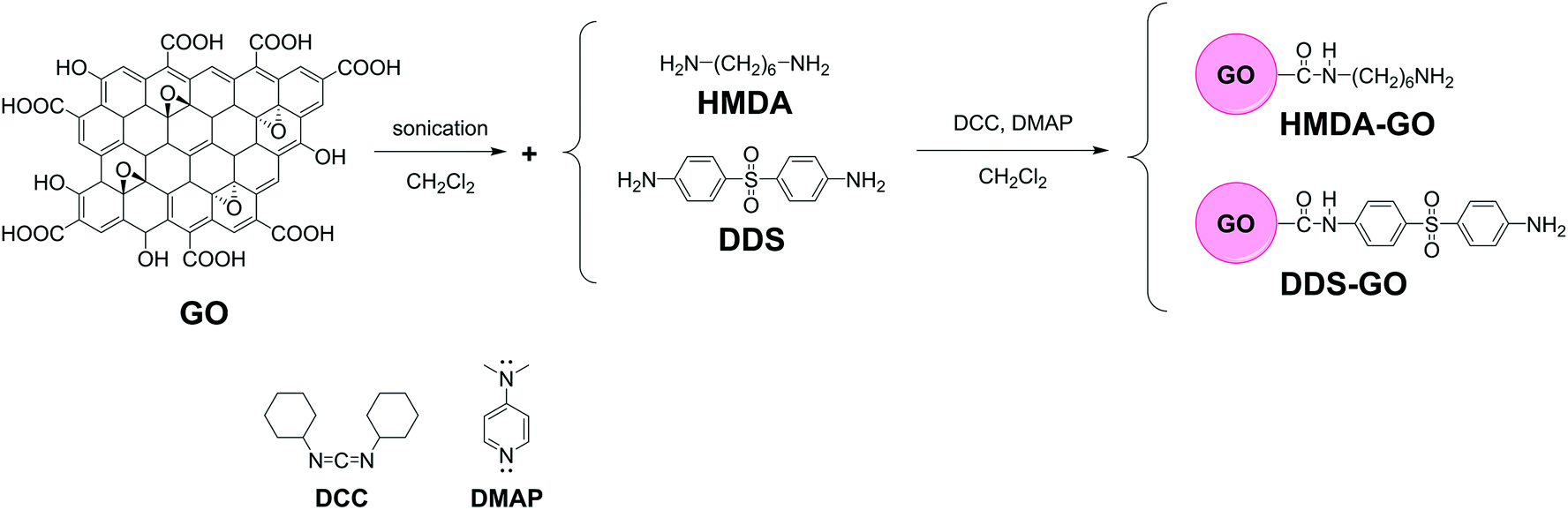 | ||
| Fig. 1 Synthetic routes for diamine-GO derivatives. | ||
General procedure for fabrication of epoxy nanocomposites
We calculated the approximately stoichiometric quantities of the curing agent required for realizing optimum properties. The EP resin used in this study (DGEBA-type) has an epoxide equivalent weight (EEW) of about 185 g eq.−1 The viscosity is about 12000 cP at 25 °C. The curing agent used in this study, DDS, has an amine hydrogen equivalent weight (AHEW) of 62 g eq.−1 Stoichiometric amounts of the EP resin (E) and curing agent (C) were calculated according to the following formula because one N–H group reacts with one epoxide group. | (1) |
For example, the stoichiometric amount of DDS for 100 g of YD-128 was calculated to be 33 g. When diamine-GO was added in the nanocomposite, the mixing amount of the curing agent reduced to 80% of the stoichiometric amount of DDS, because the amine groups on DDS–GO could also react with epoxide groups as the curing agent. The content of diamine-GO was calculated based on the entire amount of the EP resin and curing agent. EP/DDS/diamine-GO nanocomposites were prepared with different weight fractions of the diamine-GO (0.1 wt%, 0.5 wt%, and 1 wt%). More details on the mixing ratios for obtaining EP nanocomposites are shown in the ESI (Table S1†). The fabrication procedure for EP nanocomposites is as follows. Stoichiometric amounts of diamine-GO and EP resin were mixed using a planetary mixer with the mixing speed of 2000 rpm for 10 min. After adding a stoichiometric amount of DDS to the mixture of EP/amine-GO, the mixture was further mixed by stirring at 155 °C for 20 min. Then, the mixture was placed in a vacuum to remove the trapped air bubbles and poured into a mold, followed by precuring it in an oven at 170 °C for 4 h and curing at 230 °C for 2 h.
Material characterization
The structural divergence of the GO and diamine-GO was confirmed by Fourier-transform infrared spectroscopy (FT-IR; Nicolet iS 10, Thermo Scientific, USA). Thermogravimetric differential thermal analyses (TG-DTA) of the GO and diamine-GO were carried out with a TA Q50 (TA Instrument, USA) under nitrogen gas flow at a heating rate of 10 °C per minute. X-ray photoelectron spectroscopy (XPS; AXIS-NOVA, Kratos Inc., USA) was employed to discern the chemical changes on the surface of GO and diamine-GO. Al Kα (1485.6 eV) was used as the X-ray source at 14.9 keV anode voltage. 4.6 A and 20 mA were applied as the filament current and mission current, respectively. Atomic force microscopy (AFM; Veeco-Digital Instruments, USA) images of the GO and diamine-GO were recorded to study morphologies. Optical microscopy (OM; Eclipse Ni, Nikon Inc, Japan) images of the GO and diamine-GO were used to evaluate the dispersion stability of DDS–GO in the EP nanocomposites. To investigate the thermal properties of EP nanocomposites, a differential scanning calorimeter (DSC; Q20, TA Instrument, USA) and a dynamic mechanical analyzer (DMA; Q800, TA Instrument, USA) were used. The DSC scanning temperature range was from 25 to 350 °C under nitrogen gas flow with a heating rate of 10 °C per minute. The DMA scanning temperature range was from 30 to 260 °C with a scanning rate of 3 °C min−1. The mechanical properties of nanocomposites were measured by using a universal testing machine (UTM; Instron, USA). The samples for the UTM measurement were prepared with 1.5 mm thickness, 0.5 cm width, and 2.5 cm length, and the tensile strength test was performed at a testing speed of 5 mm min−1.Results and discussion
The carboxylic acid groups on GO and the amine groups were connected via amide covalent groups formed by the Steglich reaction.32 Steglich esterification is the most common reaction for condensation of carboxylic acid and alcohol groups to form an ester group, and it is also very powerful for the formation of an amide group from a carboxylic acid group and an amine group, which is more nucleophilic than the alcohol group. Successful condensation was confirmed by the appearance of amide groups determined via several analyses, as presented in Fig. 2.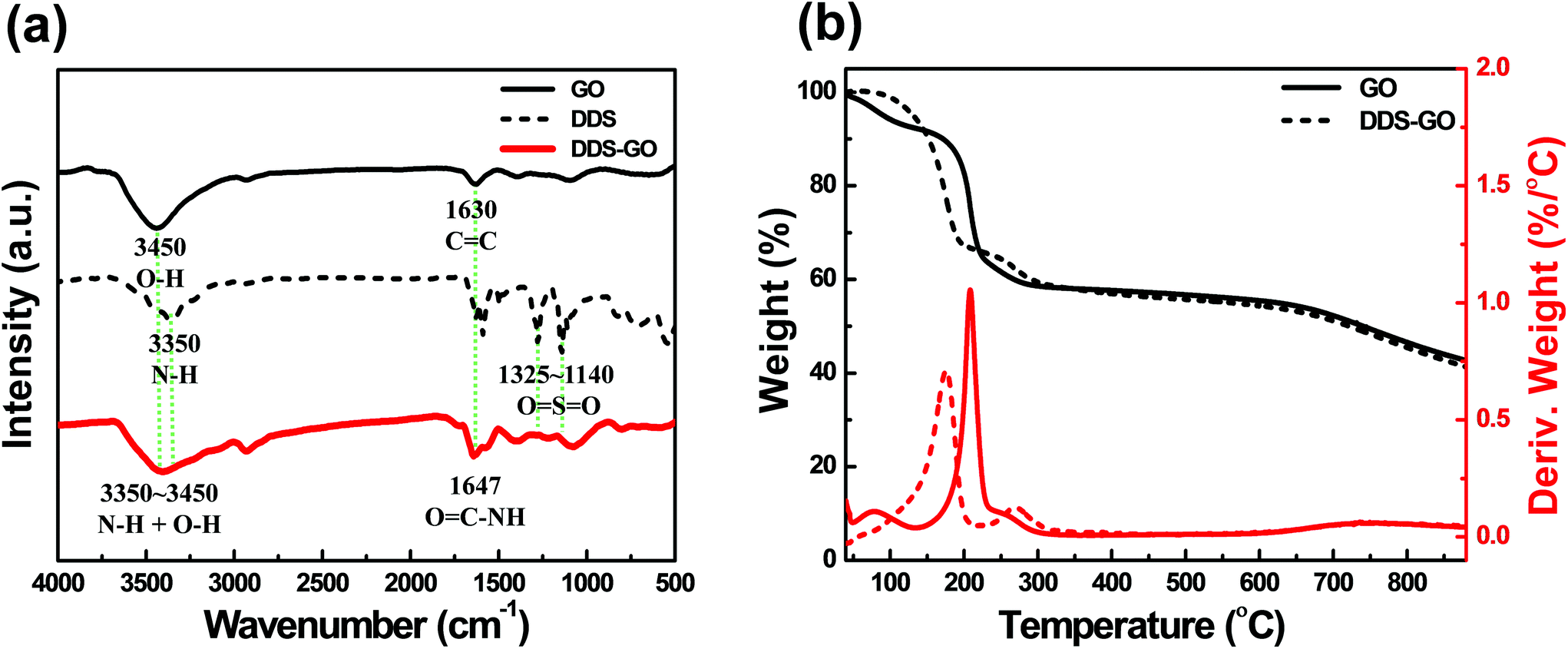 | ||
| Fig. 2 (a) FT-IR spectra of GO, DDS, and DDS-GO. (b) TG-DTA curves of GO and DDS–GO during heating up to 900 °C under a N2 atmosphere. | ||
FT-IR analysis is an important technique to study the structural change in diamine-GO. Representative FT-IR spectra of DDS–GO are presented in Fig. 2a to provide the proof of the successful synthesis of DDS–GO. FT-IR spectra of HMDA–GO are given in the ESI (Fig. S1†). There are three characteristic absorption bands in the FT-IR spectra of DDS–GO. First, an absorption band at 3450 cm−1 attributed to the O–H stretching vibration derived from O–H groups on GO.33 This absorption band was quite broad, because the absorption band at about 3350 cm−1 assigned to the N–H vibration of the amine group on DDS34 combined with the O–H stretching vibration band of GO. Absorption bands corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C skeletal vibration at 1630 cm−1 and CO stretching vibration in the spectra of GO33 and absorption bands of sulfone OSO stretching vibration at 1140–1325 cm−1 in the spectrum of DDS34 also appeared in the FT-IR spectrum of DDS–GO. Meanwhile, a strong absorption band newly appeared at 1647 cm−1 in the spectrum of DDS–GO. We assigned it to the stretching vibration of the amide group created by condensation of the carboxylic acid group at the GO edge with the amine group of DDS.19 These FT-IR analysis results are strong evidence of GO functionalization with the diamine derivatives.
C skeletal vibration at 1630 cm−1 and CO stretching vibration in the spectra of GO33 and absorption bands of sulfone OSO stretching vibration at 1140–1325 cm−1 in the spectrum of DDS34 also appeared in the FT-IR spectrum of DDS–GO. Meanwhile, a strong absorption band newly appeared at 1647 cm−1 in the spectrum of DDS–GO. We assigned it to the stretching vibration of the amide group created by condensation of the carboxylic acid group at the GO edge with the amine group of DDS.19 These FT-IR analysis results are strong evidence of GO functionalization with the diamine derivatives.
Fig. 2b shows the TG-DTA curves of GO and DDS–GO under a N2 atmosphere (for the curves of HMDA–GO, see Fig. S2 in the ESI†). In the TG-DTA curve of GO, the small weight loss peak at 80 °C was due to the loss of residual water in GO, and the main weight loss peak that appeared around 210 °C is assigned to the pyrolysis of the labile oxygen functional groups, yielding CO, CO2, and steam.35,36 On the other hand, we could observe two different major loss peaks from the TG-DTA curve of DDS–GO. The peak near 160 °C could be ascribed to the loss of the remaining oxygen functional groups on GO, which slightly shifted to a temperature lower than that of non-functionalized GO. The peak around 270 °C corresponds to the decomposition of DDS.34 From the decomposed weight, we calculated the substituted amount of DDS in DDS–GO as 7 wt%. With this method, the substituted amount of HMDA in HMDA–GO was also evaluated as 7 wt%. The results of TG-DTA analysis also indicate that diamine derivatives were successfully introduced onto GOs.
Further investigation by XPS analysis was performed to characterize diamine-GOs, and the resulting deconvoluted XPS C 1s spectra of GO and DDS–GO are shown in Fig. 3 (for the spectra of HMDA–GO, see Fig. S3 in the ESI†). In Fig. 3a, five characteristic peaks were observed at 284.1, 285.5, 286.3, 287.8, and 290.0 eV, which correspond to C–C, C–OH, C–O–C, CO, and C(O)–O, respectively. Further, an additional peak at 288.4 eV was observed, Fig. 3b, and it could be assigned to OC–NH moieties.37 Integrated areas of the peaks corresponding to each chemical bond are listed in Table S2.† From the existence of the OC–NH peak in the XPS results, we found that amide groups were newly formed in the functionalized GO.
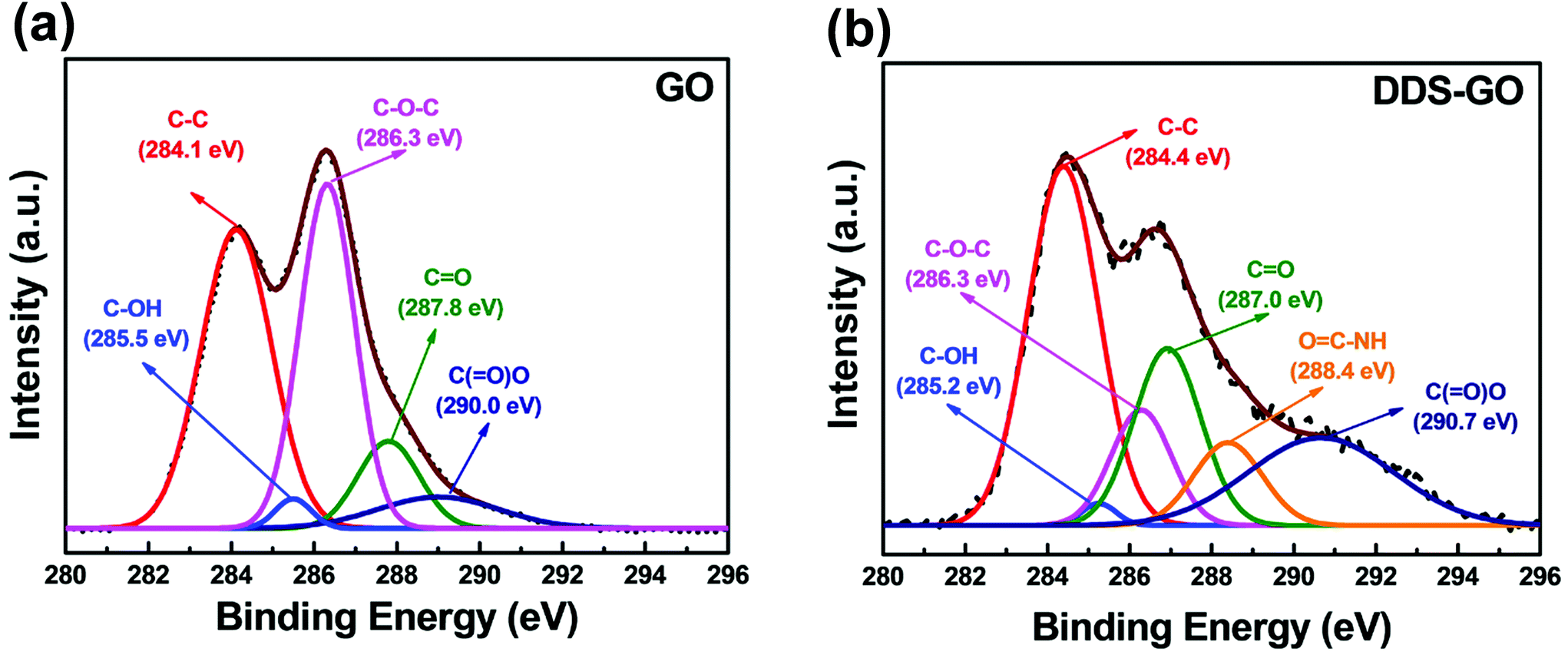 | ||
| Fig. 3 XPS C 1s spectra of (a) GO and (b) DDS–GO. Deconvoluted curves represent the theoretical XPS spectra for different chemical bonds. | ||
Thus, the results of FT-IR, TG-DTA, and XPS analyses confirmed that diamine derivatives were successfully introduced onto GOs via amide chemical bonds.
AFM analysis was employed to study the morphologies of GO and diamine-GOs, and their images are shown in Fig. 4. From these images, the presence of the nanolayer-type GO is clearly observed. The width and thickness of GO and diamine-GO were determined by the analysis results. The image of GO showed 2.0 nm average thickness and 300–1100 nm width range. Further, images of DDS–GO and HMDA–GO revealed an average thickness of 1.0 nm and a width ranging from 100–500 nm. It is considered that the additional ultrasonication process in the case of the functionalized GO would shatter the GO into smaller pieces.
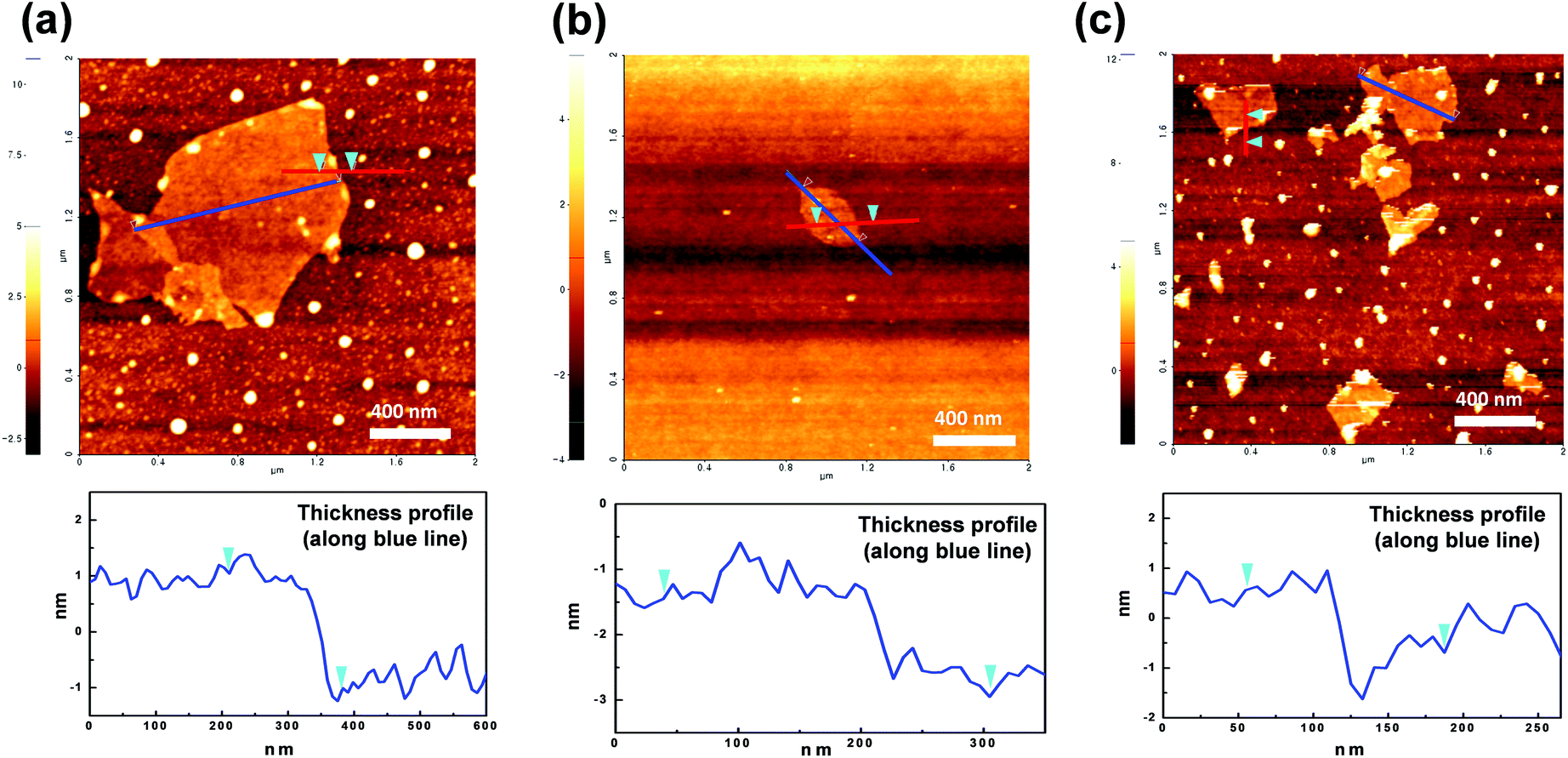 | ||
| Fig. 4 AFM images of (a) GO, (b) DDS–GO, and (c) HMDA–GO. | ||
Good dispersion of nanomaterials greatly influences and improves the properties of the EP nanocomposites. Therefore, in order to investigate the dispersion state, EP/DDS/DDS–GO nanocomposites were observed by optical microscopy. The results are shown in Fig. 5, and they indicated that the DDS–GO dispersion in the EP nanocomposites was very homogeneous when the concentration was increased to 1.0 wt%. The presence of many polar moieties such as carboxylic acid, amide, and amine groups was thought to facilitate the dispersion of DDS–GO in the EP nanocomposite because they actively interact with EP via hydrogen or covalent bonding.
 | ||
| Fig. 5 Optical microscopy photographs of EP/DDS/DDS–GO nanocomposites as a function of weight percentage of DDS–GO. (a), (b), (c), and (d) Correspond to 0, 0.1, 0.5, and 1.0 wt% of DDS–GO into the mixture of EP/DDS, respectively. | ||
Addition of nanomaterials into the EP had a significant effect on the resulting thermal properties. As described in the Experimental section, diamine-GOs were used as co-curing agents of EP nanocomposites and their amount was controlled from 0.0 to 1.5 wt%. Curing temperatures were estimated from the temperatures of exothermic peaks in the first heating DSC curves of the EP/DDS/GO and EP/DDS/diamine-GO mixtures (for the DSC curves, see Fig. S4–6 in the ESI†), and are presented in Table S3.†Tg values of the cured EP/DDS/GO and EP/DDS/diamine-GO nanocomposites were determined based on endothermic peaks of their second heating DSC curves (Fig. 6, Table S4†) (for the DSC curves, see Fig. S7–9 in the the ESI†). When the GO was added to 1.0 wt% to the EP/DDS, the Tg of the EP/DDS/DDS–GO nanocomposite decreased from 160.7 to 136.8 °C. It has been reported that the non-functionalized GOs decrease the Tg of EP nanocomposites because of the tendency of GO to agglomerate in the polymer matrix and disturb the polymer heat flow, but this tendency has not been investigated sufficiently.38 In contrast, when 1.0 wt% of HMDA–GO or DDS–GO was added to the mixture of EP/DDS, the Tg of EP/DDS/diamine-GO nanocomposites remarkably increased to 170.5 or 183.4 °C, respectively; the high values obtained by adding HMDA–GO or DDS–GO are 125 and 135% which are higher than those for EP/DDS/GO nanocomposites, respectively. This is because the amine moieties functionalized on GO could improve the interfacial affinity by reacting with the epoxide groups of the EP resin. Subsequently, the disturbance of the interface between GO and EP could be reduced. If terminal amine groups on GO react with the epoxide groups of EP, diamine-GO could participate in the formation of a polymer crosslink network like the curing agent, and EP/DDS/diamine-GO nanocomposites would show higher CD than the EP/DDS/GO nanocomposite. Further, it is known that the CD of EP nanocomposites is closely related to their thermal properties.21,22
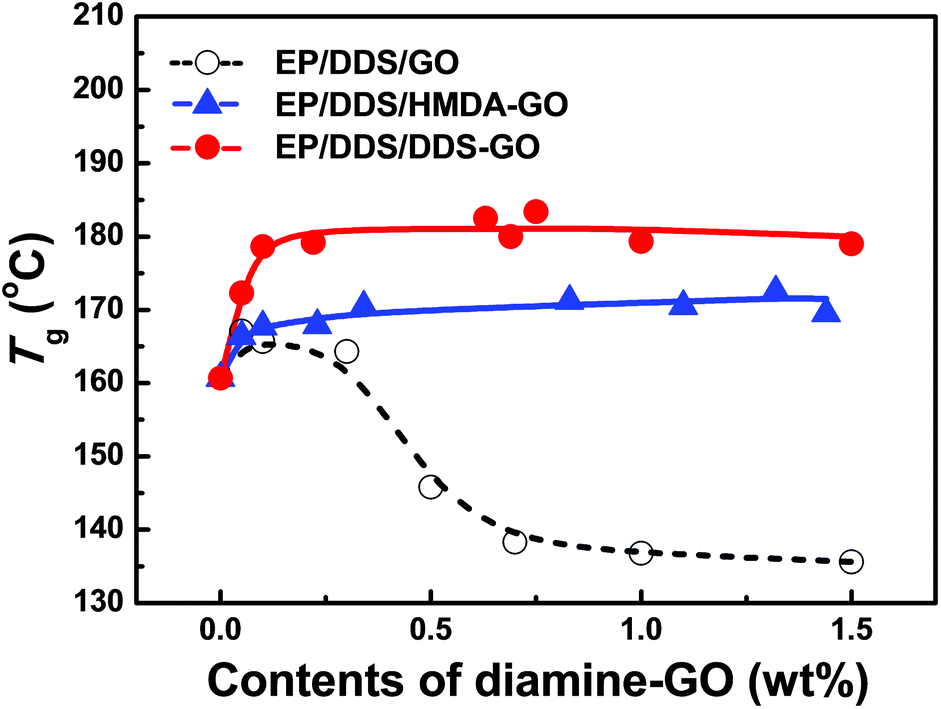 | ||
| Fig. 6 Glass transition temperatures (Tg) of EP/DDS/diamine-GO nanocomposites. Tg was estimated from the second heating DSC results of the EP/DDS/diamine-GO mixture. | ||
DMA measurements of EP/DDS/GO nanocomposites and EP/DDS/DDS–GO nanocomposites were performed to estimate the CD of EP nanocomposites. The storage modulus and tanδ of EP/DDS/DDS–GO nanocomposites are shown in Fig. 7a and b (for the DMA results of the EP/DDS/GO nanocomposite, see Fig. S10 in the ESI†). Based on the storage modulus and tanδ in the rubber region, the CD of EP nanocomposites was calculated according to the following formula:30
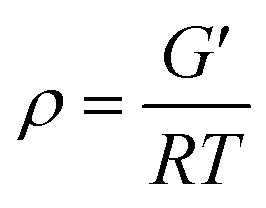 | (2) |
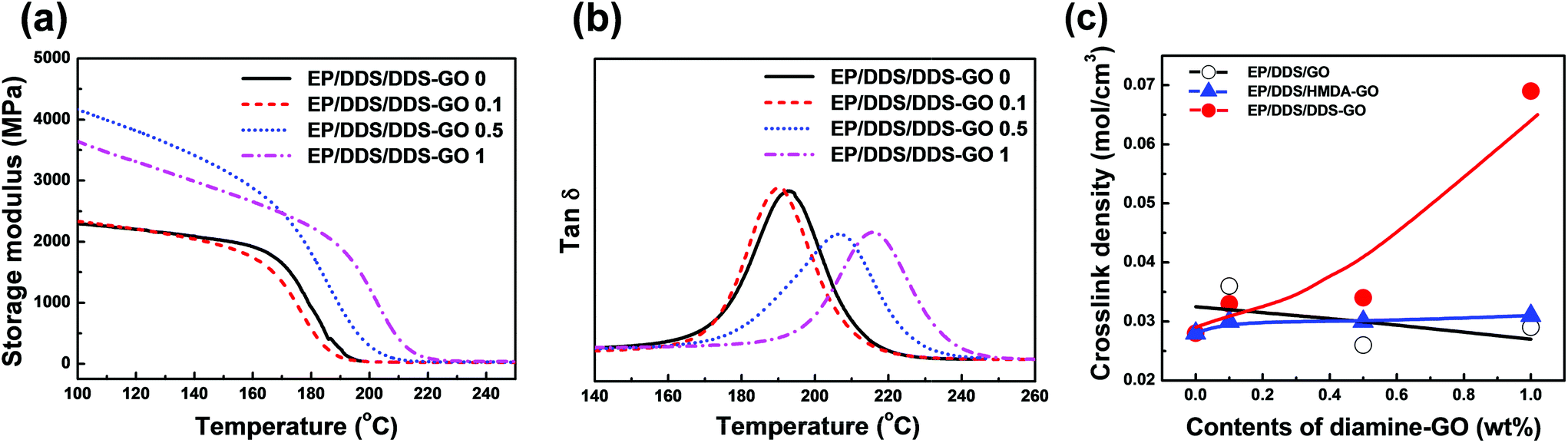 | ||
| Fig. 7 DMA results of EP/DDS/diamine-GO nanocomposites. (a) Storage modulus, (b) tanδ, and (c) crosslink density of the EP/DDS/diamine-GO nanocomposites. Samples were tested in the temperature range of 30 to 260 °C at a scanning rate of 3 °C min−1. The crosslink density was calculated using storage modulus and tanδ in the rubbery region of EP/DDS/diamine-GO nanocomposites. | ||
It has been also known that CD is not only related to the thermal properties of the EP nanocomposite, but also related to their mechanical properties.23,24 In order to investigate the effect of CD on the mechanical properties of the EP nanocomposite, the tensile strength of EP/DDS/diamine-GO nanocomposites was evaluated by UTM analysis; the analysis result is shown in Fig. 8 (for all results of UTM analysis, see Fig. S11–13 in the ESI†). Similar to the Tg, the tensile strength of the EP/DDS/DDS–GO nanocomposite was higher (110.3 MPa) than those of neat EP (87.4 MPa) and the EP/DDS/GO nanocomposite (74.3 MPa) when 0.1 wt% of DDS–GO or GO was added. When 0.1 wt% of HMDA–GO was added, the EP/DDS/HMDA–GO nanocomposite also showed higher tensile strength (90.4 MPa) than neat EP and the EP/DDS/GO nanocomposite, but the tensile strength is relatively smaller than the EP/DDS/DDS–GO nanocomposite (110.3 MPa) with 0.1 wt% of DDS–GO. In the EP/DDS/diamine-GO nanocomposites, diamine-GOs could have strong interactions with EP through covalent bonds between diamine-GOs and EP, and this is considered to be the reason for the improvement in the tensile strength of the EP/DDS/diamine-GO nanocomposite.
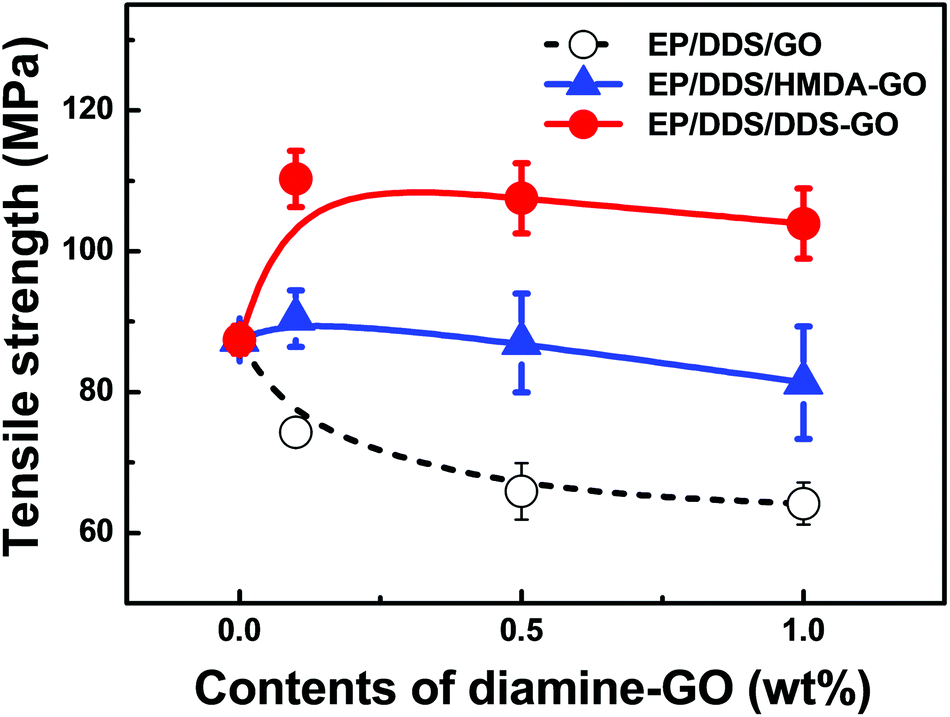 | ||
| Fig. 8 Tensile strengths of EP/DDS/diamine-GO nanocomposites. It was estimated by using a universal testing machine. Samples for measurements were prepared with 1.5 mm × 50 mm × 250 mm size, and tests were performed with a testing speed of 5 mm min−1. | ||
Meanwhile, both EP/DDS/HMDA–GO and EP/DDS/DDS–GO nanocomposites showed higher Tg and tensile strength values than the EP/DDS/GO nanocomposite, but the EP/DDS/DDS–GO nanocomposite exhibited superior properties to the EP/DDS/HMDA–GO nanocomposite. We thought that this result was due to the structural difference between HMDA, which has a flexible aliphatic structure, and DDS, which has a rigid aromatic sulfone structure. In the case of HMDA–GO, terminal amine groups are connected with GO through flexible alkyl chains, and these may cause a lower Tg and tensile strength of the EP/DDS/HMDA–GO nanocomposite than those of the EP/DDS/DDS–GO nanocomposite. In addition, terminal amine groups with different nucleophilicities exist. These terminal amine groups act as curing sites and are considered to be major factors that affect the thermal and mechanical properties of EP nanocomposites. HMDA has electron-rich aliphatic amine groups which can make the curing temperature of HMDA–GO lower than that of the main curing agent DDS. Consequently, a spatially heterogeneous curing reaction will take place in the nanocomposite, and this could negatively affect the thermal properties of EP/DDS/HMDA–GO. In contrast, DDS–GO starts to participate in curing at a similar temperature to DDS. Therefore, the curing reaction would occur more homogeneously in the EP/DDS/DDS–GO nanocomposite, and the thermal properties would be better. This hypothesis about the difference between HMDA–GO and DDS–GO was supported by the CD of EP/DDS/HMDA–GO, which was much lower than that of EP/DDS/DDS–GO. Moreover, the EP/DDS/DDS–GO nanocomposite showed increasing CD when more DDS–GO was added, while the CD of the EP/DDS/HMDA–GO nanocomposite did not change much despite the high contents of HMDA–GO.
Conclusions
We successfully prepared diamine-functionalized graphene oxides (diamine-GOs), by introducing diamine derivatives at the GO edge. The formation of amide groups was confirmed by several analyses such as FT-IR, TG-DTA, and XPS. EP nanocomposites obtained using diamine-GOs as co-curing agents of commercially available EP showed significantly improved thermal and mechanical properties compared to those of neat EP. In particular, 4,4′-diaminodiphenyl sulfone-functionalized GO (DDS–GO) was the most effective additive: 0.1 wt% content of DDS–GO enhanced the Tg of the EP/DDS/DDS–GO nanocomposite from 160.7 °C for EP to 183.4 °C and the tensile strength from 87.4 MPa for EP to 110.3 MPa. The improvement in properties was considered to be due to the good dispersion of DDS–GO in the EP/DDS/DDS–GO nanocomposite and the increase in the crosslink density (CD) of the EP/DDS/DDS–GO nanocomposite. Thus, we can improve the CD value of EP/DDS from 0.028 to 0.069 mol cm−3 by adding 1.0 wt% of DDS–GO. A higher CD value was achieved for EP/DDS/DDS–GO because DDS–GO has terminal amine groups that tend to participate in the formation of the crosslink network via direct covalent bonding with epoxide groups of EP. It is expected that the high-performance EP nanocomposite could be prepared by improving the CD of EP nanocomposites, and the method in this study could provide great potential for future industrial applications.Acknowledgements
This work was supported by a grant from the Korea Institute of Science and Technology (KIST) Institutional program (2Z04480). This work was also supported by the Ministry of Trade, Industry and Energy (MOTIE) and Korea Institute for Advancement of Technology (KIAT) through the Research and Development for Regional Industry (R0004423).References
- A. M. Atta, N. O. Shaker and N. E. Nasser, J. Appl. Polym. Sci., 2008, 107, 347 CrossRef CAS.
- V. Dixit, A. K. Nagpal and R. Singhal, Int. J. Plast. Technol., 2010, 14, 38 CrossRef CAS.
- T. Song, Z. Li, J. Liu and S. Yang, Polym. Sci., Ser. B, 2013, 55, 147 CrossRef CAS.
- B. J. P. Jansen, K. Y. Tamminga, H. E. H. Meijer and P. J. Lemstra, Polymer, 1999, 40, 5601 CrossRef CAS.
- T. V. Kosmidou, A. S. Vatalis, C. G. Delides, E. Logakis, P. Pissis and G. C. Papanicolaou, eXPRESS Polym. Lett., 2008, 2, 364 CrossRef CAS.
- M. Abdalla, D. Dean, M. Theodore, J. Fielding, E. Nyairo and G. Price, Polymer, 2010, 51, 1614 CrossRef CAS.
- M. M. Rahman, S. Zainuddin, M. V. Hosur, C. J. Robertson, A. Kumar, J. Trovillion and S. Jeelani, Compos. Struct., 2013, 95, 213 CrossRef.
- J. Zhu, S. Wei, J. Ryu, M. Budhathoki, G. Liang and Z. Guo, J. Mater. Chem., 2010, 20, 4937 RSC.
- L. Tang, Y. Wan, D. Yan, Y. Pei, L. Zhao, Y. Li, L. Wu, J. Jiang and G. Lai, Carbon, 2013, 60, 16 CrossRef CAS.
- J. A. King, D. R. Klimek, I. Miskioglu and G. M. Odegard, J. Appl. Polym. Sci., 2013, 128, 4217 CrossRef CAS.
- A. S. Wajid, H. S. T. Ahmed, S. Das, F. Irin, A. F. Jankowski and M. J. Green, Macromol. Mater. Eng., 2013, 298, 339 CrossRef CAS.
- Q. Li, Y. Guo, W. Li, S. Qiu, C. Zhu, X. Wei, M. Chen, C. Liu, S. Liao, Y. Gong, A. K. Mishra and L. Liu, Chem. Mater., 2014, 26, 4459 CrossRef CAS.
- T. Ramanathan, A. A. Abdala, S. Stankovich, D. A. Dikin, M. Herrera-Alonso, R. D. Piner, D. H. Adamson, H. C. Schniepp, X. Chen, R. S. Ruoff, S. T. Nguyen, I. A. Aksay, R. K. Prud'homme and L. C. Brinson, Nat. Nanotechnol., 2008, 3, 327 CrossRef CAS PubMed.
- T. Kuilla, S. Bhadra, D. Yao, N. H. Kim, S. Bose and J. H. Lee, Prog. Polym. Sci., 2010, 35, 1350–1375 CrossRef CAS.
- C. Bao, Y. Guo, L. Song, Y. Kan, X. Qian and Y. Hu, J. Mater. Chem., 2011, 21, 13290 RSC.
- B. Qi, S. R. Lu, X. E. Xiao, L. L. Pan, F. Z. Tan and J. H. Yu, eXPRESS Polym. Lett., 2014, 8, 467 CrossRef.
- B. Qi, Z. Yuan, S. Lu, K. Liu, S. Li, L. Yang and J. Yu, Fiber Polym., 2014, 15, 326 CrossRef CAS.
- M. M. Gudarzi and F. Sharif, eXPRESS Polym. Lett., 2012, 6, 1017 CrossRef.
- T. Liu, Z. Zhao, W. W. Tjiu, J. Lv and C. Wei, J. Appl. Polym. Sci., 2014, 131, 40236 Search PubMed.
- L. Guan, Y. Wan, L. Gong, D. Yan, L. Tang, L. Wu, J. Jiang and G. Lai, J. Mater. Chem. A, 2014, 2, 15058 CAS.
- T. Iijima, N. Yoshioka and M. Tomoi, Eur. Polym. J., 1992, 28, 573 CrossRef CAS.
- K. W. Putz, M. J. Palmeri, R. B. Cohn, R. Andrews and L. C. Brinson, Macromolecules, 2008, 41, 6752 CrossRef CAS.
- K. Kim, I. Jeon, S. Ahn, Y. Kwon and J. Baek, J. Mater. Chem., 2011, 21, 7337 RSC.
- K. Wei, G. Zhu, Y. Tang, T. Liu and J. Xie, J. Mater. Res., 2013, 28, 2903 CrossRef CAS.
- V. Patil, R. V. Dennis, T. K. Rout, S. Banerjee and G. D. Yadav, RSC Adv., 2014, 4, 49264 RSC.
- G. Z. Li, L. Wang, H. Toghiani, T. L. Daulton, K. Koyama and C. U. Pittman, Macromolecules, 2001, 34, 8686 CrossRef CAS.
- J. Park and S. C. Jana, Macromolecules, 2003, 36, 8391 CrossRef CAS.
- W. Naous, X. Yu, Q. Zhang, K. Naito and Y. Kagawa, J. Polym. Sci., Polym. Phys. Ed., 2006, 44, 1466 CrossRef CAS.
- M. S. Lakshmi, B. Narmadha and B. S. R. Reddy, Polym. Degrad. Stab., 2008, 93, 201 CrossRef CAS.
- C. Lee and J. Park, Trans. Electr. Electron. Mater., 2010, 11, 69 CrossRef.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- B. Neises and W. Steglich, Angew. Chem., Int. Ed. Engl., 1978, 17, 522 CrossRef.
- L. Shahriary and A. A. Athawale, Int. J. Renewable Energy Environ. Eng., 2014, 2, 58 Search PubMed.
- L. Jiang, Y. Huang, Q. Zhang, H. He, Y. Xu and X. Mei, Cryst. Growth Des., 2014, 14, 4562 CAS.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558 CrossRef CAS.
- X. Du, Z. Yu, A. Dasari, J. Ma, M. Mo, Y. Meng and Y. Mai, Chem. Mater., 2008, 20, 2066 CrossRef CAS.
- Y. Chen, X. Zhang, P. Yu and Y. Ma, Chem. Commun., 2009, 4527 RSC.
- N. Norhakim, S. H. Ahmad, C. H. Chia and N. M. Huang, Sains Malays., 2014, 43, 603 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Results of FT-IR spectra, TD-TGA curves, XPS spectra, DSC curves, DMA analysis, and UTM analysis of EP nanocomposites. See DOI: 10.1039/c5py01483b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2016 |