
Physical hydrogels constructed on a macro-cross-linking cationic polysaccharide with tunable, excellent mechanical performance†
Xiaomei
Ma
*ab,
Lei
Guo
a,
Quan
Ji
ab,
Yeqiang
Tan
ab,
Yacheng
Xing
a and
Yanzhi
Xia
*bc
aSchool of Chemistry and Chemical Engineering, Qingdao University, Qingdao 266071, P. R. China. E-mail: mxmalice@163.com; qdxyzh@163.com
bCollaborative Innovation Center for Marine Biomass Fibres, Materials and Textiles of Shandong Province, Qingdao 266071, P. R. China
cState Key Laboratory Cultivating Base for New Fibre Materials and Modern Textiles, Qingdao University, Qingdao 266071, P. R. China
First published on 12th October 2015
Abstract
Cationic chitosan was exploited originally as a macro-cross-linker to prepare hydrogels with superb extensibility, perfect elasticity, high toughness and fatigue-resistance by one-step free-radical polymerization. The as-developed approach can be applied to a broad range of hydrophilic monomers and unsaturated Brønsted–Lowry acids, leading to hydrogels with tunable performance.
Soft-wet polymeric hydrogels have long been attracting the attention of scientists in materials science and the biomedical field, being considered as optimal substitutes for injured tissues (such as articular cartilage, tendons, muscles and ligaments) attributed to their similarities to biological tissues. However, the effective utilization of traditional chemically crosslinked hydrogels as load-bearing structural materials is seriously hampered by the fragility and weakness arising from an inhomogeneous network structure. In the past decade, various hydrogels with novel network structures have been developed relevant to enhancing mechanical performance, such as topological hydrogels with movable crosslinks,1,2 tetra-PEG hydrogels3 with a homogeneous network, double-network (DN) hydrogels with two highly asymmetrical interpenetrating networks,4,5 and hydrogels constructed based on poly-functional “macro-cross-linkers” including nanoscale substances (such as clay,6 graphene oxide,7 hydroxyapatite,8 silica nanoparticles,9 cellulose nanocrystals10), radiation peroxidized macromolecular microspheres, graphene, micelles and polymer chains,11–14 allylic starch-based nano-spheres and vinylated micelles.15–17 Although great advances have been made concerning hydrogel mechanical performance, there remains some bottleneck problems to be addressed, such as cumbersome or circumscribed preparation procedures, lack of integrated high mechanical performance such as strength, toughness and fatigue-resistance.
The rupture of hydrogels generally occurs through crack initiation and successive expanding, correlated with strength and toughness respectively.18,19 Hydrogels with macro-cross-linkings or homogeneous/topological networks can distribute the applied force homogeneously inside the network, decreasing the probability of initial crack formation, thus enhancing the strength (fracture stress) of the hydrogels.1,3,6–17 To improve hydrogel's toughness, it is pivotal to introduce an effective energy dissipation mechanism to increase the fracture energy and resist crack expanding, such as breakage of covalent bonds in the first network of fully chemically linked DN hydrogels.18 Yet the poor fatigue-resistance resulting from permanent damage by irreversible disruption of covalent bonds remains an obstacle to their practical application. Substituting irreversible covalent bonds with reversible noncovalent association such as H-bonding, electrostatic interaction (ionic association) and hydrophobic association should be an efficacious protocol to overcome this drawback.5
Among the reversible interactions, ionic association might be the best mechanism to achieve a “hypo covalent bond” with some degree of reversibility attributed to its higher strength than H-bonding and hydrophobic association.20,21 Accordingly, to decrease the probability of crack initiating and inhibit crack propagation concurrently, it is advantageous to integrate ionic association with a “macro-cross-linker” for the construction of hydrogels with satisfactory mechanical performance.
To test and verify the protocol, biocompatible and biodegradable polycationic chitosan is engineered as a “macro-cross-linker” in this research. Chitosan is a linear copolymer composed of glucosamine and N-acetyl glucosamine units (as shown in Fig. 1a), deacetylation products of chitin – a naturally occurring polysaccharide with large abundance. As is well-known, chitosan is insoluble in water due to inter-/intra-molecular H-bonding and its crystallizing structure, but the amino group chitosan bears make it a weak base that can be dissolved in the aqueous solution of a Brønsted–Lowry acid, which protonates –NH2 groups to cationic –NH3+, forming ion-association complexes with the acid radicals simultaneously (as illustrated in Fig. 1a). Taking advantage of chitosan's intrinsic alkalinity, we planned to neutralize chitosan with an unsaturated Brønsted–Lowry acid to yield an ion-association complex bearing vinyl groups (collectively called vinylated chitosan) first, then to manufacture hydrogels through free-radical copolymerization of hydrophilic monomers (as hydrogel monomers) with the as-formed vinylated chitosan functioning as a multifunctional macro-cross-linker (see the ESI†). During polymerization, the vinyl groups electrostatically bound to chitosan react with hydrophilic monomers, forming irreversible covalent bonds with the hydrophilic monomers and becoming chain units of hydrophilic polymers. Consequently, in the resulting hydrogel, the electronic interaction between –NH3+ and acid radicals becomes the primary force linking the hydrophilic polymer chains and chitosan chains (Fig. 1b). The mechanism involved in the formation of hydrogels is simple and easy to understand, yet the approach has not been explored to prepare hydrogels with high mechanical performance. Although there have been numerous reports on chitosan-based hydrogels, all of which make use of chitosan as the substrate of hydrogels, this is the first time chitosan has been employed as a macro-cross-linker for high mechanical performance hydrogels, as far as we know.
 | ||
| Fig. 1 (a) Schematic formation of chitosan complexes, (b) hydrogel forming mechanism based on macro-crosslinking vinylated chitosan, (c) PMAA-CMAA0.22-4.17-20 hydrogels before, on and after compression, (d) PMAA-CMAA0.47-4.17-17.5 hydrogels before and after stretching. | ||
According to the aforementioned strategy, methacrylic acid (MAA) was explored as the unsaturated Brønsted–Lowry acid first. Preparation of polymethacrylic acid hydrogels was investigated systematically using MAA vinylated chitosan (CMAA) as the macro-cross-linker. The obtained hydrogels (denoted as PMAA-CMAAi-c-s, where i mol%, c mol% and s wt% stand for the initiator dosage, chitosan ratio and solids content respectively) show super stretchable, highly elastic, tough and high fatigue-resistant performance as anticipated (Fig. 1c and d).
The formation of ion-association complexes is confirmed by Fourier transform infrared (FT-IR) spectroscopy. Fig. 2 depicts the IR spectrograms of MAA, chitosan, CMAA and PMAA-CMAA0.22-4.17-20 hydrogels. The absorption band at 1697 cm−1 caused by the –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration of –COOH groups in MAA's spectrogram disappears in CMAA's spectrogram, while a new peak at 1540 cm−1 appears, attributed to asymmetric deformation of –COO−, indicating the complex formation of –COO− with –NH3+. The peak remains in PMAA-CMAA's IR curve. Compared to MAA and CMAA, the peak at 1635 cm−1 caused by the –CC– stretching vibration disappears in PMAA-CMAA as a result of polymerization.
O stretching vibration of –COOH groups in MAA's spectrogram disappears in CMAA's spectrogram, while a new peak at 1540 cm−1 appears, attributed to asymmetric deformation of –COO−, indicating the complex formation of –COO− with –NH3+. The peak remains in PMAA-CMAA's IR curve. Compared to MAA and CMAA, the peak at 1635 cm−1 caused by the –CC– stretching vibration disappears in PMAA-CMAA as a result of polymerization.
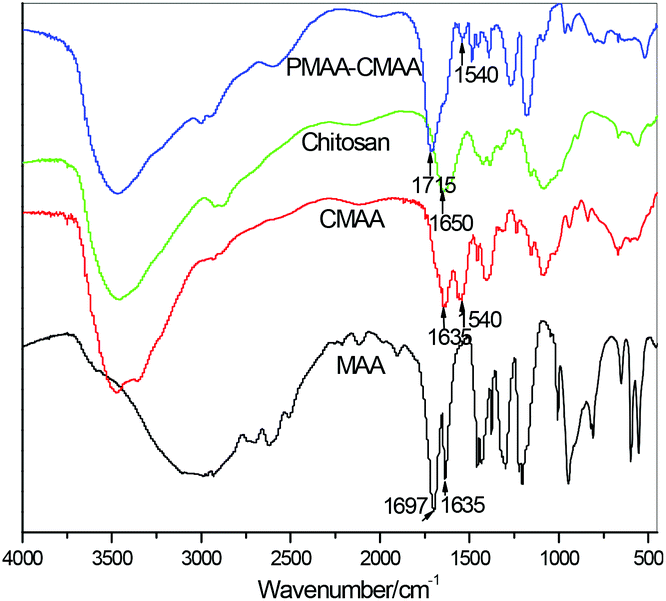 | ||
| Fig. 2 IR spectrograms of MAA, chitosan, CMAA and PMAA-CMAA0.22-4.17-20 hydrogels. | ||
The influence of the preparation conditions including the initiator dosage (rKPS, moles of the initiator to total moles of the unsaturated Brønsted–Lowry acid and hydrogel monomer, where KPS stands for potassium persulfate), chitosan ratio (rchitosan, measured by the molar ratio of –NH2 groups to hydrogel monomers) and solids content (SC) on the mechanical performance of PMAA-CMAA hydrogels was investigated systematically by mechanical testing. Uniaxial tensile measurements (Fig. SI1†) demonstrate that, when SC and rchitosan are kept constant at 20 wt% and 4.17 mol%, the tensile strength of PMAA-CMAA hydrogels decreases from 50 kPa to 17 kPa as rKPS increases from 0.10 mol% to 0.48 mol%, while the fracture elongation increases from 2070% to 6596%. At constant rchitosan (4.17 mol%) and rKPS (0.22 mol%), both tensile strength and fracture elongation increase with SC over the range of 15%–25 wt%, from 16 kPa and 2271% to 50 kPa and 4875% respectively. For hydrogels with constant SC (20 wt%) and rKPS (0.22 mol%), the tensile strength increases whereas the fracture elongation decreases monotonously with rchitosan from 2.89 mol% to 8.67 mol%. These results together with the elastic modulus and fracture energy data (Fig. SI2†) indicate super stretchable, soft but tough properties of PMAA-CMAA hydrogels. The superstretchability is demonstrated intuitively by the large strain of the hydrogel which doesn't break even if being elongated beyond 7000% when prepared at SC17.5 wt%, rchitosan 4.17 mol% and rKPS 0.47 mol% (see Fig. 1d).
Similar to tensile performance, compressive performance of PMAA-CMAA hydrogels is strongly dependent on the preparation conditions. The compressive stress of the hydrogels increases with the increase of rchitosan and SC, but decreases with the increase of rKPS (Fig. SI3†). Most of the prepared hydrogels do not fracture even if compressed to 98% strain except that cracking only occurs to the one with 8.67 mol% rchitosan, 20 wt% SC and 0.22 mol% rKPS, whose compressive strength achieves ∼17 MPa, indicating the high strength and toughness of the prepared hydrogels.
The excellent mechanical performance of PMAA-CMAA hydrogels is contrasted with PMAA hydrogels crosslinked by small molecular N,N′-methylene bisacrylamide (PMAA-BA) prepared with the same SC, rKPS and molar ratio of crosslinking groups (20 wt%, 0.22 mol% and 4.17 mol% as an example). As shown in Fig. 3a, PMAA-CMAA is compressed to 98% strain without any cracking by a stress of 9.95 MPa, while PMAA-BA fails below 60% strain with the compression strength of only ∼0.38 MPa, far less than that of PMAA-CMAA. The toughness of PMAA-CMAA (the area under the stress–strain curve) is 0.58 MJ m−3, nearly 10 times that of PMAA-BA (∼0.06 MJ m−3). PMAA-CMAA is superstretchable and can withstand more than 3500% stretch, while PMAA-BA is fragile and breaks when elongated to only ∼120% of its original length (Fig. 3b). Although PMAA-BA (with a Young's modulus of 0.032 MPa) is a bit stiffer than PMAA-CMAA (with a Young's modulus of 0.014 MPa), the tensile strength of PMAA-CMAA (0.033 MPa) is larger than that of PMAA-BA (0.023 MPa), especially the fracture energy of PMAA-CMAA (∼0.87 MJ m−3) is far larger (about 43 times) than that of PMAA (∼0.02 MJ m−3). The experimental results indicate the realization of hydrogels with high strength, super extensibility and high toughness using polyfunctional chitosan as a crosslinker in lieu of small molecular crosslinkers.
 | ||
| Fig. 3 Compressive (a) and tensile (b) stress–strain curves of PMAA hydrogels crosslinked by N,N′-methylene bisacrylamide and chitosan. | ||
The superstretchability should be ascribed primarily to extending of the curled, long polymer chains and the disruption of ionic interactions between crosslinks on loading, which distributes the applied force effectively, preventing crack propagation and thereby strengthening and toughening the formed hydrogels. However, the flexibility originating from the flexible, long polymer chains between crosslinks also contributes to soft hydrogels.
Cyclic compressive testing on PMAA-CMAA0.22-4.17-20 hydrogels without waiting under 90% strain gives almost overlapping hysteresis loops (Fig. 4a) for 5 consecutive loading–unloading measurements. The recovery rates5 for the 2nd to 5th cycles determined by the elastic modulus ratio are all around 90%, and are all above 90% determined by the energy loss ratio (Fig. 4b), indicating good resilience and recoverability as well as high fatigue-resistance of PMAA-CMAA hydrogels, which are verified further by cyclic tensile testing.
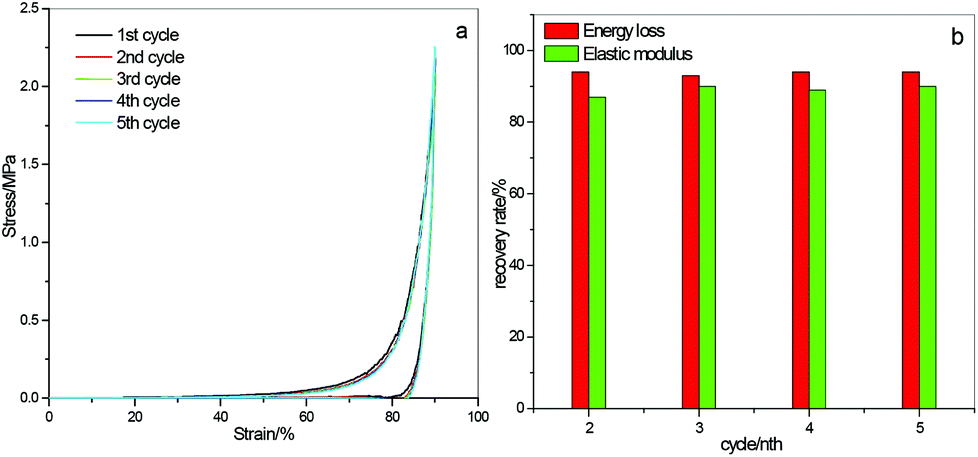 | ||
| Fig. 4 (a) Cyclic compressive stress–strain curves of PMAA-CMAA0.22-4.17-20 hydrogels with five consecutive cycles; (b) compression recovery rate of the same sample determined by the elastic modulus ratio and energy loss ratio. | ||
Fig. 5a displays three consecutive cyclic tensile loading–unloading cycles of PMAA-CMAA0.22-4.17-20 hydrogels under 500%, 1000% and 2000% strain respectively. The large hysteresis loop of the first cycle indicates effective energy dissipation during extension primarily due to the deformation and dissociation of ionic complexes under stress. A different residual strain was observed at a different stretch after unloading. The corresponding residual strains are ca. 25%, 105% and 260% respectively to 500%, 1000% and 2000% tensile strain, i.e. the larger stretch with the larger residual strain. The hysteresis loops of the second and third cycles are close and much smaller than the first cycle, meaning reducing of the energy dissipation capability after the first cycle. However, the residual strain disappeared spontaneously at room temperature rapidly without any additional stimuli. For instance, ∼8% residual strain was observed for 1000% tensile stretch after 2 h (shown in Fig. 5b), which might be caused by chain disentanglement and is difficult to recover completely. The cyclic tensile behaviour of PMAA-CMAA0.22-4.17-20 hydrogels subjected to 1000% strain after different resting times is displayed in Fig. 5c. The second cycle following the first one immediately exhibits a much smaller hysteresis loop, indicating that the deformation could not recover at once without resting. However, the hysteresis loop gets larger when the hydrogel restores for a short while. The recovery extent determined by the energy loss ratio and elastic modulus ratio attains 80% and 84% respectively after 2 hours’ resting (Fig. 5d), exhibiting better recoverability and fatigue-resistance of PMAA-CMAA hydrogels than many already reported.5,6 The good recoverability and high fatigue-resistance can mainly be attributed to reversible disruption–reforming of ion interactions between the networks. Additionally, reversible H-bondings between polymer chains also have significant effects in enhancing the hydrogel's mechanical performance (see below).
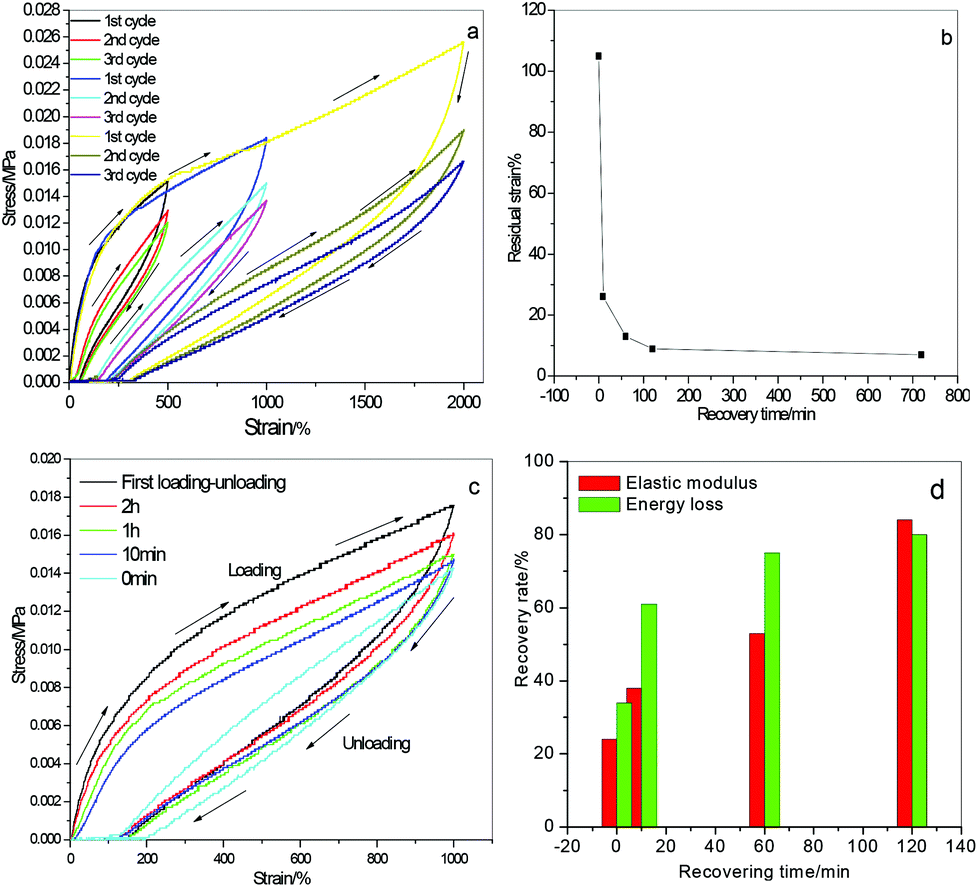 | ||
| Fig. 5 (a) Tensile stress–strain curves of PMAA-CMAA0.22-4.17-20 with three consecutive cycles under 500%, 1000% and 2000% stretch; (b) residual strain of the same sample stretched to 1000% with resting time after the first cycle; (c) tensile loading–unloading curves after different recovering times; (d) recovery rate determined by the elastic modulus ratio and energy loss ratio. | ||
Besides MAA, many other hydrophilic monomers can be exploited as the hydrogel monomers, such as acrylamide (AM), 2-hydroxyl-ethyl acrylate (HEA), 2-hydroxyethyl methacrylate (HEMA), and N,N-dimethylacrylamide (DMAM). Specifically, acrylic acid (AA) and 2-acrylamido-2-methyl-1-propanesulfonic acid (AMPS) are also suitable as the unsaturated Brønsted–Lowry acids except for MAA. The mechanical performance of the corresponding hydrogels is shown in Table 1. The difference demonstrated by different hydrogels indicates that other secondary interactions caused by the molecular structure, such as H-bonding, play an important but not a decisive role inside the network (Fig. SI4†).
| Hydrogelsa | Tensile performance | Compressive performance | |||
|---|---|---|---|---|---|
| Strength (kPa) | Fracture strain (%) | Toughness (MJ m−3) | Stress at 98% strain (MPa) | Fractured or not | |
| a All hydrogels were prepared with a feed composition of 20 wt% SC, 0.22 mol% rKPS and 4.17 mol% rchitosan. | |||||
| PAM-CMAA | 33 | 3100 | 1.05 | 5.93 | Not |
| PDMAM-CMAA | 28 | 1200 | 0.15 | 9.06 | Not |
| PHEA-CMAA | 20 | 1230 | 0.16 | 8.35 | Not |
| PHEMA-CMAA | — | — | — | 6.12 | Fractured |
| PAA-CAA | 31 | 7500 | 1.90 | 6.61 | Not |
| PAM-CAA | >31 | >9000 | >2.11 | 5.74 | Not |
| PHEA-CAA | 15 | 680 | 0.07 | 6.55 | Not |
| PAM-CAMPS | 11 | 5100 | 0.52 | 5.20 | Not |
Conclusions
In conclusion, a facile and universal approach is proposed in this communication which employs biomassed polycationic chitosan as a macro-cross-linker for the construction of hydrogels through one-step polymerization. The as-prepared hydrogels are physically linked mainly through reversible ionic association between cationic chitosan and anionic acid radicals. The comprehensive effects of ionic association and H-bonding endow the hydrogels with excellent mechanical performance, especially high fatigue-resistance. Specifically, the approach shows great flexibility in that the hydrogel network structure and the inter/intra-molecular interactions inside can be regulated by many aspects such as variation of preparing conditions (such as the initiator ratio, chitosan dosage or solids content), and alteration of hydrogel monomers or unsaturated Brønsted–Lowry acids which complex with chitosan, contributing to tunable tensile/compressive performance to meet different needs.Acknowledgements
The authors acknowledge financial support from the Collaborative Innovation Centre for Marine Biomass Fibres, Materials and Textiles of Shandong Province and the National Science Foundation of China (no. 51403113).Notes and references
- Y. Okumura and K. Ito, Adv. Mater., 2001, 13, 485 CrossRef CAS.
- N. Miyamoto, K. Shimasaki, K. Yamamoto, M. Shintate, Y. Kamachi, B. P. Bastakoti, N. Suzuki, R. Motokawa and Y. Yamauchi, Chem. – Eur. J., 2014, 20, 14955 CrossRef CAS PubMed.
- T. Sakai, T. Matsunaga, Y. Yamamoto, C. Ito, R. Yoshida, S. Suzuki, N. Sasaki, M. Shibayama and U. Chung, Macromolecules, 2008, 41, 5379 CrossRef CAS.
- J. P. Gong, Y. Katsuyama, T. Kurakawa and Y. Osada, Adv. Mater., 2003, 15, 1155 CrossRef CAS.
- Q. Chen, L. Zhu, C. Zhao, Q. Wang and J. Zheng, Adv. Mater., 2013, 25, 4171 CrossRef CAS PubMed.
- (a) K. Haraguchi and T. Takehisa, Adv. Mater., 2002, 14, 1120 CrossRef CAS; (b) M. Zhu, Y. Liu, B. Sun, W. Zhang, X. Liu, H. Yu, Y. Zhang, D. Kuckling and H. J. P. Adler, Macromol. Rapid Commun., 2006, 27, 1023 CrossRef CAS.
- R. Liu, S. Liang, X. Z. Tang, D. Yan, X. Li and Z. Z. Yu, J. Mater. Chem., 2012, 22, 14160 RSC.
- A. K. Gaharwar, S. A. Dammu, J. M. Canter, C. J. Wu and G. Schmidt, Biomacromolecules, 2011, 12, 1641 CrossRef CAS PubMed.
- L. Carlsson, S. Rose, D. Hourdet and A. Marcellan, Soft Matter, 2010, 6, 3619 RSC.
- J. Yang, C. R. Han, X. M. Zhang, F. Xu and R. C. Sun, Macromolecules, 2014, 47, 4077 CrossRef CAS.
- T. Huang, H. G. Xu, K. X. Jiao, L. P. Zhu, H. R. Brown and H. L. Wang, Adv. Mater., 2007, 19, 1622 CrossRef CAS.
- J. Q. Liu, C. F. Chen, C. C. He, J. Zhao, X. J. Yang and H. L. Wang, ACS Nano, 2012, 6, 8194 CrossRef CAS PubMed.
- C. He, K. Jiao, X. Zhang, M. Xiang, Z. Li and H. Wang, Soft Matter, 2011, 7, 2943 RSC.
- C. He, Z. Zheng, D. Zhao, J. Liu, J. Ouyang and H. Wang, Soft Matter, 2013, 9, 2837 RSC.
- Y. Tan, K. Xu, P. Wang, W. Li, S. Sun and L. Dong, Soft Matter, 2010, 6, 1467 RSC.
- C. Liu, Y. Tan, K. Xu, Y. Li, C. Lu and P. X. Wang, Carbohydr. Polym., 2014, 105, 270 CrossRef CAS PubMed.
- Y. N. Sun, G. R. Gao, G. L. Du, Y. J. Cheng and J. Fu, ACS Macro Lett., 2014, 3, 496 CrossRef CAS.
- J. P. Gong, Soft Matter, 2010, 6, 2583 RSC.
- X. Zhao, Soft Matter, 2014, 10, 672 RSC.
- K. J. Henderson, T. C. Zhou, K. J. Otim and K. R. Shull, Macromolecules, 2010, 43, 6193 CrossRef CAS.
- T. L. Sun, T. Kurokawa, S. Kuroda, A. B. Ihsan, T. Akasaki, K. Sato, M. A. Haque, T. Nakajima and J. P. Gong, Nat. Mater., 2013, 12, 932 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, Fig. SI1–SI4. See DOI: 10.1039/c5py01437a |
| This journal is © The Royal Society of Chemistry 2016 |