
Post-polymerization modification of poly(vinyl ether)s: a Ru-catalyzed oxidative synthesis of poly(vinyl ester)s and poly(propenyl ester)s†
Di
Liu
a and
Christopher W.
Bielawski
*bc
aDepartment of Chemistry, The University of Texas at Austin, Austin, TX 78712, USA
bCenter for Multidimensional Carbon Materials (CMCM), Institute for Basic Science (IBS), Ulsan 689-798, Republic of Korea. E-mail: bielawski@unist.ac.kr
cDepartment of Chemistry and Department of Energy Engineering, Ulsan National Institute of Science and Technology (UNIST), Ulsan 689-798, Republic of Korea
First published on 20th October 2015
Abstract
Poly(vinyl ester)s were readily prepared via a ruthenium catalyzed C–H oxidation of the corresponding poly(vinyl ether)s under mild conditions. The transformations were efficient and in many cases proceeded without significant chain cleavage. The method was also successfully used to prepare high molecular weight poly(propenyl ester) for the first time as well as a polyester with a relatively high content (>50%) of γ-butyrolactone repeat unit from poly(tetrahydrofuran). The polymeric products were characterized via FT-IR spectroscopy, NMR spectroscopy, gel permeation chromatography, and other techniques.
Introduction
Post-polymerization modification is a powerful method for gaining access to various types of functionally and structurally advanced macromolecules.1,2 Several industrially important polymers, such as poly(vinyl alcohol) and poly(vinyl butyral), are only accessible via post-polymerization modification since the corresponding monomers can not be isolated. For this reason, efforts have been directed toward utilizing reactive groups, such as activated esters and epoxides, as derivatization handles; as such, pre-installation of the requisite functional groups on the polymer precursor is generally required.1,3The direct transformation of relatively inert C–H bonds omnipresent in most synthetic polymers represents a potentially powerful post-polymerization modification strategy. Although there has been significant progress in the development of C–H bond functionalization methods for small molecules in recent years,4–9 only a few examples have been reported for the modification of polymeric materials.10 Elegant examples of such approaches include the regioselective functionalization of isotactic polypropylene and the main-chain modification of polysulfone via iridium or rhodium catalyzed C–H borylation.11–13
Although poly(vinyl ester)s are often obtained via the free radical polymerization of the corresponding vinyl ester monomer, the polymer produced often features ill-defined microstructures, including high degrees of head-to-head linkages and extensive branching.14 Moreover, some monomers, such as β-substituted vinyl esters and isopropenyl esters, are challenging to polymerize using free radical techniques due to steric hindrance and/or degradative chain transfer processes.15,16 For example, current methods for preparing high molecular weight (Mw > 10 kDa) poly(isopropenyl acetate) require high pressures and afford relatively low yields of polymer.17
In contrast, vinyl and propenyl ethers readily undergo cationic polymerization and well-defined polymeric materials are often obtained.18–20 Ruthenium tetroxide mediated oxidation of aliphatic ethers to their corresponding esters is a well-established and efficient transformation.21–23 We envisioned overcoming the aforementioned limitations associated with synthesizing poly(vinyl ester)s and poly(propenyl ester)s by taking advantage of established cationic polymerization process in conjunction with an efficient Ru catalyzed C–H oxidation methodology.24
Herein, we describe the synthesis of various poly(vinyl ester)s and poly(propenyl ester)s via a Ru catalyzed oxidation of the corresponding poly(vinyl ether)s and poly(propenyl ether)s. In addition, we demonstrate that the method may also be used to access aliphatic polyesters via the main chain modification of poly(tetrahydrofuran).
Results and discussion
Oxidation of poly(vinyl ether)s
Initial efforts were directed toward the oxidation of poly(butyl vinyl ether) (PBVE) using various Ru based catalysts, including RuO2·xH2O and RuCl3·xH2O. As summarized in Table 1, various oxidants and solvent mixtures commonly used in RuO4 mediated oxidation reactions were explored. Oxidants such as NaBrO3 or NaIO4 were found to be effective and resulted in high conversions of the starting materials and excellent selectivities (vide infra), as determined by NMR spectroscopy. In contrast, the use of oxone or aqueous NaClO resulted in rapid consumption of the oxidant, which may have limited the conversions observed. In addition, the use of binary mixtures of a chlorinated solvent and water gave poor results, presumably due to premature catalyst deactivation during the reaction.22 While the addition of CH3CN improved the conversion of starting material, relatively low selectivities were observed. Ultimately, a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) mixture of ethyl acetate (EtOAc):H2O was determined to be the optimal solvent.
:1 (v/v) mixture of ethyl acetate (EtOAc):H2O was determined to be the optimal solvent.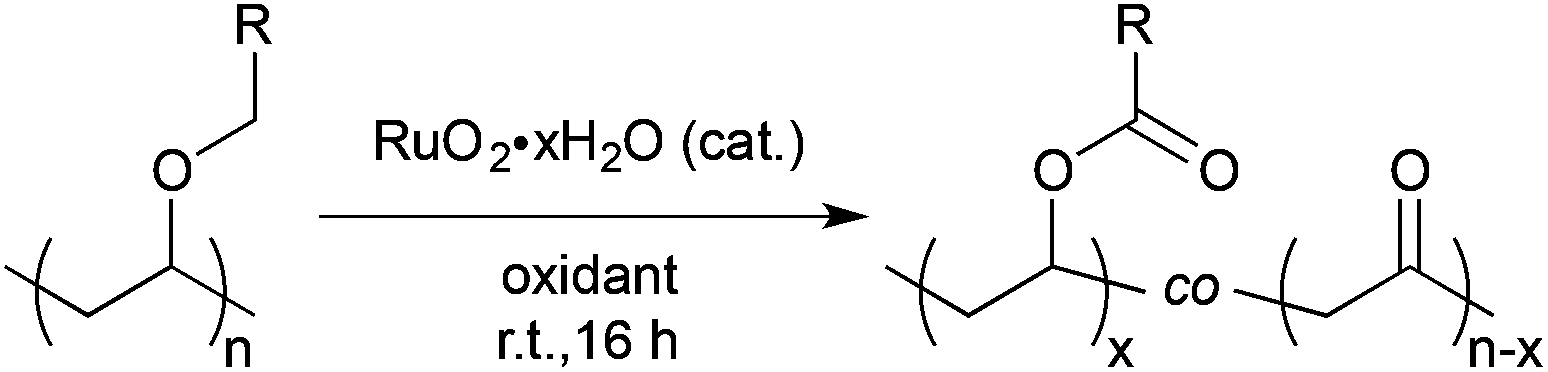 | (1) |
| Entry | R | Solventb | Oxidant | Conversionc (%) | Selectivityc,d (%) |
|---|---|---|---|---|---|
|
a See eqn (1). Conditions: 1 mg RuO2·xH2O, 10 mg poly(vinyl ether) in 2 mL of the solvent indicated, oxidant (4 equiv. with respect to the polymer repeat unit), r.t., 16 h.
b 1:1 (v/v) binary mixture.
c Calculated via1H NMR spectroscopy.
d Selectivity was defined as (x/n) × 100%.
e RuCl3·xH2O was used as the catalyst.
f [Ether repeat unit]0 = 0.3 M, 2 mmol scale, 6.5 mg Ru catalyst, 2.1 equiv. NaBrO3 with respect to the polymer repeat unit.
g 0.5 mL acetonitrile was added to the reaction mixture.
h 500 mg of NaIO4/SiO2 (20 wt% NaIO4) was used as the oxidant.25
i The reaction was run for 1 h.
|
|||||
| 1 | Pr | EtOAc/H2O | NaIO4 | 98 | 95 |
| 2 | Pr | EtOAc/H2O | NaBrO3 | 98 | 95 |
| 3 | Pr | EtOAc/H2O | NaClO | 89 | 85 |
| 4 | Pr | EtOAc/H2O | Oxone | 86 | 89 |
| 5e | Pr | EtOAc/H2O | NaBrO3 | 99 | 95 |
| 6e,f | Pr | EtOAc/H2O | NaBrO3 | 99 | 98 |
| 7e | Pr | EtOAc/H2O | NaClO | 86 | 83 |
| 8 | Pr | CH2Cl2/H2O | NaBrO3 | 37 | 75 |
| 9 | Pr | CH2Cl2/H2O | NaIO4 | 35 | 80 |
| 10 | Pr | CH2Cl2/H2Og | NaIO4 | 97 | 88 |
| 11 | Pr | EtOAc | NaIO4/SiO2h | 96 | 91 |
| 12 | i-Pr | EtOAc/H2O | NaBrO3 | 99 | 97 |
| 13 | Me | EtOAc/H2O | NaBrO3 | 89 | 89 |
| 14i | Me | EtOAc/H2O | NaBrO3 | 30 | 89 |
| 15 | H | EtOAc/H2O | NaIO4 | 39 | 60 |
The structure of the polymer obtained from the aforementioned reaction using NaBrO3 as the oxidant and EtOAc/H2O as the solvent was elucidated using FT-IR and 1H NMR spectroscopy. As shown in Fig. 1, the strong νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and νC–O signals recorded at 1176.6 and 1734.5 cm−1, respectively, were consistent with the formation of ester functional groups; these signals were not observed in the poly(vinyl ether) starting material. In addition, the IR spectrum of the product matched that of an authentic sample of poly(vinyl butyrate) (PVB). As shown in Fig. 2, the chemical shifts assigned to the C–H groups α to the ether repeat units in the main chain of the PBVE starting material (δ 3.2–3.7 ppm, CDCl3) shifted downfield to approximately 4.8 ppm in the product. The poly(vinyl ester) product also exhibited a signal near 2.2 ppm, which was attributed to the side-chain methylene units adjacent to the ester carbonyl groups. Other recorded 1H NMR signals were consistent with the structure of PVB.
O and νC–O signals recorded at 1176.6 and 1734.5 cm−1, respectively, were consistent with the formation of ester functional groups; these signals were not observed in the poly(vinyl ether) starting material. In addition, the IR spectrum of the product matched that of an authentic sample of poly(vinyl butyrate) (PVB). As shown in Fig. 2, the chemical shifts assigned to the C–H groups α to the ether repeat units in the main chain of the PBVE starting material (δ 3.2–3.7 ppm, CDCl3) shifted downfield to approximately 4.8 ppm in the product. The poly(vinyl ester) product also exhibited a signal near 2.2 ppm, which was attributed to the side-chain methylene units adjacent to the ester carbonyl groups. Other recorded 1H NMR signals were consistent with the structure of PVB.
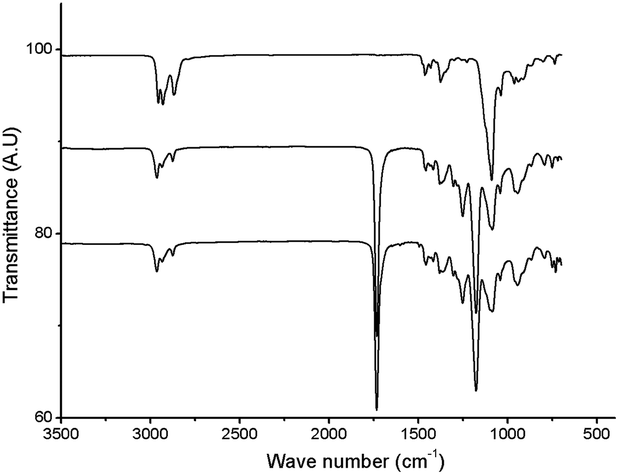 | ||
| Fig. 1 FT-IR spectra of PBVE before (top) and after (middle) oxidation (Table 1, entry 6), and (bottom) an authentic sample of PVB. | ||
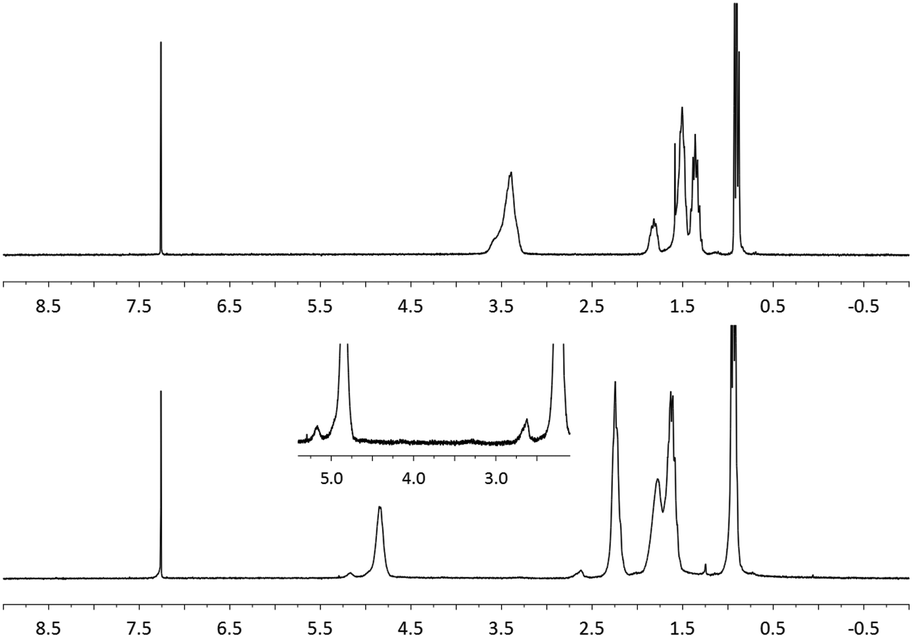 | ||
| Fig. 2 1H NMR spectra of PBVE before (top) and after (bottom) oxidation (Table 1, entry 6) (CDCl3, 400 MHz). | ||
The formation of a side product comprising main-chain ketones (i.e., the structures shown in the brackets indicated by the subscripted n–x in eqn (1)) was also observed upon close inspection of the NMR data. As shown in Fig. 2 (bottom, inset), the signal recorded at δ 2.6 ppm was attributed to the main chain methylene units adjacent to the ketone groups, which presumably formed via C–O bond cleavage followed by oxidation. Similarly, the signal recorded at 5.2 ppm was assigned to main chain methine units positioned β to the ketone groups. The assignments were further supported by a nearly constant integral ratio of the two signals (I2.6 ppm/I5.2 ppm ≈ 2) among the various samples analyzed as well as by a 1H–1H COSY experiment (see the ESI†). It has been previously shown that the oxidation chemistry displayed by RuO4 can be strongly influenced by the solvent.26 Indeed, the quantity of ketone groups observed in the products appeared to depend on the reaction conditions employed. For example, we observed up to 37% of the repeat units contained ketone units when a chlorinated solvent was used; in contrast, the use of EtOAc/H2O as the reaction medium was found to reduce ketone formation to less than 5%.
Using the optimized reaction conditions described above, a variety of poly(vinyl ether)s were explored as starting materials. As summarized in Table 1, poly(vinyl isobutyrate) and poly(vinyl acetate) were obtained from the corresponding poly(vinyl ether)s in excellent yield and selectivity. However, attempts to oxidize poly(methyl vinyl ether) resulted in relatively limited selectivity as well as incomplete conversion. Poly(tert-butyl vinyl ether) was found to be inert toward oxidization under the conditions explored (see the ESI†).
To probe whether the aforementioned oxidation reactions resulted in chain cleavage, a series of poly(vinyl ether)s and their corresponding poly(vinyl ester)s were analyzed via gel permeation chromatography (GPC). While slight changes in the polymer molecular weights were observed when NaBrO3 was used as the oxidant, no significant changes in the respective polydispersity indices (Đ) were measured. Since PVB is more polar than poly(butyl vinyl ether) (PBVE), it was expected that the former should display stronger intramolecular interactions and therefore exhibit a relatively longer retention time (and thus a low molecular polystyrene standard equivalent molecular weight) when measured by GPC.27 Nevertheless, the nearly unchanged Đ indicated that significant chain cleavage did not occur over the course of the oxidation reaction. In comparison, performing a reaction with NaIO4 as the oxidant resulted in a more pronounced reduction in molecular weights and relatively larger Đ values under otherwise identical conditions. Similar results were obtained with poly(isobutyl vinyl ether) (PIBVE); see Table 2.
| Starting material | Oxidant | M n (kDa) | Đ | M n (kDa) | Đ |
|---|---|---|---|---|---|
| Pre-oxidation | Post-oxidation | ||||
| a Reaction conditions: [ether repeat unit]0 = 0.1 M, 4 equiv. of oxidant (indicated) with respect to the polymer repeat unit, 5 wt% RuO2·xH2O, r.t., 24 h. b Determined via GPC against polystyrene standards (THF, 35 °C). | |||||
| PBVE | NaBrO3 | 21.9 | 1.3 | 19.6 | 1.4 |
| PBVE | NaIO4 | 21.9 | 1.3 | 14.1 | 1.6 |
| PIBVE | NaBrO3 | 10.4 | 1.4 | 10.1 | 1.4 |
| PIBVE | NaIO4 | 10.4 | 1.4 | 7.7 | 1.7 |
Synthesis of poly(propenyl ester)s (PPE)s
It has been reported that poly(propenyl acetate) may be prepared using a SnCl4 catalyzed acetylation of poly(tert-butyl propenyl ether).28,29 However, this method requires large quantities of a metal catalyst (40 wt%) and results in low molecular weight polymeric products ([η] = 0.05). Building on our previous results, subsequent attention shifted toward the synthesis of poly(propenyl ester)s using the aforementioned post-polymerization oxidation methodology.First, poly(butyl propenyl ether) (PBPE) was synthesized via the cationic polymerization of the corresponding monomer. Although PBPE is soluble in EtOAc, it often precipitates from solution in the presence of water. To circumvent this problem, butyl acetate was used in lieu of EtOAc as the solvent for subsequent experiments. The oxidation of PBPE using NaBrO3/RuO2·xH2O at room temperature was found to be slower than that observed with PBVE (90% conversion after 72 h), presumably due to the differences in steric bulk. However, the rate of the oxidation reaction increased after raising the temperature of the corresponding reaction mixture to 60 °C. The polymeric product from the aforementioned reaction was isolated via precipitation and then analyzed by NMR spectroscopy as well as GPC. As shown in Fig. 3, diagnostic signals were recorded between 4.5–5.4 ppm and 1.9–2.4 ppm, and assigned to methine units in the main chain and methylene units α to ester groups in the side chains, respectively. Moreover, 1H NMR signals that corresponded to the methylene and methine units positioned α to the ether repeat units (δ 3.0–3.7 ppm, CDCl3) in the starting material were not observed, consistent with a high conversion to the corresponding poly(propenyl ester) product. Similar to the results described above, weak signals were observed near 3.0 ppm, which were assigned to a ketone by-product and calculated to be present in ca. 5 mol%. Analogous results were obtained when poly(ethyl propenyl ether) (PEPE)30 or poly(ethyl isopropenyl ether) (PEIPE) was used as the starting material. Although GPC analysis indicated that chain cleavage occurred during the oxidation reaction (see Table 3), the molecular weights of the PPEs prepared as described above were relatively high when compared to those synthesized using other methodologies.
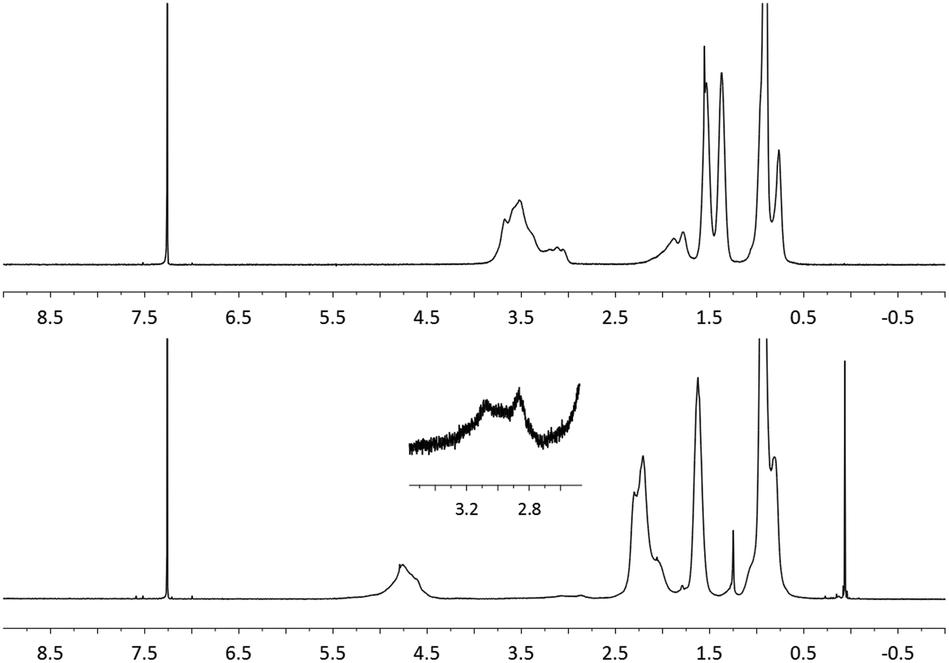 | ||
| Fig. 3 1H NMR spectra of PBPE before (top) and after (bottom) oxidative modification (CDCl3, 400 MHz). | ||
| Starting material | Conversion (%) | Selectivity (%) | M n (kDa) | Đ | M n (kDa) | Đ |
|---|---|---|---|---|---|---|
| Pre-oxidation | Post-oxidation | |||||
|
a Determined via GPC against polystyrene standards (THF, 35 °C).
b [Repeat unit]0 = 0.2 M, BuOAc/H2O (1:1 v/v), 4 equiv. NaIO4 with respect to the polymer repeat unit, 60 °C, 16 h.
c [Repeat unit]0 = 0.4 M, BuOAc/H2O (1:1 v/v), 2 equiv. NaBrO3 with respect to the polymer repeat unit, 60 °C, 16 h.
d [Repeat unit]0 = 0.5 M, EtOAc/H2O (1:1 v/v), 3 equiv. NaIO4 with respect to the polymer repeat unit, r.t., 18 h.
e Ketone formation was not observed via1H NMR spectroscopy.
|
||||||
| PBPEb | 99 | 94 | 30.1 | 2.6 | 21.8 | 2.3 |
| PEPEc | 99 | 95 | 35.2 | 2.5 | 10.9 | 2.1 |
| PEIPEd | 99 | >99e | 8.9 | 1.4 | 7.2 | 1.6 |
Modification of poly(tetrahydrofuran) (PTHF)
Aliphatic polyesters with high contents of γ-butyrolactone (γ-BL) repeat units are often difficult to prepare due to the low polymerizability of the respective monomer.31,32 Indeed, the homopolymerization γ-BL typically requires high pressures. When γ-BL is copolymerized with other cyclic esters or ethers, the yield and molecular weight of the respective copolymer often decreases sharply with increasing feed ratio of γ-BL; the upper limit appears to be approximately 50%.33Using 1 wt% of RuO2·xH2O as the catalyst and 1.1 equiv. of NaBrO3 with respect to the repeat unit of the polymeric starting material, relatively low and high molecular weight samples of poly(tetrahydrofuran) (PTHF) were independently oxidized to their corresponding poly(butyric ester)s. As shown in Table 4, GPC analyses of the products revealed that a significant reduction in molecular weight as well as the Đ had occurred over the course of the corresponding oxidation reactions. The use of buffered solutions or lower reaction temperatures did not significantly suppress the bond cleavage. Nonetheless, assuming that PTHF was randomly oxidized, three types of monomeric units are possible: (1) γ-BL, (2) 1,4-butanediol (BD) and/or (3) succinic acid (SA). All three types of units were identified in the aforementioned polymeric products via1H NMR spectroscopy (see Fig. 4) and calculated to be present in a ratio of 54:27:19 (BL:BD:SA) for low molecular weight product and 54:23:23 for the product of relatively high molecular weight. The compositions of the modified polymers were further confirmed upon saponification and subsequent spectroscopic analyses of the product mixtures (see the ESI† for additional details).34
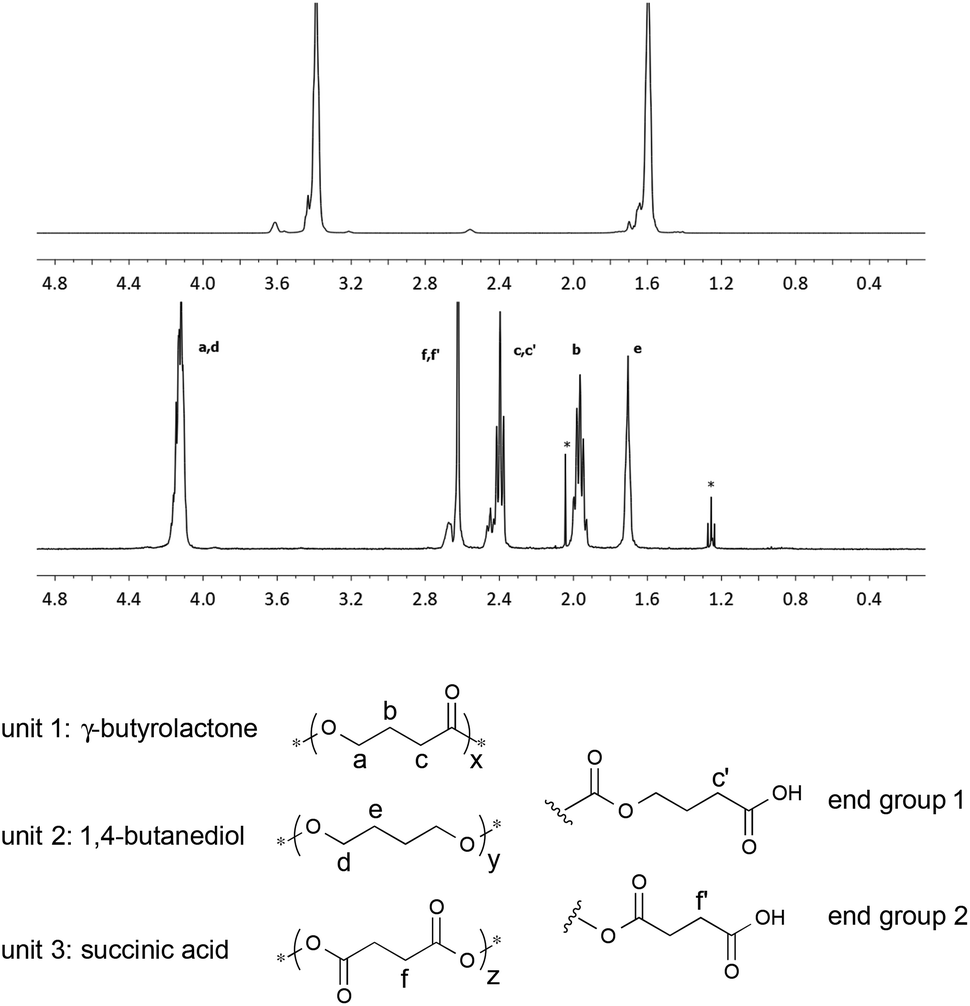 | ||
| Fig. 4 1H NMR spectra (CDCl3, 400 MHz) of PTHF (top) (Table 4, entry 1), an oxidized derivative of PTHF (middle) (Table 4, entry 1), and the corresponding structural analysis (bottom). | ||
| Polymer | M n (kDa) | Đ | M n (kDa) | Đ | γ-BL%b (mol%) |
|---|---|---|---|---|---|
| Pre-oxidation | Post-oxidation | ||||
| a Determined via GPC against polystyrene standards (THF, 35 °C). b The composition of the modified PTHF was determined by 1H NMR spectroscopy using formula: γ-BL% = 2I2.4 ppm/(2I2.4 ppm + I1.7 ppm + I2.6 ppm) × 100%. | |||||
| LMW-PTHF | 3.2 | 2.3 | 1.2 | 1.7 | 54 |
| HMW-PTHF | 146.3 | 2.0 | 14.5 | 1.7 | 54 |
Conclusion
We have demonstrated that the Ru-catalyzed C–H oxidation of poly(vinyl ether)s may be utilized to access poly(vinyl ester)s in high yield and selectivity. In addition, the methodology provided access to high molecular weight poly(propenyl ester)s which, to the best of our knowledge, are the first examples of their kind. We have also shown that polyesters with a relatively high content of γ-butyrolactone may be synthesized via the oxidation of poly(tetrahydrofuran). A limitation to the method described is inadvertent C–O cleavage/oxidation, although this side reaction may be minimized through judicious selection of the solvent. By finely controlling the quantities of oxidants and/or the reaction conditions employed, it should now be possible to synthesize a broad range of poly(vinyl ester)s and poly(propenyl ester)s as well as their ketone containing copolymers, effectively overcoming many of the limitations associated with existing (co)polymerization methodologies.Experimental
Instrumentation
1H NMR and 13C NMR spectra were recorded on a Varian Mercury 400, Varian Inova 500 or Varian DirectDrive 600 spectrometer and internally referenced to the residual solvent (CDCl3: 1H, δ = 7.26; 13C, δ = 77.2, D2O: 1H, δ = 4.80). IR spectra were recorded on a Thermo Scientific Nicolet iS5 FT-IR spectrometer equipped with iD3 ATR accessory (Ge). Intrinsic viscosity ([η]) was measured using an Ubbelohde type viscometer in benzene at 30 °C. Gel permeation chromatography (GPC) was performed in THF on a Viscotek 2001 GPC max system using a set of two columns (Viscotek MBHMW-3078 and Viscotek MBMMW-3078) thermostatted to 35 °C and operated at a flow rate of 1.0 mL min−1. The GPC system was outfitted with an RI detector (Viscotek 3580) and a light scattering/viscometer dual detector (Viscotek 270), and calibration was carried out using narrow polystyrene standards purchased from Scientific Polymer Products. Trace metal analyses were performed on an Agilent 7500ce inductively coupled plasma mass spectrometer (ICP-MS). Prior to each analysis, samples (∼5 mg) were digested with a mixture of 0.5 mL of HNO3 (15.8 N) and 0.1 mL of 30% H2O2 overnight, and then diluted to a total volume of 30 mL with deionized water followed by centrifugation.Materials
Ethyl propenyl ether (98%, cis/trans = 2:1) as well as all other vinyl ethers were purchased from commercial sources, washed with aqueous KOH and distilled twice from CaH2 before polymerization. Butyl propenyl ether (cis/trans = 3:2) was prepared via the Ru-catalyzed isomerization of butyl allyl ether by following a procedure reported in the literatue.35 Poly(ethyl vinyl ether) and poly(methyl vinyl ether) were purchased from Sigma Aldrich; poly(tetrahydrofuran) was purchased from Scientific Polymer Products. All oxidizing agents and Ru catalysts were purchased from commercial sources and used as received. All solvents used for polymerization were dried and degassed using a Vacuum Atmospheres Company solvent purification system and stored over molecular sieves in a nitrogen-filled glove box.
Cationic polymerization of butyl propenyl ether and ethyl propenyl ether
The polymerization reaction was performed under an atmosphere of dry nitrogen by adding 0.5–1 mol% BF3·Et2O to a mixture of the butyl propenyl ether and toluene (1:10 v/v) in a Schlenk flask at −78 °C. After stirring the mixture at −78 °C for 4 h, the reaction was quenched with cold methanol containing 5% (v/v) of aqueous ammonium hydroxide. The resulting mixture was then warmed to ambient temperature and poured into excess methanol. The precipitated solids were collected by filtration, washed with methanol and then dried under high vacuum to afford poly(butyl propenyl ether) as a white solid (1.32 g, 83% yield). 1H NMR (400 MHz, CDCl3): δ 3.97–2.94 (m, 3H), 2.29–1.67 (m, 1H), 1.66–1.24 (m, 4H), 1.18–0.66 (m, 6H). Mn = 30.1 kDa, Đ = 2.6. Using a similar procedure, poly(ethyl propenyl ether) was obtained as a white solid (2.24 g, 96% yield). 1H NMR (400 MHz, CDCl3): δ 3.91–2.97 (m, 3H), 2.26–1.62 (m, 1H), 1.23–1.06 (s, 3H), 1.06–0.67 (m, 3H). Mn = 35.2 kDa, Đ = 2.5.
Cationic polymerization of ethyl isopropenyl ether
The polymerization reaction was performed under an atmosphere of dry nitrogen and initiated by adding 0.5 mL of a solution of iodine ([I2]0 = 0.05 M in Et2O) to a mixture of 3 mL of monomer and 30 mL of toluene in a Schlenk flask at −78 °C.36 After stirring the mixture at −78 °C for 4 h, the reaction was quenched with cold methanol containing 5% (v/v) of aqueous ammonium hydroxide. The resulting mixture was then warmed to ambient temperature and poured into excess methanol. The precipitated solids were collected by filtration, washed with methanol and then dried under high vacuum to afford the desired polymer as a white solid (0.84 g, 37% yield). 1H NMR (400 MHz, CDCl3): δ 3.58–3.17 (s, 2H), 2.05–1.64 (m, 2H), 1.53–1.21 (m, 3H), 1.22–0.99 (m, 3H). Mn = 8.9 kDa, Đ = 1.4.Cationic polymerization of butyl vinyl ether
Using a modified procedure,37 3 mL of BVE was dissolved in 30 mL of hexane in a Schlenk flask at 0 °C. The resulting mixture was charged with 0.05 mL of a HCl solution (1 M in Et2O) and then stirred for 15 min. The polymerization was initiated by adding 0.020 mL of a ZnCl2 solution (1.0 M in Et2O) to the mixture, an then stirred for 5 h at 0 °C. The polymerization was quenched using 1 mL of cold methanol containing 5% (v/v) of aqueous ammonium hydroxide. After the resulting mixture was washed with 10% aqueous sodium thiosulfate followed by water, it was evaporated under reduced pressure, which afforded the desired product as a colorless semisolid (2.0 g, 87% yield). 1H NMR (400 MHz, CDCl3): δ 3.73–3.23 (m, 3H), 1.92–1.26 (m, 6H), 0.97–0.82 (t, 3H). Mn = 21.9 kDa, Đ = 1.3.Cationic polymerization of isobutyl vinyl ether
Using a modified procedure,38 1 mL of IBVE was dissolved in 10 mL of toluene in a Schlenk flask at 0 °C for 10 min. The mixture was then charged with 0.1 mL of an initiator (IBVE-CF3COOH adduct) solution which was freshly prepared by mixing 0.2 mL of IBVE and 0.1 mL of CF3COOH in 3 mL CCl4 at 0 °C for 15 min. The polymerization was initiated by adding 0.05 mL of a ZnCl2 solution (1.0 M in Et2O) to the aforementioned mixture and stirred for 12 h at 0 °C. The polymerization was quenched with 1 mL of cold methanol containing 5% (v/v) of aqueous ammonium hydroxide. The quenched mixture was washed with 10% of aqueous sodium thiosulfate followed by water and then evaporated under reduced pressure to afford the desired product as a pale yellow semisolid (0.66 g, 86% yield). 1H NMR (400 MHz, CDCl3): δ 3.70–2.98 (m, 3H), 1.95–1.23 (m, 3H), 1.08–0.68 (d, 6H). Mn = 10.4 kDa, Đ = 1.4.Cationic ring opening polymerization of tetrahydrofuran (THF)
In a dry box, 1 mL of BF3·Et2O was added to 10 mL of dry THF and the mixture was stirred at ambient temperature for 48 h. After pouring the resulting viscous solution into 200 mL of deionized water, the precipitated solids were collected and purified twice by dissolution/precipitation with THF/water. The final product was obtained as a white solid (0.8 g). 1H NMR (400 MHz, CDCl3): δ 3.50–3.38 (s, 4H), 1.70–1.50 (s, 4H) Mn = 146.3 kDa, Đ = 2.0.General post-polymerization oxidation procedure
In a reaction vessel, 0.1 mmol of a poly(vinyl ether) was dissolved in 1 mL of EtOAc and then mixed with 1 mL of an aqueous solution containing an oxidant (0.4 M). Afterward, mixture was charged with 1 mg RuO2·xH2O or RuCl3·xH2O, and then stirred at room temperature for 16 h. Upon settling, two layers were observed: the bottom layer was removed with a pipette and the faintly yellow colored top layer was diluted with 4 mL EtOAc and then quickly charged with 2 mL of freshly prepared 10% aqueous sodium hydrosulfite. The resulting mixture was then stirred and the bottom layer was removed. The organic layer was washed with brine, dried with anhydrous sodium sulfate and then evaporated to dryness to afford corresponding poly(vinyl ester). Larger scale reactions (>1 mmol polymer) were successfully performed at concentrations up to 0.4 M with catalyst loadings as low as 1 wt%.Acknowledgements
This material is based upon work funded by the Camille Dreyfus Teacher-Scholar Awards Program, the U. S. Army Research Office (W911NF-09-1-0446 and W911NF-09-1-0456) and the National Science Foundation (CHE-1266323). CWB is grateful to the IBS (IBS-R019-D1) and the BK21 Plus Program funded by the Ministry of Education and the National Research Foundation of Korea for their support.Notes and references
- P. Theato and H.-A. Klok, Functional polymers by post-polymerization modification: concepts, guidelines, and applications, Wiley-VCH, Weinheim, 2013 Search PubMed.
- K. A. Gunay, P. Theato and H. A. Klok, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1–28 CrossRef.
- M. A. Gauthier, M. I. Gibson and H. A. Klok, Angew. Chem., Int. Ed., 2009, 48, 48–58 CrossRef CAS PubMed.
- S. Y. Zhang, F. M. Zhang and Y. Q. Tu, Chem. Soc. Rev., 2011, 40, 1937–1949 RSC.
- K. R. Campos, Chem. Soc. Rev., 2007, 36, 1069–1084 RSC.
- J. Jin and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2015, 54, 1565–1569 CrossRef CAS PubMed.
- M. M. Diaz-Requejo, T. R. Belderrain, M. C. Nicasio, S. Trofimenko and P. J. Perez, J. Am. Chem. Soc., 2002, 124, 896–897 CrossRef CAS PubMed.
- Z. P. Li, R. Yu and H. J. Li, Angew. Chem., Int. Ed., 2008, 47, 7497–7500 CrossRef CAS PubMed.
- Y. H. Zhang and C. J. Li, Angew. Chem., Int. Ed., 2006, 45, 1949–1952 CrossRef CAS PubMed.
- N. K. Boaen and M. A. Hillmyer, Chem. Soc. Rev., 2005, 34, 267–275 RSC.
- Y. Kondo, D. Garcia-Cuadrado, J. F. Hartwig, N. K. Boaen, N. L. Wagner and M. A. Hillmyer, J. Am. Chem. Soc., 2002, 124, 1164–1165 CrossRef CAS PubMed.
- C. Bae, J. F. Hartwig, N. K. B. Harris, R. O. Long, K. S. Anderson and M. A. Hillmyer, J. Am. Chem. Soc., 2005, 127, 767–776 CrossRef CAS PubMed.
- T. S. Jo, S. H. Kim, J. Shin and C. Bae, J. Am. Chem. Soc., 2009, 131, 1656–1657 CrossRef CAS PubMed.
- P. J. Flory and F. S. Leutner, J. Polym. Sci., 1948, 3, 880–890 CrossRef CAS.
- T. Otsu, A. Shimizu and M. Imoto, J. Polym. Sci., Part A: Gen. Pap., 1965, 3, 615–618 CrossRef CAS.
- V. P. Zubov, M. V. Kumar, M. N. Masterova and V. A. Kabanov, J. Macromol. Sci., Part A: Pure Appl. Chem., 1979, 13, 111–131 CrossRef.
- T. Nishino, N. Kitamura and K. Murotani, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 754–761 CrossRef CAS.
- M. Ouchi, M. Kamigaito and M. Sawamoto, Macromolecules, 1999, 32, 6407–6411 CrossRef CAS.
- Y. Hirokawa, T. Higashimura, K. Matsuzaki and T. Uryu, J. Polym. Sci., Polym. Chem. Ed., 1979, 17, 1473–1481 CrossRef CAS.
- T. Kawaguchi, F. Sanda and T. Masuda, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 3938–3943 CrossRef CAS.
- L. M. Berkowitz and P. N. Rylander, J. Am. Chem. Soc., 1958, 80, 6682–6684 CrossRef CAS.
- P. H. J. Carlsen, T. Katsuki, V. S. Martin and K. B. Sharpless, J. Org. Chem., 1981, 46, 3936–3938 CrossRef CAS.
- A. B. Smith and R. M. Scarborough, Synth. Commun., 1980, 10, 205–211 CrossRef CAS.
- H. Ohgi, T. Sato, S. H. Hu and F. Horii, Polymer, 2006, 47, 1324–1332 CrossRef CAS.
- Y. L. Zhong and T. K. M. Shing, J. Org. Chem., 1997, 62, 2622–2624 CrossRef CAS PubMed.
- V. Piccialli, Molecules, 2014, 19, 6534–6582 CrossRef PubMed.
- Slight decreases in the polymer's intrinsic viscosities were observed upon modification (see the ESI†).
- S. H. Okamura, T. Higashimura, Y. Kitagawa and A. Mizote, JP Patent, 1969, 44-027280 Search PubMed.
- T. Higashimura, Y. Kitagawa and S. Okamura, Kobunshi Kagaku, 1967, 24, 655–661 CrossRef CAS.
- The oxidized product, poly(propenyl acetate), was found to display a [n] = 0.16.
- T. Moore, R. Adhikari and P. Gunatillake, Biomaterials, 2005, 26, 3771–3782 CrossRef CAS PubMed.
- M. Nishiura, Z. M. Hou, T. Koizumi, T. Imamoto and Y. Wakatsuki, Macromolecules, 1999, 32, 8245–8251 CrossRef CAS.
- L. Ubaghs, M. Waringo, H. Keul and H. Hocker, Macromolecules, 2004, 37, 6755–6762 CrossRef CAS.
- The polymeric products obtained via the Ru-catalyzed post-polymerization modification methodology described herein were measured by ICP-MS to contain 2–177 ppm of residual Ru, depending on the polymer analyzed (see the ESI†).
- S. Krompiec, P. Bujak, J. Malarz, M. Krompiec, L. Skorka, T. Pluta, W. Danikiewicz, M. Kania and J. Kusz, Tetrahedron, 2012, 68, 6018–6031 CrossRef CAS.
- K. Matsuzaki, S. Okuzono and T. Kanai, J. Polym. Sci., Polym. Chem. Ed., 1979, 17, 3447–3458 CrossRef CAS.
- M. Kamigaito, M. Sawamoto and T. Higashimura, Macromolecules, 1992, 25, 2587–2591 CrossRef CAS.
- M. Kamigaito, M. Sawamoto and T. Higashimura, Macromolecules, 1991, 24, 3988–3992 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR, 13C NMR, GPC, DSC, TGA and MS data. See DOI: 10.1039/c5py01409c |
| This journal is © The Royal Society of Chemistry 2016 |