
Far beyond primary poly(vinylamine)s through free radical copolymerization and amide hydrolysis†
Mathilde
Dréan
ab,
Philippe
Guégan
b,
Christine
Jérôme
a,
Jutta
Rieger
*b and
Antoine
Debuigne
*a
aCenter for Education and Research on Macromolecules (CERM), Department of Chemistry, University of Liege (ULg), Sart-Tilman, Allée de la Chimie 3, Bat. B6a, B-4000 Liège, Belgium. E-mail: adebuigne@ulg.ac.be
bSorbonne Universités, UPMC Univ Paris 06, CNRS, Institut Parisien de Chimie Moléculaire, Team “Chimie des Polymères” (LCP), 4 Place Jussieu, F-75005 Paris, France. E-mail: jutta.rieger@upmc.fr
First published on 6th October 2015
Abstract
Due to their affinity for many supports, their pH responsiveness, metal binding capacity and polyelectrolyte complexation, poly(vinylamine) derivatives have attracted attention for many applications including coatings, water purification, or gas membrane separation. Nevertheless, most of them possess only pendant primary amines despite the possible benefits of incorporating different amino groups along the chain. In this work, a straightforward and scalable synthesis route towards polymers bearing primary and secondary amines, as well as imidazole groups, is reported. The general strategy relies on the radical copolymerization of different vinylamides and vinyl imidazoles followed by the hydrolysis of the resulting poly(vinylamide) derivatives. Binary and ternary free radical copolymerizations of N-vinylacetamide (NVA), N-methyl vinylacetamide (NMVA) and 1-vinylimidazole (VIm) were investigated and the reactivity ratios for each copolymerization system were determined. Thanks to these values a series of statistical copolymers with predictable composition and low deviation over the chain distribution could then be synthesized. Finally, the acidic hydrolysis of the acetamide functions towards the corresponding amine was performed and optimized. Copolymers containing various pendant amino groups and with low dispersity in the chain composition could be obtained, which opens new perspectives for the above mentioned applications.
Introduction
Poly(vinylamine) (PVAm) is a special and highly valuable macromolecule because it exhibits the highest possible density of pendant primary amines among all vinyl polymers.1 Excellent water solubility, metal binding capacity,2,3 ionization/pH behavior,4 polyelectrolyte complexation,5 and affinity for many supports6,7 are key properties of PVAm that make it useful for a broad range of applications, such as water purification,8 paper additive and coating applications,9–11 gas membrane separation,12,13 or functionalization of macroscopic and nanoparticle surfaces.6The huge potential of this polymer has led to intensive research for the development of straightforward synthetic routes towards PVAm and its derivatives.1 Because PVAm cannot be prepared from vinylamine due to its low stability and fast conversion into the corresponding imine through tautomeric equilibrium,14 multistep procedures had to be developed (Scheme 1). The Hoffman rearrangement of a polyacrylamide precursor15 successfully yielded polymers with a high density of pendant primary amines but this approach suffers from producing non-negligible amounts of side products, such as carboxyl and urea groups along the chain.16 In this respect, the radical polymerization of N-vinylamides, mainly N-vinylformamide (NVF)17–19 or N-vinylacetamide (NVA),20,21 with subsequent hydrolysis of the amide groups is “cleaner” and more straightforward. Optimal conversion of PNVF into PVAm can be achieved upon basic treatment,17,18 whereas a high level of hydrolysis of the amide functions of PNVA was reached under acidic conditions.20 Except for the cost of the monomer (N-vinylphthalimide), hydrazinolysis of poly(N-vinylphthalimide) (NVPI) is another interesting route towards PVAm.22
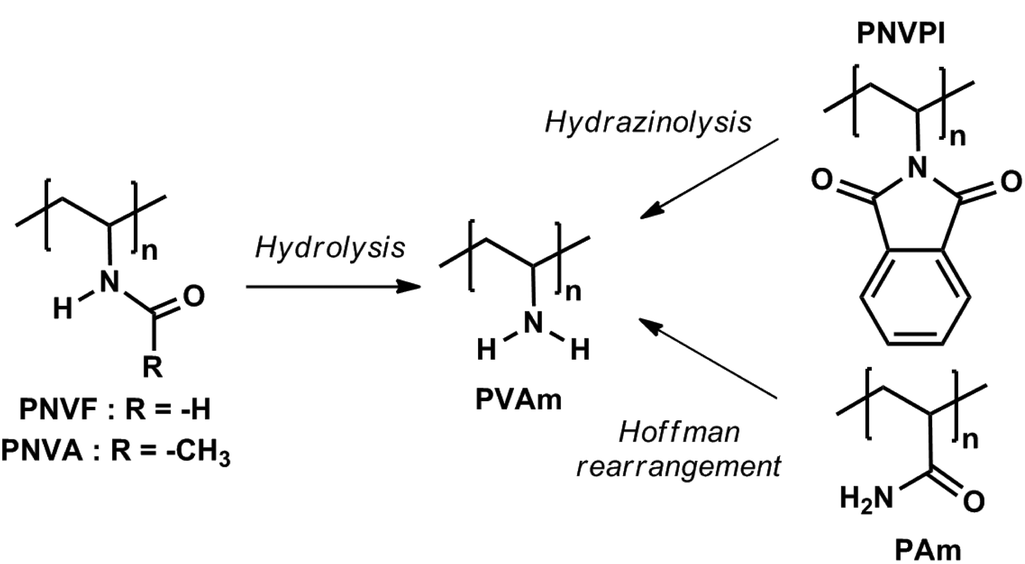 | ||
| Scheme 1 Synthetic routes towards poly(vinylamine). | ||
In order to meet the needs of today's applications, it is crucial to adjust the properties of PVAm and to provide it with additional functionalities by incorporating supplementary chemical moieties in its backbone. So far, the functionalization of PVAm is largely dominated by post-polymerization derivatization strategies arising from the ease of coupling amines with reactive molecules.1 In this way, several chemical groups have been successfully incorporated by post-treatment of PVAm including fluorinated23 and other hydrophobic groups,24 sugars,25,26 antibodies,27 dyes,28 and nucleic acids.29 Another strategy for derivatizing PVAm consists of the radical copolymerization of vinylamides with selected comonomers followed by the hydrolysis of the amides. For example, NVF was copolymerized with vinylesters, acrylates or acrylamides, followed by acid or base treatment leading to the corresponding polyvinylamine-based copolymers.30,31 Although straightforward, this approach requires a perfect knowledge of the comonomer reactivity ratios and close examination of the fate of the comonomer units during hydrolysis. Nevertheless, despite the possible benefits of incorporating various amino groups along the polymer chain32,33 poly(vinylamine)-based copolymers are generally restricted to pendant primary amines. Examples of synthesis of poly(N-methyl vinylamine) (PMVAm) from poly(N-methyl vinylacetamide) (PNMVA) are scarce34,35 due to the difficulties in hydrolyzing secondary amides. In reasonable timescales (<3 days), only 44% conversion of amides was achieved using HCl 4 N at 100 °C leading to random amines/amides containing copolymers.34
This work reports a simple synthetic approach to move on from simple primary poly(vinylamine)s towards novel copolymers bearing large and predictable amounts of pendant primary and secondary amines as well as imidazoles. The two-step strategy, depicted in Scheme 2, consists of the binary or ternary free radical copolymerization of NVA, NMVA and/or 1-vinylimidazole (VIm), followed by acidic hydrolysis. Special attention was paid to the determination of the comonomer reactivity ratios36 by the Fineman–Ross (FR),37 Kelen–Tudos (KT)38 and the non-linear (NL) least squares fitting curve39,40 methods. The Skeist's model41 was also used for controlling the chemical composition of the copolymers which changes with the monomer conversion. Based on the reactivity ratios, we also prepared a series of P(NVA-stat-NMVA-stat-VIm) terpolymers whose composition could be precisely predicted according to the Alfrey and Goldfinger's equation.42 Finally, an efficient hydrolysis procedure was applied in order to convert efficiently the N-methyl vinylacetamide into secondary amines and produce the targeted poly(vinyl amine) copolymers.
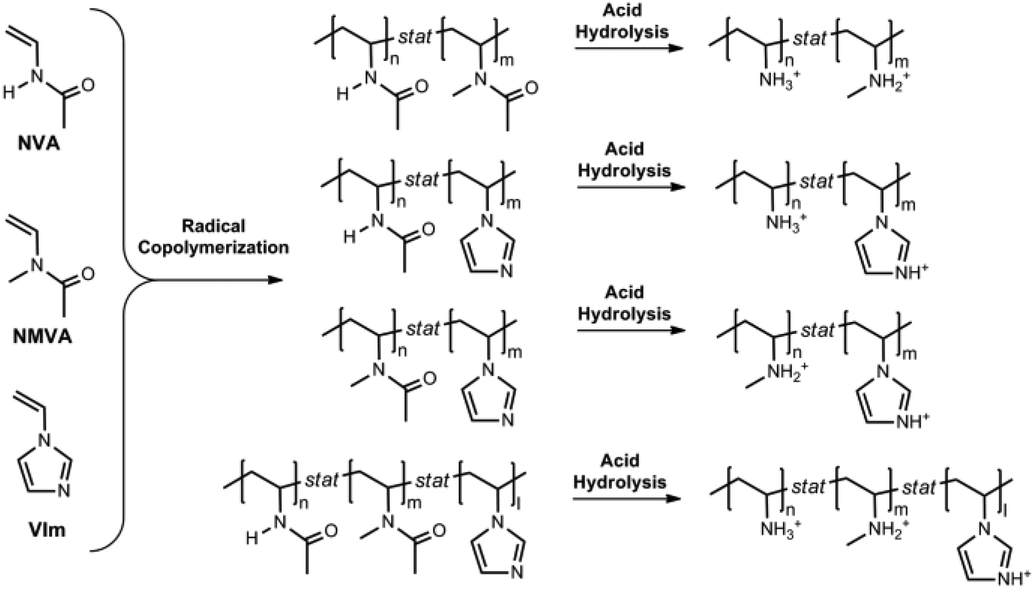 | ||
| Scheme 2 General strategy for the synthesis of poly(vinylamine)-copolymers. | ||
Experimental section
Materials
N-Vinylacetamide (NVA) (>98%, TCI) was used as received. N-Methylvinylacetamide (NMVA) (>98% Aldrich) and 1-vinylimidazole (VIm) (>99%, Aldrich) were purified by distillation under reduced pressure. Methanol was degassed by bubbling argon for 30 min. 2,2′-Azobis(4-methoxy-2,4-dimethyl valeronitrile) (V-70, t1/2 = 10 h at 30 °C) (>98%, Wako) was used as received. Dialysis membranes (cut off 1 kDa) were purchased from VWR.Characterization
The (co)polymers were characterized by size exclusion chromatography (SEC) at 55 °C in dimethylformamide (DMF) containing LiBr (0.025 M) using a flow rate of 1 mL min−1. SEC curves were recorded with a Waters chromatograph equipped with three columns (Waters Styragel pss gram 1000 Å (×2), 30 Å), a dual λ absorbance detector (Waters 2487) and a refractive index detector (Waters 2414). Multi-angle laser light scattering (MALLS) measurements were performed with a Dawn Heleos apparatus from Wyatt Technology to determine the absolute average number molar masses (Mn) and average weight molar masses (Mw). Data were processed with Astra V software (Wyatt Technology). In this case, dn/dc values were measured by refractometry analysis using a Wyatt Optilab rEX refractive index detector (λ = 658 nm). Mn and molar mass dispersities (Đ) were also determined using a polystyrene calibration.The poly(vinylamine) (PNVAm) and poly(N-methyl vinylamine) (PNMVAm) homopolymers were analyzed by SEC in water containing NaCl (0.1 M) and TFA (0.1 vol%) at 30 °C (flow rate: 1 ml min−1) using a Waters SEC equipped with a pre-column (PSS NOVEMA Max analytical 10 micron, 8.0 × 50 mm) and a linear column (PSS NOVEMA Max analytical linear S micron 8.0 × 300 mm). Poly(2-vinylpyridine) (P2VP) standards were used to generate the calibration plot.
Infrared spectra were recorded with a Thermo Scientific Nicolet IS5 apparatus.
1H NMR spectra were recorded at 298 K with a Bruker spectrometer (400 MHz) and treated with MestraNova software. Heteronuclear Single Quantum Correlation (HSQC) spectra were recorded at 298 K on a Bruker Avance (500 MHz) instrument in D2O and chemical shifts are reported in δ values (ppm) relative to internal TMS. 2D spectra were treated with TopSpin. Elementary analyses were carried out by Service de Microanalyse ICSN – CNRS (Gif sur Yvette, France) to determine the C/N ratios in the copolymers before and after acidic treatment in order to calculate the hydrolysis level of the amide functions.
General copolymerization procedure for reactivity ratio determination
For each pair of comonomers (NVA–NMVA, NVA–VIm and NMVA–VIm), at least seven copolymerization experiments were carried out under the same conditions with various feed compositions (molar fraction (f) ranging from 0.05 and 0.95) (see15382 Table S1†). Typically, copolymerizations were performed under argon in a Schlenk tube by dissolving V70 (0.02 equivalents compared to the comonomers) and various amounts of comonomers (NVA, NMVA and VIm) in MeOD (8.8 mmol of comonomers per mL of MeOD) containing traces of dimethylsulfoxide (VDMSO/VMeOD = 0.05). MeOD was used as a solvent in order to easily evaluate the initial and instantaneous composition of the mixture by 1H NMR whereas small quantities of DMSO served as an internal calibration for monitoring the monomer conversion along the polymerization. The reaction medium was heated at 30 °C and the polymerization was stopped below 20% of monomer conversion. The copolymers were purified by precipitation (in acetone for P(NVA-stat-VIm) and P(NVA-stat-NMVA) and in diethylether for P(NMVA-stat-VIm), collected by filtration and dried under vacuum at 40 °C. The molar fraction of each comonomer in the copolymer (F) was determined by 1H NMR in MeOD. Polymerization time, conversion, initial feed composition and copolymer composition are presented in Table S1.† These data were used to determine the reactivity ratios according to the Fineman–Ross (FR),37 Kelen–Tudos (KT),38 Extended Kelen–Tudos (EKT)43 and the non-linear (NL)39,40 least squares fitting curve methods. Compositional drifts were also evaluated according to the Skeist's equation.41![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100 g mol−1, Mn cal PS = 38160 g mol−1, Đcal PS = 1.73, FNVA = 0.68, FNMVA = 0.32).
000 g mol−1, Mn cal PS = 20060 g mol−1, Đcal PS = 2.37, FNMVA = 0.54, FVIm = 0.46).
700 g mol−1, Mn cal PS = 25470 g mol−1, Đcal PS = 1.75, FNVA = 0.32, FNMVA = 0.34, FVIm = 0.34) (see Table 3). In general, the compositions of the terpolymers were successfully predicted based on the Alfrey–Goldfinger (AG)42 equation.
300 g mol−1, Mn cal PS = 31300 g mol−1, Đcal PS = 1.69).
650 g mol−1, Mn cal PS = 17780 g mol−1, Đcal PS = 1.79).
100 g mol−1, Mn cal PS = 38160 g mol−1, Đcal PS = 1.73, FNVA = 0.68, FNMVA = 0.32).
000 g mol−1, Mn cal PS = 20060 g mol−1, Đcal PS = 2.37, FNMVA = 0.54, FVIm = 0.46).
700 g mol−1, Mn cal PS = 25470 g mol−1, Đcal PS = 1.75, FNVA = 0.32, FNMVA = 0.34, FVIm = 0.34) (see Table 3). In general, the compositions of the terpolymers were successfully predicted based on the Alfrey–Goldfinger (AG)42 equation.
300 g mol−1, Mn cal PS = 31300 g mol−1, Đcal PS = 1.69).
650 g mol−1, Mn cal PS = 17780 g mol−1, Đcal PS = 1.79).
General procedure for the hydrolysis of the amide-containing (co)polymers
The copolymer (400 mg) was dissolved in HCl 6 N (5.5 mL). The resulting solution of the (co)polymer (7% w/v) was then placed in an acid digestion Parr vessel and heated at 120 °C for 64 h. After cooling, the solution was dialyzed against pure distilled water for one night. During the last two hours of dialysis, the pH was maintained at 7 (pH controlled with a pH meter and adjusted by addition of NaOH 0.1 N or HCl 1 N). The (co)polymers were recovered upon lyophilization. The (co)polymer was analyzed by 1H NMR in D2O, IR and EA. Hydrolysis yields were determined by EA comparing the C/N content ratio in the (co)polymer before and after acidic treatment. Aqueous SEC experiments were also performed on the PNVAm and PNMVAm samples. In addition, the hydrolysis of PNVA and PNMVA was also tested under milder conditions (5% w/v, 2 N HCl, 120 °C, 14 h).Results and discussion
Binary copolymerization
The synthesis of copolymers bearing various pendant amines starts with the free radical copolymerization of the vinylamides, NVA, NMVA and VIm (see Scheme 2). In order to predict the monomer composition in the final copolymers and the comonomer sequence along the backbone, we investigated the free radical copolymerization of these comonomers in pairwise combinations and measured their reactivity ratios. For this purpose, at least seven copolymerization experiments were conducted for each binary system with different feed compositions ranging from 0.10 to 0.95 (Table S1†). Typically, copolymerizations were performed at 30 °C in MeOD using V70 as a radical initiator. A deuterated solvent was used in order to determine precisely the initial comonomer feed by 1H NMR and to monitor the conversion by following the loss of intensity of the olefinic protons of each monomer. Low monomer conversions (<20%) were targeted to prevent significant composition drifts. After purification of the copolymers, their compositions were analyzed by 1H NMR. The representative spectra of P(NVA-stat-NMVA), P(NVA-stat-VIm) and P(NMVA-stat-VIm) are provided in Fig. S1.† The molar fractions of the comonomers in the copolymer (F) were then plotted as a function of their molar fraction in the feed (f) and fitted using the Mayo–Lewis equation (Fig. 1).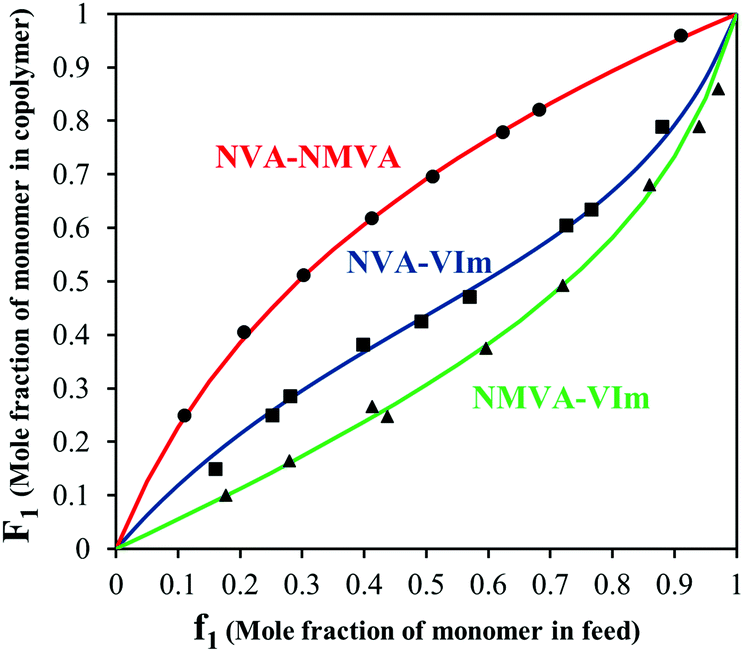 | ||
| Fig. 1 Relationship between the molar fraction of the monomer in the feed and in the copolymer for poly(N-vinylacetamide-stat-N-methyl vinylacetamide) (●), poly(N-vinylacetamide-stat-1-vinylimidazole) (■) poly(N-methyl vinylacetamide-stat-1-vinylimidazole) (▲). The full lines represent the fitting curves obtained by the least squares method. | ||
Based on these data, the reactivity ratios of each monomer pair were measured by different methods. First, the classical Fineman–Ross calculation was used (eqn (1)).37 For all copolymerizations, we obtained straight lines whose slope and intercept with the ordinate (Y-axes) corresponds to r1 and −r2, respectively (Fig. 2A). The reactivity ratio values are presented in Table 1.
| f(F − 1)/F = r1(f2/F) − r2 | (1) |
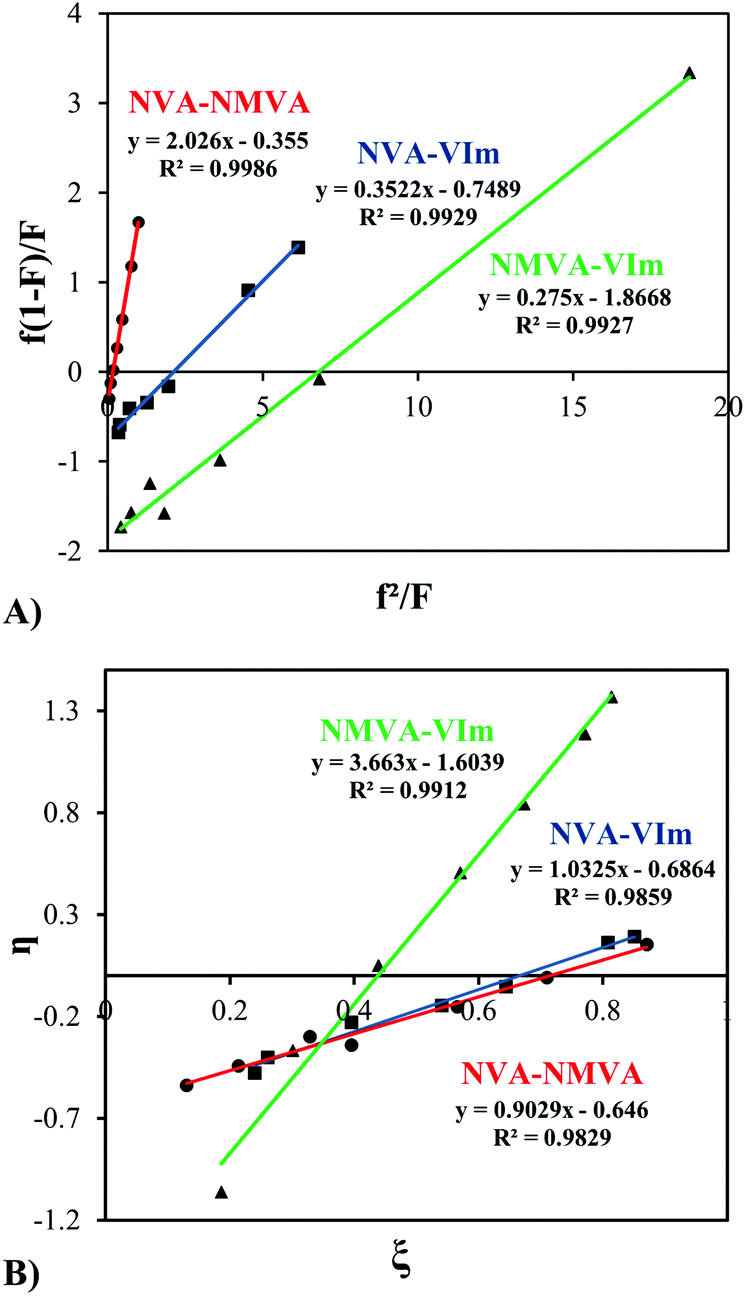 | ||
| Fig. 2 Fineman–Ross (A) and Kelen–Tudos (B) plots for the copolymerization at 30 °C of N-vinylacetamide and N-methylvinylacetamide) (●), N-vinylacetamide and 1-vinylimidazole (■), N-methylvinylacetamide and 1-vinylimidazole) (▲). | ||
| NVA–NMVA | NVA–VIm | NMVA–VIm | ||||
|---|---|---|---|---|---|---|
| Method | r NVA/NMVA | r NMVA/NVA | r NVA/VIm | r VIm/NVA | r NMVA/VIm | r VIm/NMVA |
| Fineman–Ross | 2.03 | 0.36 | 0.35 | 0.75 | 0.28 | 1.87 |
| Kelen–Tudos | 2.06 | 0.36 | 0.35 | 0.74 | 0.26 | 1.80 |
| Nonlinear | 1.98 | 0.33 | 0.35 | 0.74 | 0.27 | 1.85 |
| Average | 2.02 | 0.35 | 0.35 | 0.74 | 0.27 | 1.84 |
In addition, a second linearization method, proposed by Kelen and Tudos,38 was tested. The latter involves parameters η and ζ, mathematical functions of the mole ratios in the monomer feed (f) and in the copolymer (F) and of a parameter α calculated on the basis of the lowest and highest values of (f2/F). Fig. 2B shows the linear plots of η as a function of ζ leading to r1 and (−r2/α) via the intercepts at ξ = 1 and ξ = 0, respectively.
| η = (r1 + (r2/α))ζ − (r2/α) | (2) |
Finally, reactivity ratios were determined by a non-linear least squares fitting curve39,40 method according to the Mayo–Lewis equation (eqn (3)) in order to avoid inaccuracy inherent to the linearization process. The fitting curves for each comonomer pair are presented in Fig. 1 and the resulting r1 and r2 values calculated accordingly are presented with those obtained by the FR and KT methods in Table 1.
| F1 = (r1f12 + f1f2)/(r1f12 + 2f1f2 + r2f22) | (3) |
Although monomer conversions are low, the Extended Kelen–Tudos (EKT)43 method was also considered in order to take into account the possible composition drift along the polymerization (Table S2 and Fig. S2†) but very similar reactivity ratios were obtained as compared to the other three methods (compare Table 1 and S2†).
The reactivity ratios clearly suggest that the monomers are preferentially consumed according to the following trend: VIm > NVA > NMVA. In the case of the N-vinylamide copolymerization, the insertion of NVA within the chain is favored compared to NMVA. The slightly higher steric hindrance of the latter might contribute to this trend but little electronic effects cannot be excluded. The incorporation of NMVA is also disfavored in its copolymerization with VIm, for which the content of VIm in the resulting copolymer is always superior to the one of the feed. Finally, the NVA–VIm copolymerization is slightly in favor of VIm and exhibits the highest tendency for alternation as indicated by the rather low r1 × r2 value (∼0.25) and reactivity ratios below one. Hydrogen bonding between the N–H proton of the acetamide group and the imidazole ring might account for this alternation propensity.
Considering the important differences in reactivity ratios, the comonomers are consumed at different rates in the copolymerizations, and consequently the monomer feed composition changes significantly in the course of the copolymerization. As chains are created and terminate continuously in free radical polymerization, this results in an inhomogeneity in the composition of the chain distribution. In order to get a good compositional picture of the copolymer distribution, the instantaneous and the cumulative copolymer compositions were predicted on the whole range of the monomer conversion with the Skeist's model (SK) (eqn (4)) using the average reactivity ratios determined beforehand. The cumulative composition is given by eqn (5).
| Conv = 1 − (M/M0) = (f1/f°1)α(f2/f°2)β [(f°1 − δ)/(f°1 − δ)]γ | (4) |
| F1 cumul = (f°1 − f1 (1 − conv.))/conv. | (5) |
As an illustration, Fig. 3 shows the evolution of the instantaneous feed and copolymer composition as well as the cumulative copolymer composition as a function of the overall conversion when starting from a near-stoichiometric comonomer feed composition. All copolymerizations were performed at 30 °C in methanol and samples were withdrawn regularly from the medium along the polymerization. The cumulative copolymer compositions measured by 1H NMR at different conversions fitted well with the predicted cumulative composition curve (eqn (5)), which confirms the validity of the model and the determined reactivity ratios. For the NVA–NMVA and NMVA–VIm copolymerizations, the instantaneous composition changes only slightly up to 25% of monomer conversion, but then evolves drastically. In contrast, the instantaneous composition of the P(NVA-stat-VIm) copolymers changes little up to almost 75% of conversion. This model thus provides important information on the polymer sequence, particularly on the homogeneity of the composition over the chain distribution that is worth to be taken into account when considering the synthesis of these types of copolymers. Interestingly, in the case of a controlled radical copolymerization (CRP), a composition drift of the feed during the polymerization leads to gradient copolymers showing an evolution of their composition along the backbone. Since reactivity ratios are often very similar for conventional and controlled radical copolymerizations performed under the same experimental conditions, the compositional drifts determined above by the Skeist's method are also useful to predict the gradient composition of the same copolymers prepared by controlled radical polymerization techniques. Of course, such calculations could be performed for any initial comonomer feed ratio in order to anticipate and prevent significant inhomogeneity in the composition of the chains. It is important to note that, controlling the (co)polymerization of N-vinylamides and VIm remains hitherto quite a challenge. As an example, the organometallic-mediated radical polymerization (OMRP) was proved to be very efficient for controlling the polymerization of several N-vinylamides,44–47 except NVA, whereas the OMRP of VIm has not been reported. To the best of our knowledge, no CRP method can manage the simultaneous control of these three monomers.
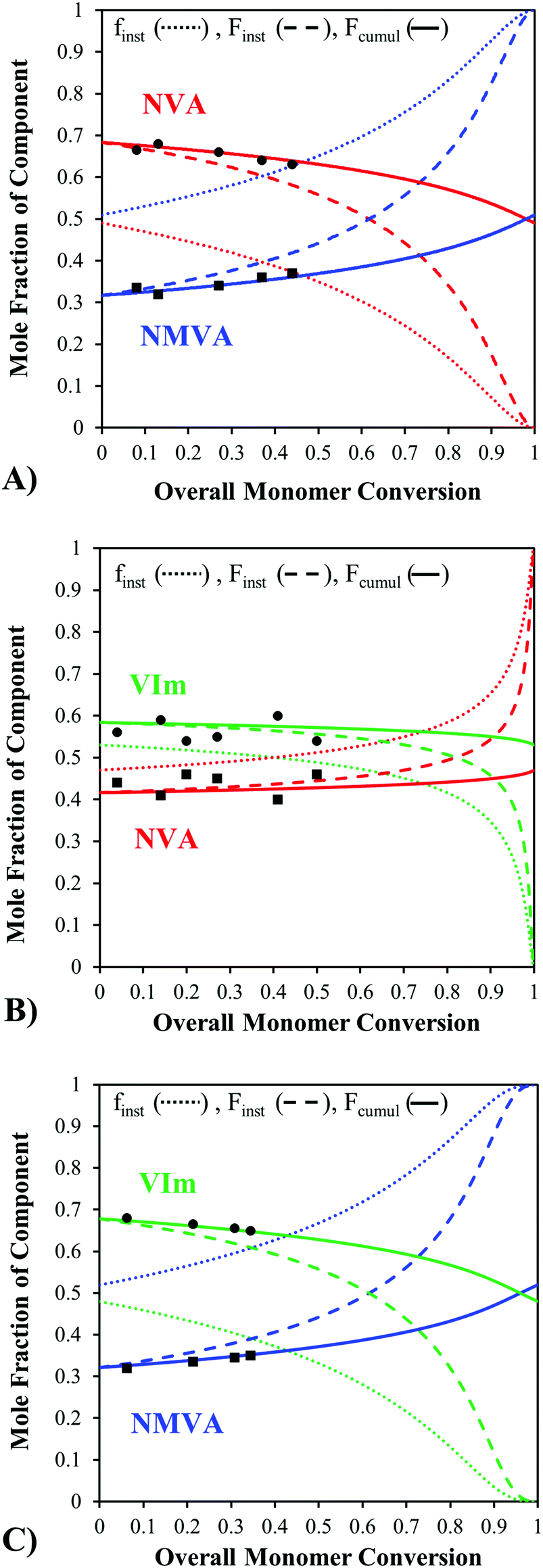 | ||
| Fig. 3 Instantaneous mole fraction of the monomer in the feed (finst ⋯) as well as instantaneous (Finst ---) and cumulative (Fcumul —) mole fractions in the copolymer vs. the overall molar monomer conversion calculated based on the Skeist's model when starting from near equimolar mixtures of comonomers. Experimental data of the average copolymer composition (●, ■) were determined by 1H NMR. | ||
Ternary copolymerization
After studying the binary copolymerization systems, we investigated the terpolymerization of NVA, NMVA and VIm under similar experimental conditions. Copolymerizations were initiated with V-70 in methanol at 30 °C and the monomer conversions were kept below 20%. Table 2 compares the actual and theoretical copolymer compositions for different comonomer feed compositions. The predicted compositions were calculated based on the Alfrey–Goldfinger (AG)42 equation (eqn (6)), using the average reactivity ratios summarized in Table 1.| d[M1]:d[M2]:d[M3] = [M1] {[M1]/r31r21 + [M2]/r21r32 + [M3]/r31r23}{{[M1] + [M2]/r12 + [M3]/r13}}:[M2] {{[M1]/r12r31 + [M2]/r12r32 + [M3]/r32r13}{{[M2] + [M1]/r21 + [M3]/r23}}}:[M3] {{[M1]/r13r21 + [M2]/r23r12 + [M3]/r13r23}{{[M3] + [M1]/r31 + [M2]/r32}} | (6) |
| Feed compositiona | Terpolymer composition | |||||||
|---|---|---|---|---|---|---|---|---|
| Measuredb | Calculatedc | |||||||
| f NVA | f NMVA | f VIm | F NVA | F NMVA | F VIM | F NVA | F NMVA | F VIm |
| Polymerization at 30 °C.a Determined by 1H NMR of the reaction mixture at t0 in MeOD.b Determined by 1H NMR in MeOD of the isolated terpolymers.c Calculated by using the Alfrey and Goldfinger's equation. | ||||||||
| 0.19 | 0.64 | 0.17 | 0.32 | 0.34 | 0.34 | 0.29 | 0.34 | 0.37 |
| 0.33 | 0.31 | 0.35 | 0.40 | 0.07 | 0.53 | 0.37 | 0.12 | 0.51 |
| 0.36 | 0.54 | 0.10 | 0.50 | 0.26 | 0.24 | 0.51 | 0.24 | 0.25 |
| 0.41 | 0.58 | 0.01 | 0.65 | 0.33 | 0.02 | 0.69 | 0.27 | 0.03 |
| Entry | Initial copolymer compositiona | Hydrolysis levelb (%) | Hydrolyzed copolymer compositionc | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| F NVA | F NMVA | F VIM | NVA | NMVA | F NVA | F NVAm | F NMVA | F NMVAm | F VIM | |
| Hydrolysis conditions: HCl 6 N/120 °C/64 h.a Determined by 1H NMR in D2O.b Determined by elementary analysis (see Table S3 for raw data and calculations).c Calculated by taking into account the initial composition determined by 1H NMR and the hydrolysis level determined by elementary analysis.d Set to 94% according to the result for PNVA homopolymers. | ||||||||||
| 1 | 1 | 0 | 0 | 94 | — | 0.06 | 0.94 | 0 | 0 | 0 |
| 2 | 0 | 1 | 0 | — | 81 | 0 | 0 | 0.19 | 0.81 | 0 |
| 3 | 0.68 | 0.32 | 0 | 94d | 90 | 0.04 | 0.64 | 0.03 | 0.29 | 0 |
| 4 | 0.52 | 0 | 0.48 | 91 | — | 0.05 | 0.47 | 0 | 0 | 0.48 |
| 5 | 0 | 0.54 | 0.46 | — | 83 | 0 | 0 | 0.09 | 0.45 | 0.46 |
| 6 | 0.32 | 0.34 | 0.34 | 94d | 75 | 0.02 | 0.30 | 0.08 | 0.26 | 0.34 |
Generally, the compositions of the terpolymers measured by 1H NMR were close to the prediction. The slight discrepancies between the values might be due to complex penultimate effects that are not taken into account in the used theories. Nevertheless, the prediction tool is valid and of practical importance for this ternary copolymerization. Moreover, the data collected in Table 2 confirm the trends observed in the binary systems: VIm and NMVA are the most and the least reactive monomers, respectively. Overall, the compositional prediction and thus control of the P(NVA-stat-NMVA-stat-VIm) terpolymers are now possible. Indeed, comonomer reactivity ratios combined with the AG model allow the determination of the amount of each comonomer to be introduced in the initial monomer feed in order to target a tercopolymer with a specific composition. Overall, this study makes the synthesis of such copolymers highly efficient and paves the way for further innovation based thereon.
Hydrolysis of poly(vinylamide) derivatives
The hydrolysis of poly(N-vinylamide)s has been previously investigated in the literature as a route to the corresponding pendant amine polymers used in many industrial applications.8–10,12,13 Among the PVAm precursors, PNVF is certainly the most popular because of the ease of hydrolysis of the formamide moieties.17–19 Nevertheless, as reported by Akashi et al.,20,21 near complete removal of the acetamide groups of PNVA can also be achieved by hydrochloric acid treatment (HCl 2 N) at 120 °C for 12 h. In contrast, the conversion of PNMVA into the corresponding pendant secondary amine polymer is more difficult and requires extremely long reaction times (around 150 h).35 For shorter timescales (60 h) only moderate hydrolysis of PNMVA (44%) was achieved upon treatment of PNMVA with HCl 4 N at 100 °C.34In order to produce polymers with high amine contents, we first optimized the hydrolysis conditions of PNVA and PNMVA homopolymers prior to investigating the transformation of the binary and ternary copolymers prepared in the former section. Two sets of hydrolysis conditions were tested. The first method is similar to the one reported by Akashi et al.,20,21i.e. HCl 2 N/120 °C/14 h. Secondly, harsher conditions were considered (HCl 6 N/120 °C/64 h). The latter are inspired by the hydrolysis method reported by Okatova et al.34 (4 N/100 °C/60 h) using a prolonged reaction time and a higher acid concentration except for that we increased the hydrochloric acid concentration to 6 N in order to reach beyond the 44% of hydrolysis yield reported for PNMVA.34
In all cases, the copolymer concentration in the HCl solution was rather low (5–7% w/v). For PNVA, both procedures afforded high levels of hydrolysis, as suggested by the disappearance of signal c at 2.0 ppm corresponding to the methyl protons of the acetamide group on the 1H NMR spectra of the purified product (Fig. 4; left spectra). In agreement with previously reported NMR characterization of PVAm, a shift of the methylene protons from 1.6 ppm (a) to 2.0 ppm (a′) was observed upon hydrolysis.48 It is worth mentioning that the amine proton resonance cannot be detected because of rapid proton exchange with D2O. The conversion of PNVA into PNVAm was quantified by elementary analysis (EA) by comparison of the C/N content ratio in polymers before and after acidic treatment (Table S3†). This comparative method based on the C/N ratio rather than on absolute C or N mass percentages prevents mistakes inherent to the hygroscopic character of these polymers. PNVA hydrolysis rates as high as 90% and 94% were recorded, when using the milder (HCl 2 N, 120 °C, 14 h) and the harsher (HCl 6 N, 120 °C, 64 h) hydrolytic conditions, respectively (Table 3, entry 1). In other words, no significant difference in the PNVA hydrolysis is observed in the two sets of experiments.
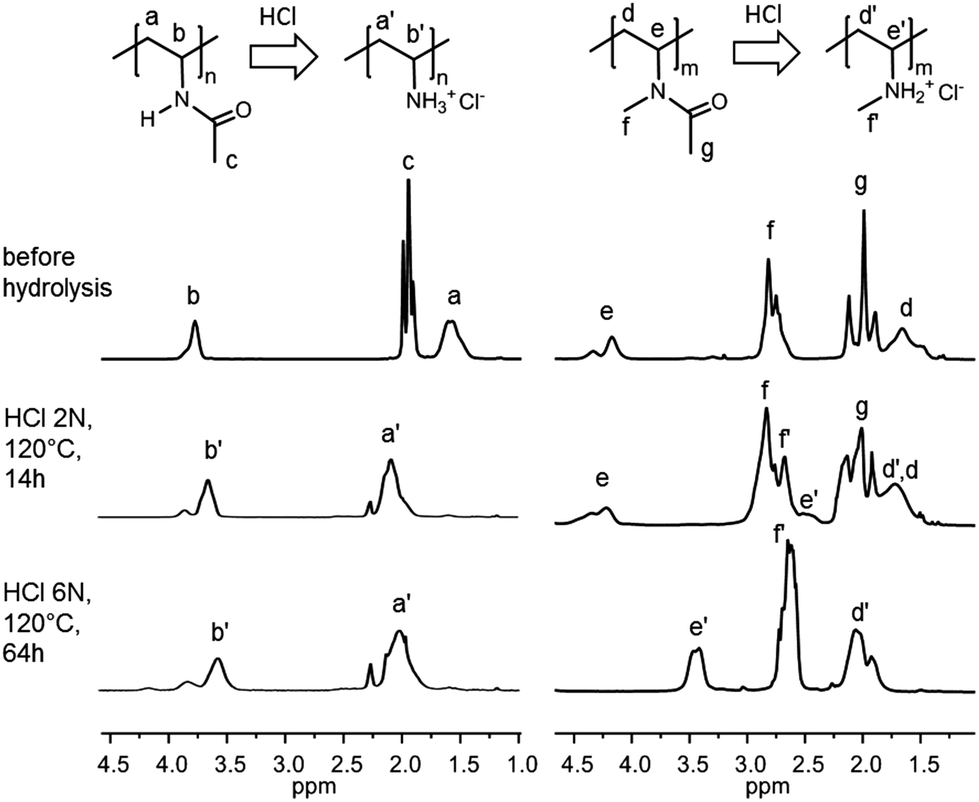 | ||
| Fig. 4 1H NMR of the PNVA (left) and PNMVA (right) before and after acidic hydrolysis and purification by dialysis under various conditions. Spectra were recorded at 298 K in D2O. | ||
In contrast, the hydrolysis of PNMVA was only effective under harsh conditions (HCl 6 N/120 °C/64 h). Indeed, the 1H NMR spectrum after the treatment of PNMVA for 14 h at 120 °C with HCl 2 N still exhibits an intense signal g at 2.0 ppm characteristic of the residual acetamide (Fig. 4, right spectra). 1H–13C Heteronuclear Single-Quantum Correlation (HSQC) spectroscopy experiments (Fig. S3†) confirmed the low level of PNMVA hydrolysis under mild acidic conditions. Comparison of the relative intensities of the acetamide (g) and methylene (d, d′) signals before and after hydrolytic treatment indicated that about 17% of the amides were hydrolyzed. This value was confirmed by elementary analysis (19% of hydrolysis). In contrast, the severe conditions (HCl 6 N/120 °C/64 h) allowed almost complete conversion of PNMVA into poly(N-methyl vinylamine) (PNMVAm). In addition to the apparent decrease in the intensity of peak g, the signals for the methine (e) and methylene protons (d) significantly shifted on the 1H NMR and HSQC spectra (Fig. 4 and S3†). According to EA analysis, the hydrolysis reached 81% within 64 h (entry 2, Table 3), which makes this approach valuable for the preparation of polymers containing pendant secondary amines. Finally, the integrity of the poly(vinylamine) backbone following hydrolysis was confirmed by SEC in aqueous media: monomodal size exclusion chromatograms were recorded for PNVAm and PNMVAm even after the harshest acidic treatment (Fig. S4†).
The optimized hydrolysis conditions (6 N/64 h/120 °C) were then applied to a series of binary and ternary copolymers containing NVA, NMVA and VIm units (entries 3–6, Table 3). For each copolymer, the efficiency of hydrolysis was quantified by EA taking into account the change in the C/N ratio upon hydrolysis. For copolymers containing both NVA and NMVA, the hydrolysis percentage of NVA was fixed at 94%, the value determined for homoPNVA hydrolyzed under the same conditions (entry 1, Table 3). Based on this approximation, the conversion of the NMVA units in the P(NVA-stat-NMVA) (entry 3, Table 3) copolymer was 90%, a value consistent with the results of P(NMVA) hydrolysis. As illustrated by the 1H NMR (Fig. S1†) and EA analyses (Table S3†) , successful hydrolysis of the amide functions was achieved for all binary copolymers including those with pendant imidazole moieties (entries 3–5, Table 3). In addition, the drastic decrease or disappearance of the carbonyl peak at 1700 cm−1 in the infrared spectra confirmed the efficient removal of the acetamide groups in the copolymers (Fig. S5†). Finally, the hydrolysis procedure involving HCl 6 N was applied to a ternary copolymer composed of similar molar fractions of NVA, NMVA and VIm (entry 6, Table 3), producing the desired P(NVAm-stat-NMVAm-stat-VIm) multi-amine copolymer.
Conclusion
The two-step strategy developed here is particularly efficient for the preparation of pendant primary, secondary amines and/or imidazoles bearing copolymers with predictable compositions. The latter consists of the synthesis of N-vinylamides and N-vinylimidazole-containing copolymers followed by the hydrolysis of the pendant amide moieties. The free radical copolymerization of NVA, NMVA and/or VIm was studied first and the reactivity ratios of each binary system were measured by different methods, i.e. Fineman–Ross, the regular and extended Kelen–Tudos as well as the nonlinear approach. The four calculation methods provided similar reactivity ratios, which demonstrates the accuracy of the values. Although these reactivity ratios were determined with a free radical polymerization system, these data could also be very helpful for future investigations aiming at controlling the molecular parameters and architectures of the copolymers through living/controlled radical copolymerization mechanisms. The reactivity of the monomers was determined to be in the following order: VIm > NVA > NMVA. The composition drift phenomenon was also evaluated by the Skeist's model and was found significant for all copolymerizations when high monomer conversions were targeted. However, stopping the copolymerizations at moderate monomer conversion allowed preventing a strong inhomogeneity of the chemical composition over the chain distribution. The compositions of the P(NVA-stat-NMVA-stat-VIm) terpolymers were also successfully predicted using the Alfrey–Goldfinger equations on the basis of the reactivity ratios determined from the binary copolymerizations. Good agreement was found between the predicted and the experimentally determined values. Thanks to this copolymerization in-depth study, a series of statistical binary and ternary copolymers with known compositions were prepared and treated with acids in order to generate the pendant primary and/or secondary amine derivatives. Optimization of the hydrolysis conditions notably allowed reaching high levels of conversion of the N-methyl acetamide moieties within manageable reaction times, which is essential for the design of copolymers containing high densities of pendant secondary amines. Overall, we demonstrated the efficiency of this method for the preparation of statistical amine-rich copolymers containing primary and secondary as well as imidazole functions in adjustable proportions.These achievements represent a great opportunity to finely tune the properties of PVAms, in order to improve their applications and to develop new ones. Indeed, the introduction of secondary amines and/or imidazole groups along the PVAm backbone will undoubtedly modify its degree of ionization depending on pH, its metal/macromolecule complexation ability, and surface adhesion capacity, which are key properties of this class of materials. For example, micro- and nanohydrogels49 formed with such unrivaled polyamines should exhibit unique pH-responsiveness50,51 that could be tuned accurately for drug release applications by adjusting the content of each pendant amino group. In addition, the presence of various amines and imidazoles with specific metal binding capacities along the same polymer backbone opens new opportunities notably in the field of water purification.52,53 Previous studies also evidenced the antibacterial properties of PVAm derivatives54 and polyvinylimidazoles,55 as well as the dependence of the antimicrobial efficiency on the charge density and hydrophobicity.54 Finally, these novel polyamines might also outperform primary PVAm in applications like paper strengthening or coating applications.9–11 In other words, much can be expected from this poly(vinylamine) based synthetic platform.
Acknowledgements
The authors are grateful to the European Community in the frame of the Erasmus Mundus International Doctoral School IDSFunMat, the UPMC University, the Belgian National Funds for Scientific Research (F.R.S.-FNRS) and the Belgian Science Policy (PAI VII-05) for their financial support. A. D. is a Research Associate of the FRS-FNRS. The authors also thank P. De Tullio (ULg) and C. Troufflard (UPMC) for skillful NMR assistance.Notes and references
- R. K. Pinschmidt, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2257–2283 CrossRef CAS.
- S. Kobayashi, K. Do Suh and Y. Shirokura, Macromolecules, 1989, 22, 2363–2366 CrossRef CAS.
- A. Toutianoush, A. El-Hashani, J. Schnepf and B. Tieke, Appl. Surf. Sci., 2005, 246, 430–436 CrossRef CAS.
- A. Katchalsky, J. Mazur and P. Spitnik, J. Polym. Sci., 1957, 23, 513–532 CrossRef CAS.
- R. Merindol, S. Diabang, O. Felix, T. Roland, C. Gauthier and G. Decher, ACS Nano, 2015, 9, 1127–1136 CrossRef CAS PubMed.
- R. Pelton, Langmuir, 2014, 30, 15373–15382 CrossRef CAS PubMed.
- C. Geffroy, M. Labeau, K. Wong, B. Cabane and M. Cohen Stuart, Colloids Surf., A: Physicochem. Eng. Aspects, 2000, 172, 47–56 CrossRef CAS.
- Q. Chen and L. Zhu, Appl. Mech. Mater., 2012, 130–134, 1507–1510 CrossRef CAS.
- S. Wang, M. Wang and F. Chen, BioResources, 2014, 10, 750–759 Search PubMed.
- H. Kroener, Wochenbl. Papierfabr., 2009, 137, 172–175 CAS.
- R. Pelton and J. Hong, Tappi J., 2002, 1, 21–25 CAS.
- S. Yuan, Z. Wang, Z. Qiao, M. Wang, J. Wang and S. Wang, J. Membr. Sci., 2011, 378, 425–437 CrossRef CAS.
- P. Li, Z. Wang, Y. Liu, S. Zhao, J. Wang and S. Wang, J. Membr. Sci., 2015, 476, 243–255 CrossRef CAS.
- B. M. Novak and J. T. Cafmeyer, J. Am. Chem. Soc., 2001, 123, 11083–11084 CrossRef CAS PubMed.
- M. Mullier and G. Smets, J. Polym. Sci., 1957, 23, 915–930 CrossRef CAS.
- A. El Achari, X. Coqueret, A. Lablache-Combier and C. Loucheux, Makromol. Chem., 1993, 194, 1879–1891 CrossRef CAS.
- L. Gu, S. Zhu and A. N. Hrymak, J. Appl. Polym. Sci., 2002, 86, 3412–3419 CrossRef CAS.
- E. Witek, M. Pazdro and E. Bortel, J. Macromol. Sci., Part A, 2007, 44, 503–507 CrossRef CAS.
- M. Zhu, E. B. Radcliffe, D. W. Ragsdale, I. V. MacRae and M. W. Seeley, Agric. For. Meteorol., 2006, 138, 192–202 CrossRef.
- M. Akashi, S. Nakano and A. Kishida, J. Polym. Sci., Part A: Polym. Chem., 1996, 34, 301–303 CrossRef CAS.
- M. Akashi, S. Saihata, E. Yashima, S. Sugita and K. Marumo, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 1153–1160 CrossRef CAS.
- Y. Maki, H. Mori and T. Endo, Macromol. Chem. Phys., 2007, 208, 2589–2599 CrossRef CAS.
- A. Sekiya, M. Tamura, H. Ishida and M. Watanabe, Chem. Lett., 1988, 1223–1224 CrossRef CAS.
- X. Chen, Y. Wang and R. Pelton, Langmuir, 2005, 21, 11673–11677 CrossRef CAS PubMed.
- J. Zhu and R. E. Marchant, Biomacromolecules, 2006, 7, 1036–1041 CrossRef CAS PubMed.
- H. Mokhtari, R. Pelton and L. Jin, J. Colloid Interface Sci., 2014, 413, 86–91 CrossRef CAS PubMed.
- S. Rupp, M. von Schickfus, S. Hunklinger, H. Eipel, A. Priebe, D. Enders and A. Pucci, Sens. Actuators, B, 2008, 134, 225–229 CrossRef CAS.
- A. Scorilas, A. Bjartell, H. Lilja, C. Moller and E. P. Diamandis, Clin. Chem., 2000, 46, 1450–1455 CAS.
- C. G. Overberger and S. Kikyotani, J. Polym. Sci., Polym. Chem. Ed., 1983, 21, 525–540 CrossRef CAS.
- R. K. Pinschmidt, L. A. Wasowski, G. G. Orphanides and K. Yacoub, Prog. Org. Coat., 1996, 27, 209–218 CrossRef CAS.
- R. K. Pinschmidt, W. L. Renz, W. E. Carroll, K. Yacoub, J. Drescher, A. F. Nordquist and N. Chen, J. Macromol. Sci., Part A, 1997, 34, 1885–1905 CrossRef.
- Y. Yuan, F. Gong, Y. Cao, W. Chen, D. Cheng and X. Shuai, J. Biomed. Nanotechnol., 2015, 11, 668–679 CrossRef CAS PubMed.
- C. L. Gebhart and A. V. Kabanov, J. Controlled Release, 2001, 73, 401–416 CrossRef CAS PubMed.
- O. V. Okatova, I. I. Gavrilova, N. N. Ul'yanova, E. F. Panarin and G. M. Pavlov, Russ. J. Appl. Chem., 2012, 85, 1239–1246 CrossRef CAS.
- H. C. W. M. Buys, F. F. Vercauteren, A. van Elven and A. H. A. Tinnemans, Recl. Trav. Chim. Pays-Bas, 2010, 108, 123–127 CrossRef.
- G. Odian, Principles of Polymerization, 4th edn, 2004 Search PubMed.
- M. Fineman and S. D. Ross, J. Polym. Sci., 1950, 5, 259–262 CrossRef CAS.
- T. Kelen and F. Tudos, J. Macromol. Sci., Part A: Chem., 1975, 9, 1–27 CrossRef.
- A. Wamsley, B. Jasti, P. Phiasivongsa and X. Li, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 317–325 CrossRef CAS.
- J. M. Ting, T. S. Navale, F. S. Bates and T. M. Reineke, ACS Macro Lett., 2013, 2, 770–774 CrossRef CAS.
- I. Skeist, J. Am. Chem. Soc., 1946, 68, 1781–1784 CrossRef CAS PubMed.
- T. Alfrey and G. Goldfinger, J. Chem. Phys., 1944, 12, 322 CrossRef CAS.
- T. Kelen, F. Tüdöus, B. Turcsányi and J. P. Kennedy, J. Polym. Sci., Polym. Chem. Ed., 1977, 15, 3047–3074 CrossRef CAS.
- A. Debuigne, A. N. Morin, A. Kermagoret, Y. Piette, C. Detrembleur, C. Jerome and R. Poli, Chem. – Eur. J., 2012, 18, 12834–12844 CrossRef CAS PubMed.
- K. Nakabayashi and H. Mori, Eur. Polym. J., 2013, 49, 2808–2838 CrossRef CAS.
- A. Kermagoret, C.-A. Fustin, M. Bourguignon, C. Detrembleur, C. Jerome and A. Debuigne, Polym. Chem., 2013, 4, 2575–2583 RSC.
- A. Kermagoret, K. Mathieu, J.-M. Thomassin, C.-A. Fustin, R. Duchene, C. Jerome, C. Detrembleur and A. Debuigne, Polym. Chem., 2014, 5, 6534–6544 RSC.
- M. Akashi, E. Yashima, T. Yamashita, N. Miyauchi, S. Sugita and K. Marumo, J. Polym. Sci., Part A: Polym. Chem., 1990, 28, 3487–3497 CrossRef CAS.
- B. H. Tan, J. P. K. Tan and K. C. Tam, in Hydrogel Micro and Nanoparticles, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2012, pp. 81–115 Search PubMed.
- K. Yamamoto, T. Serizawa, Y. Muraoka and M. Akashi, Macromolecules, 2001, 34, 8014–8020 CrossRef CAS.
- L. Jin, Q. Sun, Q. Xu and Y. Xu, Bioresour. Technol., 2015, 197, 348–355 CrossRef PubMed.
- A. K. Kushwaha, N. Gupta and M. C. Chattopadhyaya, Desalin. Water Treat., 2014, 1–13 Search PubMed.
- R. Wang, J. Men and B. Gao, Clean: Soil, Air, Water, 2012, 40, 278–284 CrossRef CAS.
- E.-H. Westman, M. Ek, L.-E. Enarsson and L. Wågberg, Biomacromolecules, 2009, 10, 1478–1483 CrossRef CAS PubMed.
- H. El-Hamshary, M. M. G. Fouda, M. Moydeen, M. H. El-Newehy, S. S. Al-Deyab and A. Abdel-Megeed, Int. J. Biol. Macromol., 2015, 72, 1466–1472 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional copolymerization data, 1H NMR and HSQC spectra, size exclusion chromatography and infra-red analyses. See DOI: 10.1039/c5py01325a |
| This journal is © The Royal Society of Chemistry 2016 |