DOI:
10.1039/C5PY01343G
(Paper)
Polym. Chem., 2016,
7, 54-62
Chemical vapour deposition of soluble poly(p-xylylene) copolymers with tuneable properties†
Received
22nd August 2015
, Accepted 6th October 2015
First published on 7th October 2015
Abstract
Chemical vapour deposition (CVD) is a technique widely applied for the synthesis of thin and highly conformal polymer coatings. The copolymerization of poly(1,4-xylylene)s (PPX), with 2-hydroxyethyl methacrylate (HEMA) via CVD revives a hardly investigated field of PPX chemistry. The use of alkyl substituted [2.2]paracyclophanes leads to soluble PPX-copolymers. Proof of copolymerization was obtained by correlation of both monomer units in 1H,13C-HMBC 2D-NMR spectra. Next, insoluble p(PX-N-co-HEMA) copolymers were synthesized in a modified commercially available SCS Labcoater. The availability of the hydroxy ester functionality of HEMA on the film surface was indirectly confirmed by the reduction of water contact angles down to 65°. Preliminary studies using human umbilical vein endothelial cells (HUVEC) indicate the cytocompatibility of as deposited p(PX-N-co-HEMA) films.
Introduction
The preparation of conformal polymer coatings by CVD allows coating of sensitive substrates with complex surface geometries. W. F. Gorham established an extremely efficient CVD coating method by vapour phase pyrolysis of [2.2]paracylclophane to 1,4-quinodimethane, subsequent deposition and spontaneous polymerization to PPX.1 The applications for coatings obtained by CVD of PPX range from protective coatings to highly functionalized bioactive surfaces.2,3 Although a variety of PPX derivatives with lateral substituents are known4 their utilization in the biomedical field demands that they not only possess good biocompatibility but also demonstrate controlled and favourable interaction with the biological environment.5 In recent years several functionalized PPX derivatives have been developed on a laboratory scale for the generation and engineering of (bio)functional surfaces including alkyne-modified PPX6 for azide–alkyne cycloadditions (click reactions), anhydride and ester-modified PPX,7 amine functionalized PPX,8 and PPX carrying initiator groups for controlled radical polymerizations.9 However, the functionalization of [2.2]paracyclophanes, the starting material for PPX synthesis via CVD, can involve several reaction steps and can often be time consuming. Towards functionalized PPX, copolymerization of [2.2]paracylophanes and conventional monomers in the CVD process is a potentially powerful tool for the generation of polymer coatings that combine the outstanding properties of PPX with reactive and functional moieties. The introduction of these groups via easily available monomers could obviate the need for time consuming and expensive [2.2]paracyclophane functionalization. Despite the promising concept, the copolymerization of [2.2]paracyclophanes with double bond containing monomers or other reactive molecules has received little attention. Apart from some publications in the early days of PPX chemistry,10,11 there are only two groups that have reported such CVD copolymers of PPX in the 1990s. Copolymers of [2.2]paracyclophane or its mono and bichlorinated derivatives were obtained with maleic anhydride,12 4-vinylpyridine,12N-vinylpyrrolidone,12 styrene,12 9-vinylanthracene,13 4-vinylbiphenyl,13 perfluorooctyl methacrylate,13–15 and N-phenyl maleimide.16 The major drawbacks of these copolymers were compositional inhomogeneity, the need for post-deposition purification, and limited methods for polymer analysis due to insolubility. The most recent interest in PPX-copolymers focused on the surface functionalization of PPX by deposition on reactive monomer liquids.17,18 These studies have evoked further theoretical interest in the copolymerization mechanism and reactivity of p-xylylene moieties with conventional double bond containing monomers.19,20
In this work, we copolymerized alkyl substituted [2.2]paracyclophanes, which are known to yield soluble PPX-homopolymers,21 with 2-hydroxyethyl methacrylate (HEMA) as comonomer. To our knowledge, this is the first report of any soluble PPX-copolymers obtained via CVD and their analysis by NMR spectroscopy. Furthermore, we used a modified commercially available CVD reactor to synthesize copolymer films of [2.2]paracyclophane and HEMA free of unreacted comonomer, and analysed their potential for biomedical applications by testing their in vitro cytocompatibility using human umbilical vein endothelial cells (HUVEC).
Experimental section
Materials
[2.2]Paracyclophane and dichloro-[2.2]paracyclophane (Speciality Coatings Systems, SCS), 2-hydroxyethyl methacrylate (HEMA) (Aldrich, 97%) and chloroform (Fischer, p.a. grade) were used as received. Tetrahydrofuran and methanol were distilled prior to use. 4,12-Di(n-alkyl)[2.2]paracyclophanes were synthesized from dichloro[2.2]paracyclophane according to Bier et al.21 For cell culture tests Vasculife EnGS Basal Medium (LifelineCellTech), penicillin–strepomycin–amphotericin B (Pan Biotech), 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) (Sigma Aldrich), phosphate buffered saline (PBS) (gibco), RPMI-1640 medium (Pan Biotech), paraformaldehyde (PFA) (Applichem), 4′,6-diamidino-2-phenylindole (DAPI) (Sigma Aldrich), Phalloidin-FITC (Invitrogen) and a combined calcein AM and ethidium homodimer-1 staining kit (live/dead assay) (life technologies) were used as received.
Instrumentation
CVD reactors for PPX copolymerization.
All copolymers were prepared according to the CVD procedure described by Gorham1 with an additional comonomer inlet. The CVD was carried out either in a homemade glass apparatus or in a modified Labcoater PDS2010 (Specialty Coatings Systems) (Fig. 1). The reactor consisted of a sublimation chamber, and a pyrolysis tube (fused quartz glass, length 90 cm, diameter 2.5 cm) connected to the deposition chamber. An additional T-piece for mixing with the comonomer vapour was used for the copolymerization. During the reaction the pyrolysis tube and the precursor sublimation chamber were placed in a tubular three-zone furnace (Pyrolus AT) with adjustable temperatures for each individual zone. The glass tubes outside the furnace were wrapped with heating tape (set to 150 °C) to prevent monomer deposition. The product was deposited either parallel (Fig. 1c) or perpendicular (Fig. 1d) to the monomer flow direction and recovered from the glass walls or deposition target. The temperature of the cooling jacket, the deposition target and the comonomer evaporation vessel were controlled with individual thermoregulation units. Reduced pressure was achieved with an Edwards S two-stage oil pump connected to the pyrolysis reactor. The base pressure of 2.1 × 10−2 mbar was controlled with an Edwards Pirani gauge 1002 (PRL 10) behind the cooling trap. For copolymerization in the commercially available setup the Labcoater was modified with an additional stainless steel comonomer inlet into the deposition chamber. The comonomer evaporation temperature was controlled with a thermoregulation unit.
 |
| | Fig. 1 Schematic illustration of CVD setups for PPX copolymerization. Self-designed reactor for p(PX-co-HEMA) synthesis (a), with deposition chambers for deposition parallel (b) and perpendicular (c) to the monomer flow direction, and modified Labcoater PDS2010 (d). | |
Gel permeation chromatography (GPC).
The analysis of molecular weights and distributions was performed by GPC using tetrahydrofuran as eluent. Samples were analysed after purification with a flow rate of 0.8 ml min−1 with a setup containing two SDV columns (10 μm, 60 × 0.8 mm2), calibrated with polystyrene standard (both purchased from PSS). The elugrams were recorded with UV and RI detectors, RI was used for molecular weight determination.
Water contact angle (CA).
Static water contact angle measurements were carried out using a G10 measurement system (Krüss) and the droplet images were captured with a USB camera (eScope). The contact angles were determined with DropSnake22 plugin for ImageJ (Version 1.46r). For each sample twelve images were acquired and evaluated, the highest and the lowest value were discarded and the average was calculated from the remaining ten.
Scanning electron microscopy (SEM).
The SEM images were recorded with JSM-7500 F (JEOL) and Zeiss 1530 instruments, at an acceleration voltage of 2–3 kV. Samples were sputtered with platinum prior to analysis.
Nuclear magnetic resonance (NMR).
The NMR spectra were recorded at room temperature in CDCl3 as solvent (set to 7.26 ppm), on Bruker AVANCE 300 and DRX 400 instruments.
Thermal characterization.
The thermal properties of the polymers were characterized using a differential scanning calorimeter (DSC) 821e module and a TG 851 thermobalance (both Mettler Toledo). For DSC 5–8 mg sample were analysed at a heating rate of 20 °C min−1, and thermogravimetric analyses (TGA) were performed on a 5–12 mg sample, from 25–800 °C (10 °C min−1) under nitrogen atmosphere.
Infrared spectra (IR).
The IR spectra were recorded on a Digilab IR-spectrometer (Excalibur series) equipped with an ATR-unit containing a ZnSe crystal.
Cell culture.
All polymer coated glass slides and control glass slides were sterilized by immersion in 70% ethanol and washed twice with PBS buffer solution. The HUVECs were cultured on glass slides in agarose coated well plates in Vasculife EnGS Basal Medium supplemented with 1% penicilin–strepomycin–amphotericin B in a humidified incubator at 5% CO2 and 37 °C, the seeding density was 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per cm2. Cell viability tests (MTT assays) were performed on cell-seeded glass slides. After incubation, the culture medium was removed and the cells were incubated for 3 h with MTT containing RPMI (0.25 mg ml−1). The resultant formazan crystals were dissolved in DMSO and the absorbance at 550 nm was measured on a Synergy HT plate reader (BioTek). A Live/dead assay was performed on cell-seeded glass slides incubated for 48 h. The staining was carried out using a combined Calcein AM and Ethidium homodimer-1 staining kit. After incubation the cell culture medium was replaced by medium containing Calcein AM (0.5 μM) and Ethidium homodimer-1 (1 μM) and incubated at 37 °C for 30 min. Samples were examined with a Cell Observer Z1 fluorescence microscope (Zeiss). The Cell morphology was analysed on cell-seeded glass slides incubated for 48 h. After washing with PBS, the adhered cells were fixed with 4% paraformaldehyde (PFA) for 10 min at 0 °C, stained with DAPI and Phalloidin-FITC according to manufacturer's protocol (http://tools.lifetechnologies.com/content/sfs/manuals/mp03224.pdf) and examined on a Cell Observer Z1 fluorescence microscope (Zeiss).
000 cells per cm2. Cell viability tests (MTT assays) were performed on cell-seeded glass slides. After incubation, the culture medium was removed and the cells were incubated for 3 h with MTT containing RPMI (0.25 mg ml−1). The resultant formazan crystals were dissolved in DMSO and the absorbance at 550 nm was measured on a Synergy HT plate reader (BioTek). A Live/dead assay was performed on cell-seeded glass slides incubated for 48 h. The staining was carried out using a combined Calcein AM and Ethidium homodimer-1 staining kit. After incubation the cell culture medium was replaced by medium containing Calcein AM (0.5 μM) and Ethidium homodimer-1 (1 μM) and incubated at 37 °C for 30 min. Samples were examined with a Cell Observer Z1 fluorescence microscope (Zeiss). The Cell morphology was analysed on cell-seeded glass slides incubated for 48 h. After washing with PBS, the adhered cells were fixed with 4% paraformaldehyde (PFA) for 10 min at 0 °C, stained with DAPI and Phalloidin-FITC according to manufacturer's protocol (http://tools.lifetechnologies.com/content/sfs/manuals/mp03224.pdf) and examined on a Cell Observer Z1 fluorescence microscope (Zeiss).
Synthesis
4,12-Di(n-alkyl)[2.2]paracyclophane synthesis.
4,12-Di(n-alkyl)[2.2]paracyclophanes 1b–e were synthesized from 4,12-dichloro[2.2]paracyclophane via an established synthesis route reported by Bier et al.21
General route for p(PX-co-HEMA) copolymers synthesis in self-designed CVD reactor.
In a typical synthesis run, 300 mg of precursor 1a–e was placed in the sublimation chamber and 5 ml comonomer 2 in the comonomer evaporation vessel. The vessel was evacuated briefly to remove air, but just long enough to avoid evaporation of the comonomer. The deposition chamber was connected to the temperature controller and set to the respective deposition temperature. All other parts of the reactor were evacuated for 1 h before the furnace was preheated to the respective pyrolysis temperature (1a: 650 °C, 1b: 600 °C, 1c: 640 °C, 1d: 550 °C, 1e: 520 °C) and the transportation zone temperature was set to 300 °C. At the same time the comonomer vessel was heated to the respective evaporation temperature in a preheated temperature bath. After 0.5 h of temperature equilibration, the valve behind the furnace was closed prior to heating the sublimation zone in order to prevent p-xylylene radicals from entering the deposition chamber. The sublimation zone was heated to the respective temperature (80–130 °C). Afterwards the precursor and the monomer valve were opened simultaneously. The polymerization was terminated after 2–5 h (depending on the amount of precursor and sublimation/vaporization temperatures) by flooding the apparatus with ambient air. The copolymer product was either washed with methanol or dissolved in chloroform and precipitated in methanol for removal of unreacted HEMA comonomer.
P(PX-N-co-HEMA) 3a.
IR (ν/cm−1): 3430 (m), 3006 (w), 2922 (m), 2854 (w), 1722 (s), 1513 (s), 1450 (m), 1416 (w), 1384 (w), 1259 (w), 1158 (m), 1077 (m), 1021 (m), 952 (w), 898 (w), 821 (s), 751 (w). Data on IR analysis of p(PX-co-HEMA) 3b–e are given in ESI.†
P(PX-propyl-co-HEMA) 3d.
1H-NMR (300 MHz, CDCl3, δ/ppm): 7.15 H11 (m), 7.02 H8,12 (m), 6.91 Har* (m), 4.20 H15 (m), 3.77 H16 (m), 3.04 H5*,6* (m), 2.85 H5,6, (bs), 2.72 H5*,6* (m), 2.59 H3 (bs), 2.27–1.09 H13 (m), 1.61 H2 (bs), 1.23 H14 (bs), 0.99 H1 (bs).
P(PX-butyl-co-HEMA) 3e.
1H-NMR (300 MHz, CDCl3, δ/ppm): 7.15 H11 (m), 7.03 H8,12 (m), 6.93 Har* (m), 4.22 H15 (m), 3.79 H16 (m), 3.04 H5*,6* (m), 2.86 H5,6, (bs), 2.73 H5*,6* (m), 2.62H4 (bs), 1.57 H3 (bs), 2.41–1.17 H13 (m), 1.42 H2 (bs), 1.24 H14 (bs), 0.96 H1 (bs).
13C-NMR (100 MHz, CDCl3, δ/ppm): 177.8–176.9 C18, 140.5 C9, 140.2 Car*, 139.8 C7, 138.0 Car*, 137.3 C10, 134.9 Car*, 131.1 Car*, 129.3 C8, 129.1 C11, 128.4 Car*, 127.6 Car*, 125.9 H12, 66.3 C15, 61.2 C16, 48.2–47.7 C17, 45.5 C5*,6*, 41.7–40.8 C13, 37.5 C6, 34.3 C5, 33.6 C3, 32.6 C4, 29.7 C14, 22.9 C2, 20.6 C14, 14.1 C1.
General route for p(PX-N-co-HEMA) 3a copolymer synthesis in Labcoater PDS2010
In a typical synthesis run, 5 g precursor 1a and 5–8 ml HEMA comonomer 2 were used. After preheating the pyrolysis furnace to 650 °C, evacuation to base pressure (about 60 mbar) and preheating the comonomer to the respective evaporation temperature, the precursor sublimation furnace was set to 150 °C and as soon as the temperature reached 90 °C, the comonomer valve was opened. The synthesis was terminated before HEMA was completely evaporated (5–8 h) to ensure the availability of both monomers during the synthesis by flooding the reactor with ambient air.
Results and discussion
Copolymers synthesized in a reactor
Copolymers of different alkyl substituted [2.2]paracyclophanes 1a–e with HEMA 2 were successfully synthesized in a custom built reactor (Fig. 1) via CVD as depicted in Scheme 1.
 |
| | Scheme 1 General reaction schemes and CVD parameter for p(PX-co-HEMA) 3a–e synthesis. | |
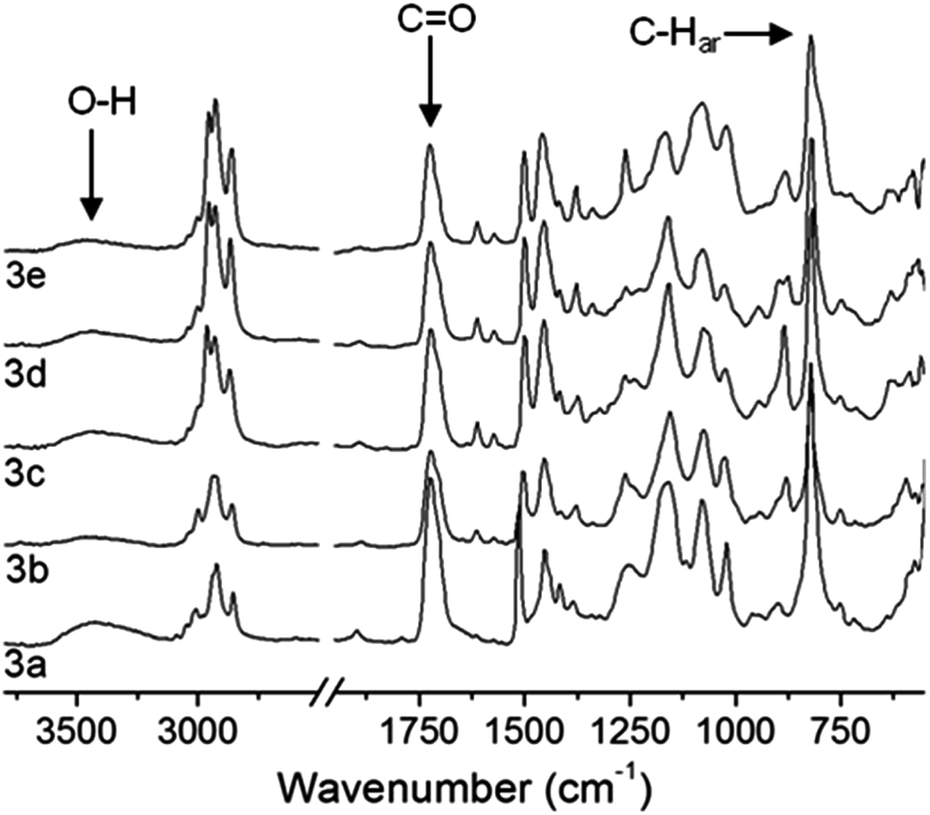
For all p(PX-co-HEMA) copolymers 3a–e characteristic vibration modes of both monomers units were identified by ATR-IR spectroscopic analysis after purification (Fig. 2). The carbonyl ester group stretching vibration around 1720 cm−1 and the broad absorption around 3700–3100 cm−1 of the hydroxy group stretching vibration confirmed that the hydroxy ester side group of HEMA was retained during the CVD process while the strong absorption around 820 cm−1, which is characteristic for the aromatic C–H vibration (C–Har) of the p-xylylene unit was clearly evident.
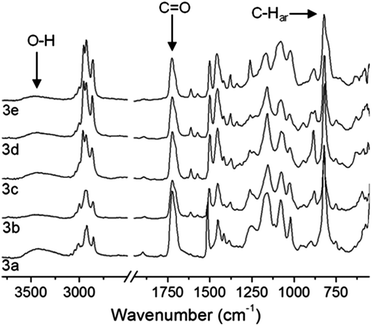 |
| | Fig. 2 ATR-IR transmission spectra of p(PX-co-HEMA) copolymers 3a–e. The integral ratio of the carboxy and the C–Har vibrations were used to monitor the relative HEMA content of the copolymer films depending on the position in the deposition chamber (see Fig. 1). | |
Deposition parallel to the monomer flow direction.
For copolymers deposited parallel to the monomer flow direction, as illustrated in Fig. 1c, the composition of the copolymer changed with the position in the deposition chamber. As depicted in Fig. 3, the composition change was monitored by the ATR-IR integral ratio of the HEMA carbonyl group and the p-xylylene aromatic C–H vibration. With increasing distance into the chamber the HEMA content in the p(PX-butyl-co-HEMA) 3e and p(PX-propyl-co-HEMA) 3d copolymers increased. However, no such clear trend was observed for P(PX-N-co-HEMA) 3a. Along with the composition gradient, the films showed a decrease in thickness with increasing distance into the chamber. Gradients for composition and film thickness, due to differences in condensation behaviour of the monomer components and monomer consumption, were reported by Gaynor et al. for different copolymers of [2.2]paracyclophanes with vinylic comonomers.13
 |
| | Fig. 3 Composition changes for p(PX-co-HEMA) copolymers deposited parallel to the monomer flow (deposition chamber Fig. 1c). Copolymer films with gradient were obtained for p(PX-propyl-co-HEMA) 3d and p(PX-butyl-co-HEMA) 3e. p(PX-N-co-HEMA) 3a showed a less pronounced trend but was far more inhomogeneous. | |
Deposition perpendicular to the monomer flow direction.
Copolymers obtained from a deposition target placed perpendicular to the monomer flow (Fig. 1d) showed an increase in HEMA content with increasing HEMA evaporation temperature for different copolymer deposition temperatures, as shown in Fig. 4. Regardless of the deposition chamber model or HEMA evaporation temperatures, the obtained copolymer films contained unreacted liquid HEMA comonomer.
 |
| | Fig. 4 Copolymer samples of p(PX-propyl-co-HEMA) 3d show a dependence in HEMA content with increasing HEMA evaporation temperature (Tsub(DPX-pr) = 120 °C, Tp(DPX-pr) = 550 °C for all reactions. This underlines the importance of reactor geometry for tunable copolymer composition. | |
Solution NMR analysis of p(PX-alkyl-co-HEMA) copolymers.
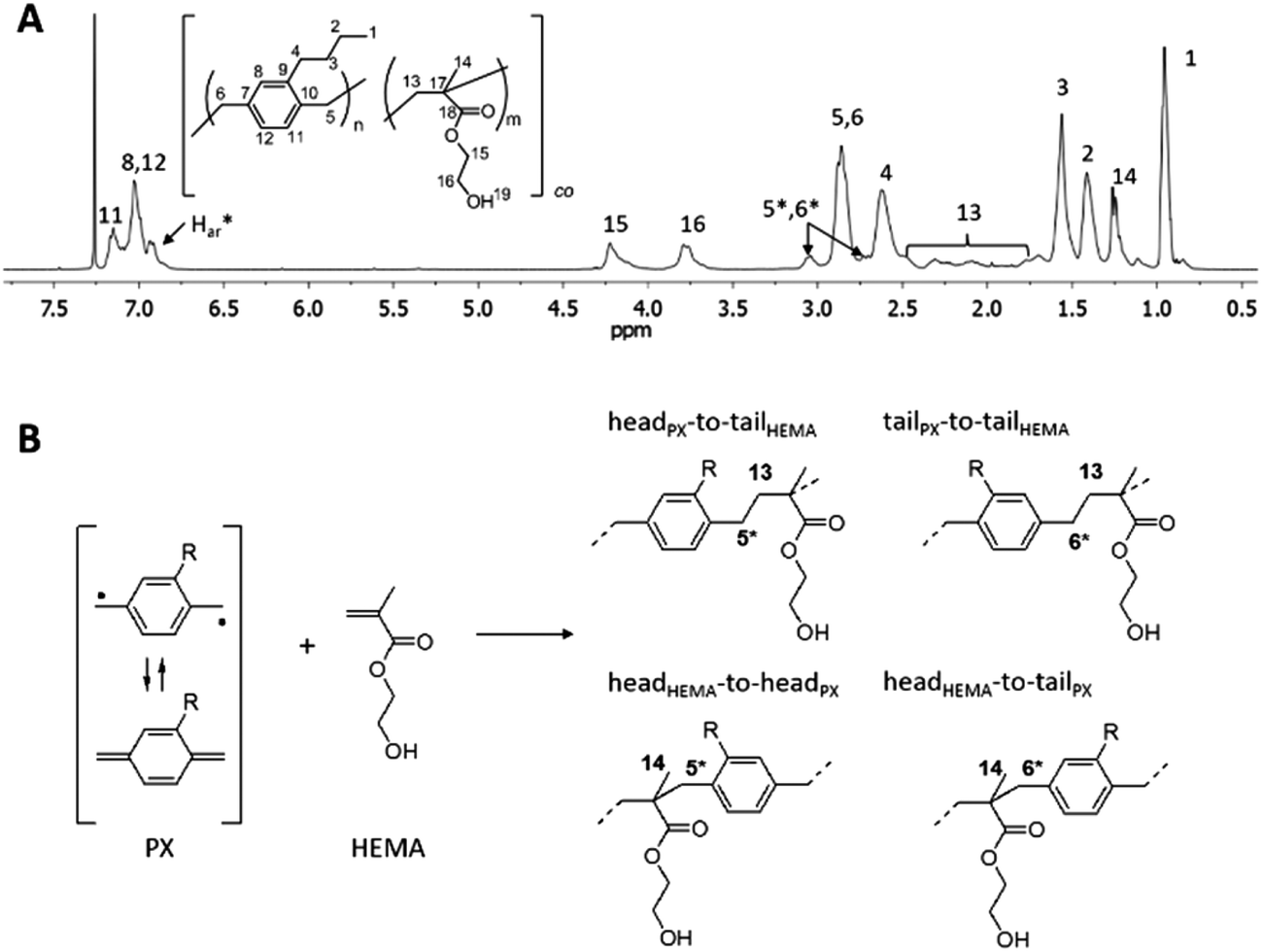
Similar to what has been reported for PPX-alkyl homopolymers,21 substituents longer than propyl on the aromatic rings of the [2.2]paracyclophane led to copolymers soluble in common organic solvents like tetrahydrofuran and chloroform. For the first time, this enabled solution-NMR analysis of PPX-copolymers prepared by the CVD method. Fig. 5a shows 1H-NMR spectra of PPX-butyl copolymers with HEMA, p(PX-butyl-co-HEMA) 3e. The characteristic hydrogen signals of the different monomer units in p(PX-butyl-co-HEMA) were identified by comparison with the homopolymers PPX-butyl21 and PHEMA23 and designated accordingly. Additional resonance signals at 6.94 ppm (Har*) and 3.05 ppm (H5*,6*) were attributed to p-xylylene units directly bound to HEMA molecules in the copolymer, as depicted in Fig. 5b. 13C-NMR analysis also showed all signals expected from both monomer units, together with resonances around 45.5 ppm (C5*,6*) and several signals in the region of aromatic carbon atoms (Car*) (Fig. 6). The splitting observed for several carbon resonances is due to head-to-tail isomerism and known for PPX-alkyl homopolymers.21 In the copolymer the number of possible magnetically different environments for individual carbon atoms is further increased. The broad distribution of the HEMA carbon resonance signals for backbone carbons C13 and C14 is also influenced by the isomerism of head-to-tail attachment for the unsymmetrically substituted alkyl-p-xylylene units. The presence of multiple C18 carbonyl peaks in the region of 177.8 to 176.7 ppm (Fig. 6a) suggests random copolymer architecture with the absence of longer HEMA blocks.
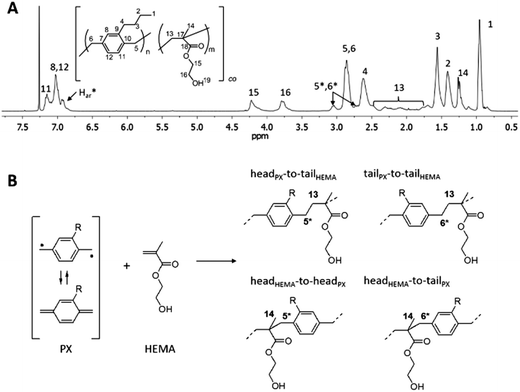 |
| | Fig. 5 (A) 1H-NMR spectra (in CDCl3) of copolymer p(PX-butyl-co-HEMA) 3e, (B) possible structures of PX-HEMA moieties due to “head-to-tail” isomers resulting from the reaction of a substituted p-xylylene diradical with HEMA. | |
 |
| | Fig. 6
13C-NMR spectrum of p(PX-butyl-co-HEMA) 3e in CDCl3. Areas of special interest are shown in magnification: (A) splitting of HEMA carbonyl C18, (B) aromatic region with additional resonances for p-xylylene units bound to HEMA units, and (C) aliphatic carbons resonances. | |
The cross peaks in 1H,13C-HSQC (heteronuclear single quantum coherence) spectra were used to confirm hydrogen and carbon peak assignment (Fig. 7). The final proof for the covalent linkage between butyl-p-xylylene units and HEMA units was given by the 1H,13C-HMBC (heteronuclear multiple bond correlation) NMR technique which clearly showed the correlation between carbon and hydrogen atoms of both monomer types. As an example, HSQC spectra confirmed the designation of carbon resonances around 45.5 ppm (C5*,6*) and methylene hydrogen atoms H5*,6* at 3.05 ppm and 2.72 ppm. HMBC spectra showed that these carbons (C5*,6*) were correlated to hydrogen atoms H14 from HEMA units (Fig. S4†). Furthermore, carbons C14 and C17 from HEMA showed correlations to hydrogen atoms H5*,6* which indeed confirmed the vicinity of both monomer units. Further proof for copolymerization was found e.g. by the correlation of the additional aromatic carbon signals (Car*) with hydrogen atoms H13 from HEMA, and the correlation of the HEMA carbonyl carbon C18 with hydrogen atoms H5*,6* (Fig. S3†).
 |
| | Fig. 7
1H,13C-HSQC spectrum of p(PX-butyl-co-HEMA) 3e in CDCl3, resonance signals were assigned with the help of 1H,13C-HMBC spectra. Regions of special interest are shown in magnification. | |
Copolymer properties
Various copolymers of 2,14-dialkyl[2.2]paracyclophane 1a–e with HEMA 2 were synthesized by changing CVD process parameters like paracyclophane sublimation temperature, comonomer evaporation temperature, or deposition temperature in order to influence the partial pressure of the respective monomer in the reactor and copolymerization behaviour. Maximum HEMA contents of 50 to 66 mol% were achieved for insoluble p(PX-co-HEMA) copolymers 3a–c (determined by elemental analysis). The soluble copolymers p(PX-propyl-co-HEMA) 3d and p(PX-butyl-co-HEMA) 3e were synthesized with maximum HEMA contents of 37 mol% and 27 mol%, respectively (determined by 1H-NMR) Table 1 shows detailed reaction parameters for p(PX-butyl-co-HEMA) 3e synthesis. The solubility of these copolymers allowed a molecular weight analysis by gel permeation chromatography (GPC) in THF. The copolymers showed higher polydispersities, and molecular weights ranging from 64000 to 101000 Da (relative to polystyrene standard, see ESI Fig. S6† for GPC traces), which were lower than those generally obtained for PPX-butyl homopolymers as reported by Bier et al.21 The incorporation of HEMA into PPX-butyl led to a decrease in thermal stability (Fig. S5†) and an increase in the glass transition temperature compared to PPX-butyl homopolymer. Exemplarily, Fig. 8 shows DSC thermograms of the second heating curve of two different p(PX-butyl-co-HEMA) copolymers compared to PPX-butyl. The homopolymer (curve a) has a glass transition temperature (Tg) of −4 °C, an exotherm at 63 °C, and an endothermal transition at 115 °C. The comparison of thermograms from PPX-N and p(PX-N-co-HEMA) copolymers, that showed overall higher HEMA content than p(PX-butyl-co-HEMA), showed that the copolymers polymorphic transitions normally observed for PPX-N were inhibited. These results showed that the thermal properties were changed by copolymerization, but also indicated the presence of longer PPX-butyl blocks in some copolymers. One explanation for this was the sample's compositional inhomogeneity due to reactor geometry, as already explained (Fig. 3). The presence of longer PPX-blocks in the copolymers is also supported by quantum calculations on reactivity ratios of p-xylylene units with vinylic/acrylic comonomers in CVD copolymerizations made by Bobrowski et al.19 They showed that p-xylylene is far more reactive compared to the comonomer, and therefore is the dominant species in the copolymer chains over a wide range of monomer feed ratios. The occasionally random distribution of comonomer units along the chain, as described by Bobrowski et al., is also supported by the presence of multiple carbonyl resonances in 13C-NMR, as discussed above. However, the HEMA contents determined in this study are surprisingly high considering these assumptions.
 |
| | Fig. 8 DSC thermograms (second heating curve) of two different p(PX-butyl-co-HEMA) 3e copolymers (b: 15 mol% HEMA, c: 8 mol% HEMA) compared to PPX-butyl homopolymer (a). | |
Table 1 Data on p(PX-butyl-co-HEMA) 3e synthesized via CVD in self-designed reactor, deposited parallel to the monomer flow direction, as illustrated in Fig. 1. All runs were performed at pyrolysis temperature Tp = 520 °C, transportation temperature Tt = 300 °C
| Run |
T
sub DPX/°C |
T
vap HEMA/°C |
T
dep/°C |
mol% HEMA |
GPC (RI detector)a |
| 105Mn |
105Mw |
PDI |
|
Molecular weights determined relative to polystyrene standards, n. d. not determined.
|
| 1 |
128 |
30 |
5 |
6 |
n.d. |
| 2 |
128 |
40 |
5 |
7 |
0.64 |
1.82 |
2.8 |
| 3 |
120 |
30 |
5 |
8 |
1.01 |
4.37 |
4.0 |
| 4 |
113 |
30 |
5 |
12 |
0.94 |
2.23 |
2.4 |
| 5 |
120 |
45 |
5 |
13 |
0.67 |
2.54 |
3.8 |
| 6 |
110 |
40 |
5 |
15 |
0.69 |
2.56 |
3.7 |
| 7 |
110 |
40 |
5 |
24 |
n.d. |
| 8 |
125 |
45 |
10 |
27 |
0.75 |
1.59 |
2.1 |
Synthesis in Labcoater
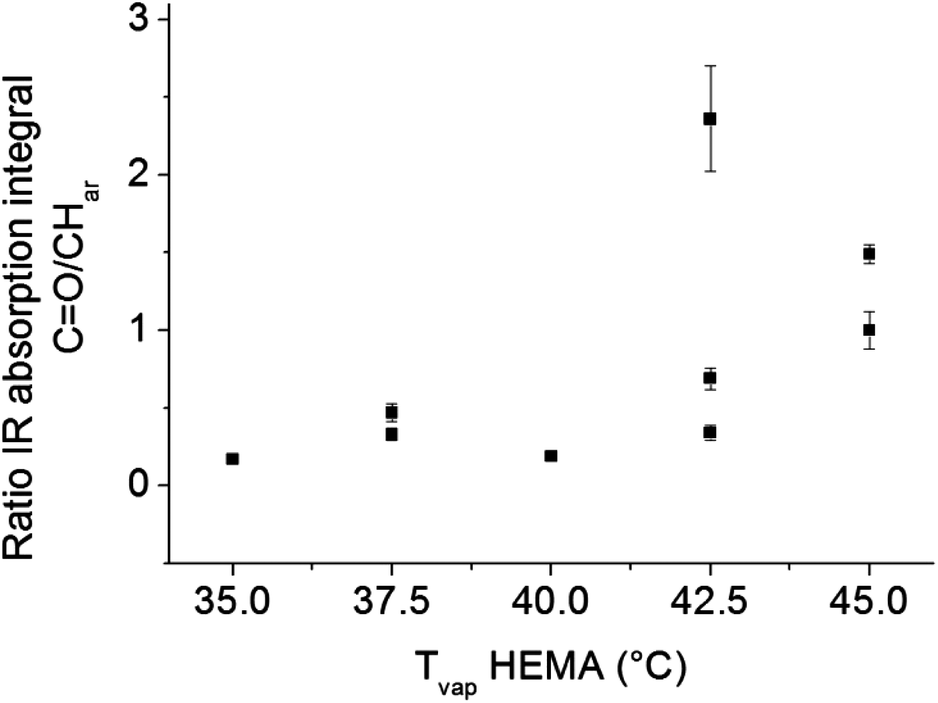
The problems of spatial inhomogeneity in film composition were overcome by copolymerization in a modified Labcoater PDS 2010 with rotating sample holder plate. The relative HEMA content of the films was monitored by ATR-IR analysis. As shown in Fig. 9, with increasing HEMA evaporation temperature, the relative HEMA content in the copolymer increased. P(PX-N-co-HEMA) copolymer films obtained by CVD from the modified Labcoater showed a decrease in water contact angle with increasing HEMA evaporation temperature, which corresponds to an increase in the relative HEMA content in the copolymers (Fig. 10). This confirmed the availability of the hydroxy groups on the copolymers film surface. Furthermore, those films exhibited a smoother surface compared to p(PX-N-co-HEMA) films deposited parallel to the monomer flow direction in the custom built CVD reactor (Fig. 1), as shown in Fig. 11. Films from the custom built reactor showed a pronounced surface roughness in form of mounds with diameters around 2–3 μm, as illustrated by SEM images in Fig. 11a and b. These surface structures have their origins in the shielding effects in the CVD process that prevent homogeneous film growth at certain angles between the substrate surface and the monomer flow direction,24 depending on deposition rate and time.25 The rotating sample holder plate in the Labcoater in combination with a different deposition direction and process pressure helps to prevent shielding effects and minimized surface structures.
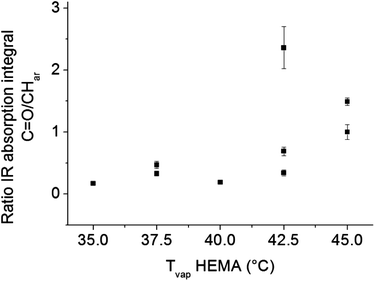 |
| | Fig. 9 P(PX-N-co-HEMA) 3a copolymer films prepared in Labcoater show increasing IR absorption signal ratio of HEMA carbonyl groups C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O to aromatic C–H vibration of p-xylyene with increasing HEMA evaporation temperature in the synthesis process. This confirms that the final copolymer composition depends on the process parameter and HEMA content in the copolymer can be tuned. O to aromatic C–H vibration of p-xylyene with increasing HEMA evaporation temperature in the synthesis process. This confirms that the final copolymer composition depends on the process parameter and HEMA content in the copolymer can be tuned. | |
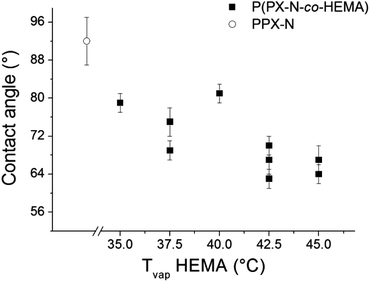 |
| | Fig. 10 Water contact angles on p(PX-N-co-HEMA) 3a surfaces (prepared in Labcoater) decrease with increasing HEMA evaporation temperature in the synthesis process. This indirectly confirms the increasing HEMA content in the polymer as well as hydroxy group availability on the film surface. | |
 |
| | Fig. 11 Scanning electron microscopy images of the surface of p(PX-N-co-HEMA) 3a films. The films synthesized in the homemade CVD reactor (A, B) deposited parallel to the monomer flow show a globular surface structure, such features were not observed for polymer films synthesized in Labcoater. | |
Cytocompatibility studies
In biomedical applications cell adhesion can be either beneficial or unfavourable depending on the desired application. However, in the case of materials for cell contacting devices, a lack of cytotoxicity is highly desirable. PPX and PHEMA homopolymers are known to be biocompatible and are widely applied. However, in the case of PPX-copolymerization via CVD monomer residue in the as-deposited films is a known problem which can hamper the utilizations of these coatings without further treatment. Since one of the potential applications of the PPX copolymer is in cardiovascular applications such as stents or vascular prostheses, cytotoxicity and cell adhesion potential were assessed in vitro in preliminary studies using human umbilical vein endothelial cells (HUVEC). The HUVECs were seeded on surfaces coated with p(PX-N-co-HEMA) and the commercially available PPX derivative PPX-N and cell viability was then assessed using the MTT assay. After 3 days of incubation, the HUVECs on both materials tested showed a decrease in metabolic activity of 65% for both PPX-N and the copolymer relative to the glass control. In order to examine whether a potential cytotoxic activity of the polymer films was the reason for the reduced cell numbers, a live/dead assay was carried out (Fig. 12a–c). In general, cell numbers were considerably lower on the polymer films versus the glass control and the absence of a significant number of dead cells (stained red) on the polymer films suggested that reduced cell adhesion might be the underlying cause of the differences observed. The reduced adhesion of HUVECs on the polymer films is most likely due the increase in hydrophilicity of the polymer surfaces compared to PPX-N homopolymers (water contact angles of 85–75°). In order to verify this hypothesis, further MTT assays with p(PX-co-HEMA) coated surfaces were conducted and the cell numbers after different time spans of incubation were compared to the controls (Fig. S-2†). It could be shown that after 3 h of incubation the number of adhered HUVECs on the PPX-copolymer surface was 50% relative to the glass control. After 24 h and 48 h the cell numbers on the polymer surfaces relative to the control lay within the same range (between 40% and 50% of the control), indicating that reduced cell adhesion on the PPX-copolymer-coated surfaces rather than a pronounced cytotoxic activity of the polymer films accounts for the difference in cell numbers detected.
 |
| | Fig. 12 HUVEC stained with live/dead assay (A–C) and DAPI/Phalloidin-FITC staining to reveal cell morphology (D–F). | |
In addition, the morphology of HUVECs on the different surfaces was examined. As observed in the viability tests, the cells showed lower adhesion on the polymer surfaces compared to the glass control and the cells appeared more dissociated with the absence of cell islands and a prevalence of single cells. Furthermore, actin localization towards the cell edges and the formation of filopodia could be observed in HUVECs on both PPX-copolymer and PPX-N surfaces, suggesting the acquisition of a more motile phenotype on those surfaces.
Conclusion
[2.2]Paracyclophanes were copolymerized with HEMA to give PPX copolymers with high comonomer content. The use of alkyl-substituted [2.2]paracyclophanes led to soluble copolymers. Two-dimensional NMR analysis of CVD copolymers obtained from 4,12-di(n-butyl)[2.2]paracyclo-phanes and HEMA proved for the first time the direct vicinity of the two monomer units and thus the copolymer formation. The copolymerization in different CVD reactors underlined the importance of reactor design and process parameters to obtain homogeneous copolymer films free of unreacted comonomer. With adequate reactor geometry and sample positioning the amount of incorporated HEMA comonomer can be influenced by CVD process parameters. The retention of the hydroxy ester during the CVD process was indirectly confirmed by ATR-IR analysis and its availability on the surface confirmed by water contact angle measurements. The technique presented simplifies and broadens methods of PPX modification by using commercially available double bound containing or other reactive molecules as comonomers. Furthermore, initial tests with HUVECs confirmed that as deposited PPX-copolymer films do not exhibit cytotoxic potential caused by toxic monomer residues, and therefore have great potential for application as coating materials in biomedical applications.
Acknowledgements
The authors thank SCS Speciality Coatings systems for kindly providing paracyclophane donation and provisioning of SCS Labcoater PDS2010. VPS wishes to thank the excellence initiative of the German Federal and State Governments Grant EXC 294 (Centre for Biological Signalling Studies) for funding.
Notes and references
- W. F. Gorham, J. Polym. Sci., Part A1, 1966, 4, 3027–3039 CrossRef CAS.
- X. Deng and J. Lahann, J. Appl. Polym. Sci., 2014, 131, 40315 Search PubMed.
- L. Alexandrova and R. Vera-Graziano, Polym. Mater. Encycl., 1996, 9, 7180–7189 Search PubMed.
- A. Greiner, S. Mang, O. Schäfer and P. Simon, Acta Polym., 1997, 48, 1–15 CrossRef CAS.
- H. Y. Chen and J. Lahann, Langmuir, 2011, 27, 34–48 CrossRef CAS PubMed.
- H. Nandivada, H.-Y. Chen, L. Bondarenko and J. Lahann, Angew. Chem., Int. Ed., 2006, 45, 3360–3363 CrossRef CAS PubMed.
- J. Lahann, M. Balcells, T. Rodon, J. Lee, I. S. Choi, K. F. Jensen and R. Langer, Langmuir, 2002, 18, 3632–3638 CrossRef CAS.
- J. Lahann, D. Klee, W. Pluester and H. Hoecker, Biomaterials, 2001, 22, 817–826 CrossRef CAS PubMed.
- X. Jiang, H. Y. Chen, G. Galvan, M. Yoshida and J. Lahann, Adv. Funct. Mater., 2008, 18, 27–35 CrossRef CAS.
- L. A. Errede and M. Szwarc, Q. Rev., 1958, 301–322 RSC.
- J. R. Schaefgen, J. Polym. Sci., 1955, 15, 203–219 CrossRef CAS.
- V. Sochilin, K. Mailyan, L. Aleksandrova, A. Nikolaev, A. Pebalk and I. Kardash, Dokl.Akad. Nauk SSSR, 1991, 319, 173–176 CAS.
- J. F. Gaynor and S. B. Desu, J. Mater. Res., 1994, 9, 3125–3130 CrossRef CAS.
- J. F. Gaynor and S. B. Desu, J. Mater. Res., 1995, 11, 236–241 CrossRef.
- J. F. Gaynor, S. B. Desu and S. S. Senkevich, Macromolecules, 1995, 28, 7343–7348 CrossRef CAS.
- J. F. Gaynor, J. J. Senkevich and S. B. Desu, J. Mater. Res., 1996, 11, 1842–1850 CrossRef CAS.
- M. Naddaka, F. Asen, S. Ferea, M. Bobrowski, P. Skurski, E. Laux, J. Charmet, H. Keppner and J. P. Lellouche, J. Polym. Sci., Part A: Polym. Chem., 2011, 517, 4583–4586 Search PubMed.
- A. Bolognesi, C. Botta, A. Andiscova, U. Giovanella, S. Arnautov, J. Charmet, E. Laux and H. Keppner, Macromol. Chem. Phys., 2009, 210, 2052–2057 CrossRef CAS.
- M. Bobrowski, S. Freza and P. Skurski, Macromolecules, 2012, 45, 8532–8546 CrossRef CAS.
- M. Bobrowski, P. Skurski and S. Freza, Chem. Phys., 2011, 382, 2026 CrossRef.
- A. K. Bier, M. Bognitzki, J. Mogk and A. Greiner, Macromolecules, 2012, 45, 1151–1157 CrossRef CAS.
- A. F. Stalder, G. Kulik, D. Sage, L. Barbieri and P. Hoffmann, Colloids Surf., A, 2006, 286, 92–103 CrossRef CAS.
- R. K. Bose and K. K. S. Lau, Chem. Vap. Deposition, 2009, 15, 150–155 CrossRef CAS.
- M. Cetinkaya, N. A. Malvadakar and G. Demirel, J. Polym. Sci., Part B: Polym. Phys., 2008, 46, 640–648 CrossRef CAS.
- R. Tao and M. Anthamatten, Langmuir, 2012, 28, 16580–16587 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional information from ATR-IR analysis, 1D and 2D NMR spectra, GPC traces and biocompatibility tests. See DOI: 10.1039/c5py01343g |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.