
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Titania modification with a ruthenium(II) complex and gold nanoparticles for photocatalytic degradation of organic compounds
Shuaizhi Zheng*ab,
Zhishun Weia,
Kenta Yoshiiriac,
Markus Braumüllerb,
Bunsho Ohtaniac,
Sven Raub and
Ewa Kowalska*ac
aInstitute for Catalysis, Hokkaido University, N21, W10, 001-0021 Sapporo, Japan. E-mail: xishuai423@hotmail.com; kowalska@cat.hokudai.ac.jp
bInstitute of Inorganic Chemistry 1, Ulm University, Albert-Einstein-Allee 11, 89081 Ulm, Germany
cGraduate School of Environmental Science, Hokkaido University, N10, W5, 060-0810 Sapporo, Japan
First published on 14th December 2015
Abstract
Titania of fine anatase nanoparticles (ST01) was modified successively with two components, i.e., a ruthenium(II) complex with phosphonic anchoring groups [Ru(bpy)2(4,4′-(CH2PO3H2)2bpy)]2+ bpy = 2,2′-bipyridine (RuIICP) and gold nanoparticles (Au). Various compositions of two titania modifiers were investigated, i.e., Au, Au + RuIICP, Au + 0.5RuIICP, RuIICP, 0.5RuIICP and 0.25RuIICP, where Au and RuIICP correspond to 0.81 mol% and 0.34 mol% (with respect to titania), respectively. In the case of hybrid photocatalysts, the sequence of modification (ruthenium(II) complex adsorption or gold deposition) was investigated to check its influence on the resultant properties and thus photocatalytic performance. Diffuse reflectance spectroscopy (DRS), X-ray photoelectron spectroscopy (XPS) and scanning transmission electron microscopy (STEM) were applied to characterize the structural properties of the prepared photocatalysts, which confirmed the successful introduction of modifiers of the ruthenium(II) complex and/or gold NPs. Different distributions of gold particle sizes and chemical compositions were obtained for the hybrid photocatalysts prepared with an opposite sequence. It was found that photocatalytic activities depended on the range of used irradiation (UV/vis or vis) and the kind of modifier in different ways. Gold NPs improved the photocatalytic activities, while RuIICP inhibited the reactions under UV/vis irradiation, i.e., methanol dehydrogenation and acetic acid degradation. Oppositely, RuIICP greatly enhanced the photocatalytic activities for 2-propanol oxidation under visible light irradiation.
Introduction
TiO2 semiconductor materials have been widely used in the area of photocatalysis for decomposition of organic pollutants and clean fuel production,1,2 due to their strong photocatalytic activity, high chemical stability, low cost and relatively non-toxic properties.3,4 However, remaining challenges in TiO2, such as rapid recombination of photogenerated electron/hole pairs, fast backward reactions (in the absence of efficient charge carrier scavengers) and inability to utilize the visible range of the solar spectrum, still need to be overcome to improve the photocatalytic efficiency and develop visible light-responsive materials.3 Various modifications of titania, like noble metal loading, transition metal ion doping, nonmetal doping, sensitizer attachment, and composition with carbon nanomaterials have been extensively investigated in the past few years.1–3,5–8 Among these modifiers, gold nanoparticles (NPs)9,10 and ruthenium(II) dyes11,12 are excellent candidates to broaden the visible light performance of titania. Spherical Au NPs show surface plasmon absorption at around 500–700 nm (until the IR range for different shapes of gold nanostructures, such as nanorods, nanocages, nanoshells)13,14 and ruthenium(II) complexes have the 1MLCT (metal to ligand charge transfer) band centered at around 450 nm. An additional advantage of Au NPs in comparison with other noble metals (Ag and Cu) is their chemical stability, resistance to oxygen, and ability to serve as co-catalytic sites.15–17 Gold NPs incorporated in DSSCs (dye-sensitized solar cells) were reported to enhance the solar conversion efficiency.18–20 Meanwhile, ruthenium(II) complexes were also successfully applied towards visible light activation in DSSCs,21 where the photoexcited electrons could be injected from ruthenium sensitizers to the conduction band of titania. The adaption of two modifiers to titania may lead to a synergetic effect, which might improve the photocatalytic performance.22–24 Recently, we have investigated hybrid TiO2 photocatalysts composed of different kinds of titania (anatase, rutile and their mixture), plasmonic NPs (Au and Ag) and a [Ru(bpy)2(4,4′-(PO3H2)2bpy)]2+ sensitizer.25 Different photocatalytic outcomes of enhancement or inhibition have been observed for titania modified with two components, possibly due to the interaction between two modifiers. Therefore, in this paper, titania with different compositions of gold NPs and ruthenium(II) was applied to gain further insight into their roles during the photocatalytic reactions. Commercial titania (ST01) of anatase form and a high specific surface area (to provide the high capability of ruthenium(II) immobilization) was selected for this study.25 Gold in the amount of 2 wt% to titania was applied based on our former results, which would provide an optimal light absorption when deposited on small anatase titania.26 The ruthenium(II) complex of [Ru(bpy)2(4,4′-(CH2PO3H2)2bpy)]2+ possessing an additional methylene unit between a phosphonate anchoring group and bpy was tested in the present study. The methylene spacer lengthens the electron transfer distance between the ruthenium(II) core and TiO2 surface,27,28 therefore would potentially increase the lifetime of the excited states and change the electron transfer kinetics,29 which are important factors governing the whole photocatalytic process. In addition, two deposition methods of inverse sequences (Au/RuIICP or RuIICP/Au) were adopted purposely to check whether it influences the resultant properties and thus the photocatalytic activities, as was suggested in the previous paper,25i.e., it was proposed that ruthenium(II) could be adsorbed also on the metallic NPs instead of titania, especially in the case of positively charged silver NPs. On the other hand, it is also suspected that deposition of gold on ruthenium(II) modified titania should result in the formation of different sizes of gold NPs than in the case of direct gold deposition on a bare titania surface.Results and discussion
Preparation of photocatalysts
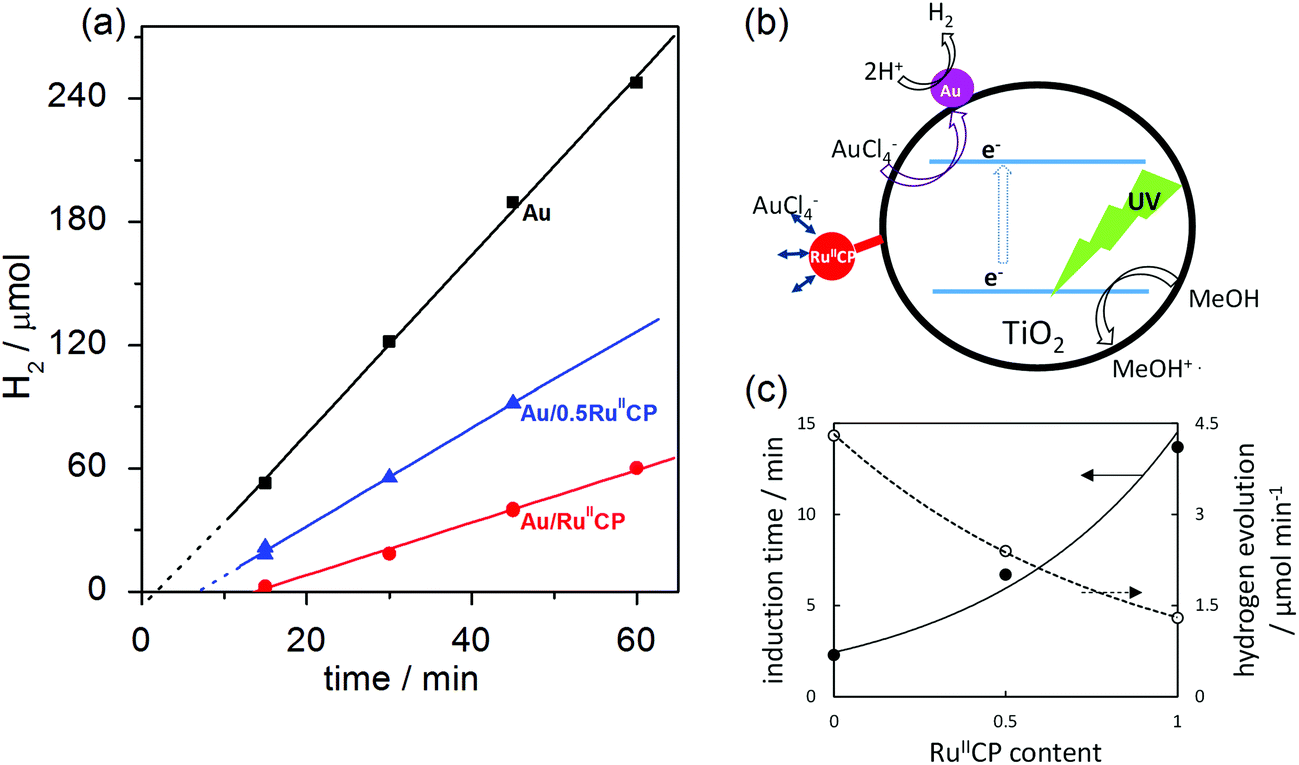 | ||
| Fig. 1 Methanol dehydrogenation during photodeposition of gold on bare and RuIICP-modified titania (ST01): (a) hydrogen evolution with time, (b) proposed mechanism, (c) influence of RuIICP amount on induction time and hydrogen evolution rate. | ||
It was found that with the increase in the amount of RuIICP on titania (0, 0.5, 1), the induction period of hydrogen generation became longer (2.3 min, 6.7 min and 13.7 min, respectively), as shown in Fig. 1(c). Similar results were obtained for deposition of gold on pre-modified titania with silver NPs, where silver hindered Au+3 reduction.16 It is suggested that the charge on the titania surface from RuIICP affects the following gold photodeposition, i.e., ruthenium(II) complexes attached to titania with an overall negative charge, which contributed to the deprotonated phosphonate groups are repulsive to the AuCl4− ions. In addition, the steric hindrance caused by the ruthenium(II) complex could hinder the gold ions to reach the titania surface and thus their reduction by photogenerated electrons, as shown in the scheme presented in Fig. 1(b). Besides the two times longer induction time with the increase in the RuIICP amount from 0.5 to 1, the rate of hydrogen generation also reduced by nearly half. It is expected that the adsorbed RuIICP influences light harvesting by titania. The excited state properties of RuIICP binding to titania showed the redox potential of −0.93 V (RuIII/II*), which would be sufficient to inject electrons to the conduction band of titania of −0.3 V under similar conditions.30 It is well known that the ruthenium complexes could inject electrons into the conduction band of titania under visible light irradiation. However, in the presence of UV light, the ruthenium complexes may also undergo ligand exchange with the solvent, decomposition or desorption, etc., which might improve or inhibit the electron injection processes, e.g., by adjusting the energy levels between the LUMO of the dye and the conduction band of titania.31 Herein, two kinds of disturbances in titania photoabsorption properties could be considered, i.e., (i) RuIICP competes with titania for light absorption to excite the electrons from the bpy ligands, and/or (ii) the “inner filter effect” due to the dark color of ruthenium(II)-adsorbed TiO2, which can result in hindrance of light penetration through the titania suspension. It is expected that the latter one could be rejected since gold modified titania possessing the highest photocatalytic activity exhibited similar darkness (photoabsorption properties) as Au/RuIICP (as shown in Fig. 3 and 4). Furthermore, the existence of induction time for RuIICP modified titania suggests that the single modified titania with only RuIICP should not possess meaningful activity (similar to bare titania samples) for hydrogen evolution.
As has been already mentioned, after the accomplishment of Au NP deposition, the hydrogen evolution processes started. Under UV/vis irradiation of titania, the electron is excited from the valence band (VB) to the conduction band (CB) leaving a hole in the VB. Herein, the Au NPs operate as the electron sinks, which trap and store the electrons from the CB, and then act as the reduction sites for hydrogen generation. For all the samples, the amount of gold was identical, i.e., 2 wt% to titania. With the increase of RuIICP content on titania, the rates of hydrogen evolution decreased (4.3 μmol min−1, 2.4 μmol min−1 and 1.3 μmol min−1). Such a phenomenon might reveal (in addition to the already mentioned competition for light absorption) that the second modifier (RuIICP) inhibits the desirable electron transfer steps for hydrogen formation. Considering all the electron transfer steps, it is highly possible that the Au NPs might either interact with RuIICP, or RuIICP works also as an electron sink for the CB electrons, which has been already suggested by time-resolved microwave conductivity (TRMC) measurements.25 The obtained data, i.e., the induction periods and hydrogen evolution rates, during gold deposition, imply that RuIICP inhibits the methanol dehydrogenation under UV/vis irradiation.
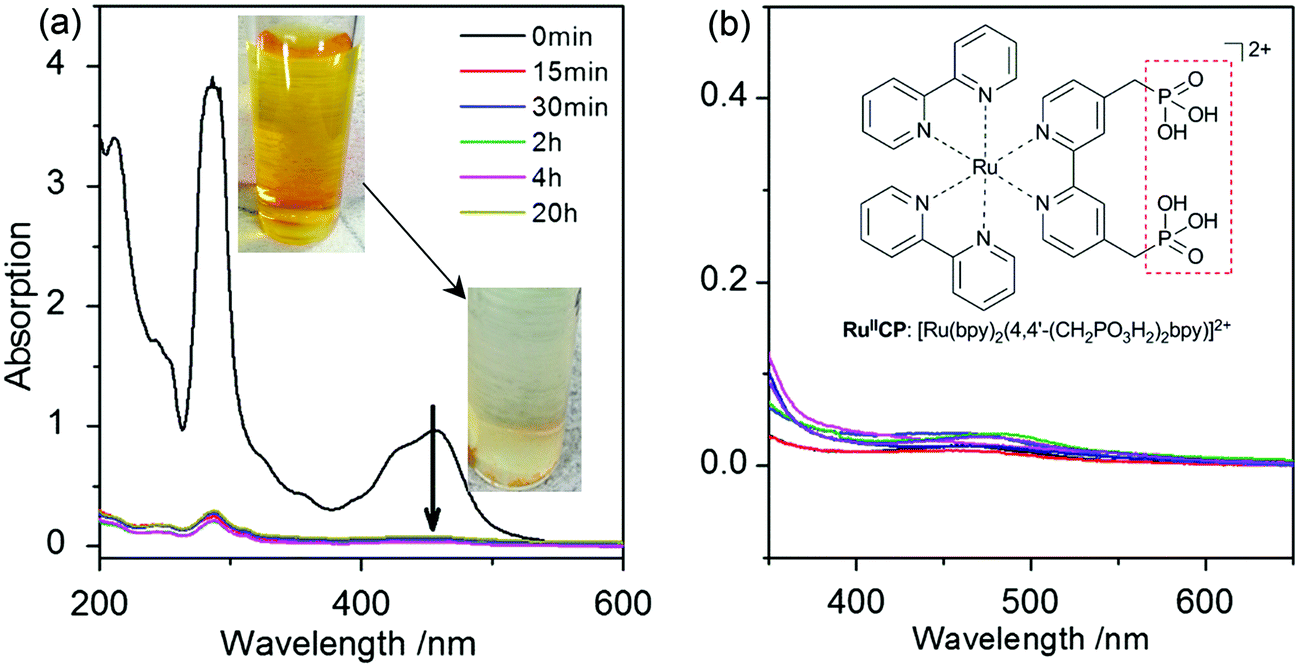 | ||
| Fig. 2 (a) RuIICP adsorption behavior on bare titania ST01 in aqueous solution; the inset: photographs of bright orange color of RuIICP aqueous solution used for adsorption, and colorless supernatant after RuIICP mixing with titania; (b) absorption spectra of all photocatalysts after 90 min UV/vis irradiation during methanol dehydrogenation (colors of curves are respective to samples presented in Fig. 4); the inset: the structure of RuIICP: [Ru(bpy)2(4,4′-(CH2PO3H2)2bpy)]2+. The absorption spectra were taken from the liquid phase of the suspension after removal of particles by centrifugation. | ||
The stability of attachment was investigated during long-lasting stirring of ruthenium(II) adsorbed titania in an aqueous suspension. It was found that even 24 h of stirring did not affect the RuIICP attachment. Moreover, even after 90 min strong UV/vis irradiation in MeOH/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), a negligible amount of ruthenium could be found in the liquid phase for all the adsorbed RuIICP-titania samples, which indicated its stable chemical binding to titania, as shown in Fig. 2(b). Thus, it was shown that the RuIICP adsorbed titania ST01 was robust under highly intensive energy of UV/vis irradiation conditions. It is important to address the stability, due to the requirement of applicable photocatalysts in industry.
:1), a negligible amount of ruthenium could be found in the liquid phase for all the adsorbed RuIICP-titania samples, which indicated its stable chemical binding to titania, as shown in Fig. 2(b). Thus, it was shown that the RuIICP adsorbed titania ST01 was robust under highly intensive energy of UV/vis irradiation conditions. It is important to address the stability, due to the requirement of applicable photocatalysts in industry.
Characterization of photocatalysts
The adsorption of RuIICP on white titania led to the preparation of yellowish/orange powders of different color intensities depending on the amount of used RuIICP, as shown in Fig. 3. Single gold modified titania had violet color, typical for small, spherical gold NPs, due to localized surface plasmon resonance (LSPR), while the samples with co-adsorbed RuIICP and Au NPs possessed purple brownish colors. | ||
| Fig. 3 Images of bare and modified titania (ST01) photocatalysts and their abbreviations. | ||
The absorption properties of the samples measured by diffuse reflectance spectroscopy (DRS) are shown in Fig. 4. The absorption of photocatalysts could be divided into three regions, i.e., (1) band-gap absorption of titania in UV range, (2) MLCT of ruthenium(II) at ca. 420–480 nm, and (3) LSPR of gold NPs at ca. 520–620 nm. It must be pointed out that the DRS data showed not only photoabsorption, but also scattering. In this regard, a broader shoulder at longer wavelengths detected for titania modified with Au/RuIICP than with RuIICP/Au and Au indicates the presence of larger gold NPs, mainly due to enhanced scattering. It was reported that intense scattering, detected as a broad shoulder at longer wavelengths than LSPR, was observed with an increase in the gold NP size from 10 nm to 50 nm.41
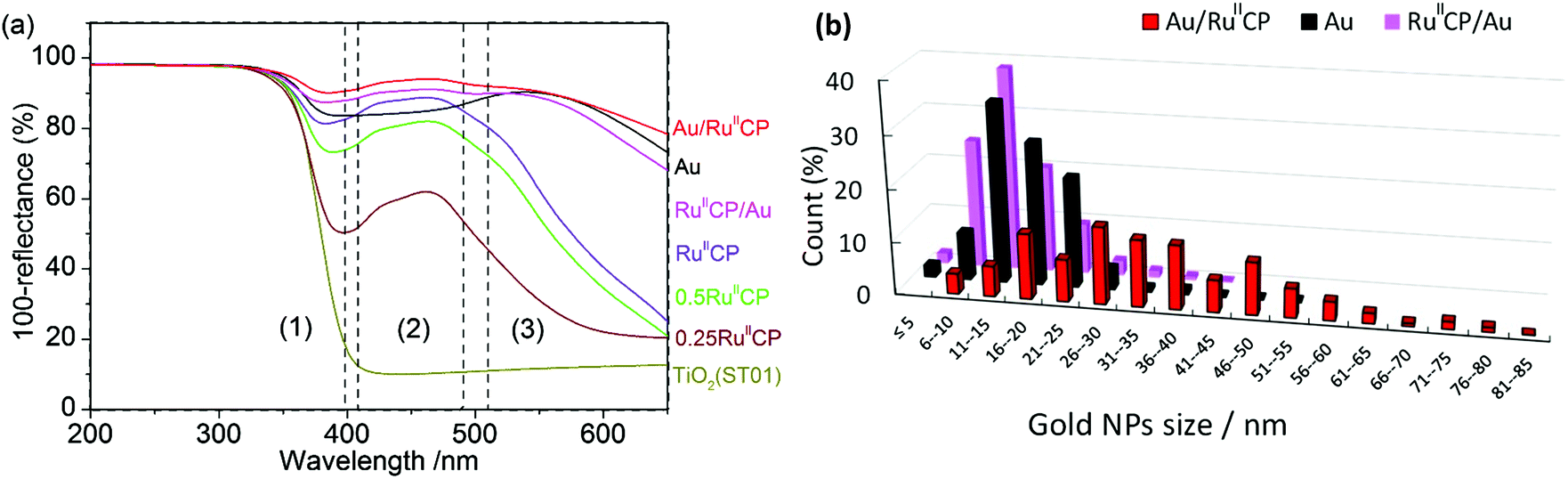 | ||
| Fig. 4 (a) DRS spectra of bare and modified titania (ST01) photocatalysts with marked ranges of photoabsorptions for: (1) titania band-gap, (2) MLCT of RuIICP and (3) LSPR of Au NPs; (b) distribution of gold NP sizes based on STEM measurements (shown in Fig. 5). | ||
In this regard, it is expected that the pre-adsorbed RuIICP disturbed the Au deposition and formation of fine gold NPs, and thus probably aggregates of gold NPs were formed on RuIICP adsorbed titania. To check this hypothesis, STEM observations were performed for titania modified with Au/RuIICP, RuIICP/Au and Au, and the exemplary STEM images with histograms of gold NP sizes are shown in Fig. 5.
 | ||
| Fig. 5 STEM images with respective histograms of gold NP sizes for titania (ST01) samples modified with Au (a and b), RuIICP/Au (c and d) and Au/RuIICP (e and f); scale bar: 100 nm. | ||
The comparison of gold NP sizes is shown in Fig. 4(b). The obtained data confirmed that the presence of ruthenium(II) highly influenced the aggregation of gold NPs. It was found that the mean size of gold NPs was in the range of 11–15 nm for titania modified with Au and RuIICP/Au, while in the case of Au/RuIICP much larger NPs were formed of 26–30 nm. In addition, it must be pointed out that in the case of gold deposition on the pre-modified titania with RuIICP, gold NPs possessed much broader distribution of their sizes (from 5 to 85 nm) and shapes (even nanorods and nanotriangles). Surprisingly, adsorption of RuIICP on gold modified titania caused a slight change in the distribution of gold sizes and a larger amount of small NPs of 6–10 nm were detected than in the case of single modified titania with Au, probably due to reshaping. Gold reshaping could be influenced by thermal treatment, a gold immobilized material,42 the presence of a surfactant, an inorganic salt,43etc. It is possible that the introduction of a second modifier could result in the change of the titania environment, like surface charge. The distribution of gold NP sizes correlates well with photoabsorption properties for all samples containing gold, i.e., the larger the gold NPs are, the broader is the shoulder of the DRS spectrum at longer wavelengths.
Three STEM modes were used for the observation of the morphology of samples, i.e., secondary electron (SE), Z-contrast (ZC) and bright-field (TE) modes. Titania ST01 possesses very fine NPs of anatase of less than 10 nm (8 nm from XRD measurements),44 and thus very fine anatase nano-crystallites in the form of aggregates are clearly observed in all the images shown in Fig. 5. It is difficult to distinguish gold from titania by simple SE mode, as shown in Fig. 5(a). Therefore for calculation of gold size distribution, TE and ZC modes were used, in which gold NPs are observed as darker and lighter spots, respectively, as clearly presented in Fig. 5(c) and (d), which show the same view of the RuIICP/Au sample. It is easily observable even without histograms that much larger gold NPs were formed in the case of gold deposition on RuIICP-modified titania (Fig. 5e and f).
To characterize the composition of photocatalysts and to check the possibility of self-co-adsorption of modifiers, i.e., RuIICP on Au or Au on RuIICP, X-ray photoelectron spectroscopy (XPS) analysis was performed, and the obtained data are shown in Fig. 6 and Table 1. The presence of gold was confirmed in all titania samples containing gold, i.e., Au, RuIICP/Au and Au/RuIICP, and its amount slightly exceeded that which was used for photodeposition (0.81 mol%) reaching 1.23, 0.88 and 1.08 mol%, respectively. Though, the differences between gold amounts are small, the lowest amount of gold (0.88 mol%) in the RuIICP/Au sample suggests that the ruthenium(II) complex could partly adsorb on the gold surface.
 | ||
| Fig. 6 XPS data of titania (ST01) modified with Au (top), RuIICP/Au (middle) and Au/RuIICP (bottom) for titanium 2p3/2 (left), gold 4f7/2 (center) and oxygen 1s (right). | ||
| Sample | Ti (mol%) | O (mol%) | O:Ti |
C (mol%) | Au (mol%) | Au:Ti (mol%) |
|---|---|---|---|---|---|---|
| Titania (ST01) | 4.04 | 27.81 | 6.9 | 68.14 | — | — |
| RuIICP | 7.44 | 30.12 | 4.1 | 62.44 | — | — |
| Au | 12.99 | 35.14 | 2.7 | 51.71 | 0.16 | 1.23 |
| RuIICP/Au | 14.74 | 38.95 | 2.6 | 46.18 | 0.13 | 0.88 |
| Au/RuIICP | 24.93 | 45.85 | 1.8 | 28.95 | 0.27 | 1.08 |
Similar to our previous findings25 gold was mainly in the zero-charged form in all the tested samples (91–100%). A small content (<3%) of positively charged gold (Auσ+) in the Au/TiO2 sample could result from Au–O–Ti linkages formed at the metal–support interface as has already been proposed for Au–TiO2 aerogels.45 This slight positive charge of gold could support the hypothesis of partial ruthenium(II) complex adsorption on the gold surface instead of titania. Similar data were reported for preferential CO adsorption on Auσ+ of Au/TiO2(P25) photocatalyst.45 It should be mentioned that various surface charges of gold NPs deposited on titania were reported, i.e., mainly zero, but also negatively (as the result of the transfer of electron density from the oxide support to Au NP)46 and positively charged.45 Our previous data for gold deposited on the other titania (P25) photocatalyst containing two crystalline (anatase and rutile) and amorphous titania forms (ca. 78, 14 and 8%, respectively)47 showed the existence of gold mainly in zero charged form (89%) with small amounts of both negative (9%) and positive (2%) states.25 Therefore, it is suggested that the support properties significantly influence the resultant properties of deposited metallic NPs. While, the lower amount of gold in Au than in Au/RuIICP modified titania could be caused by smaller particle sizes of gold in this sample, where more gold surface was covered by fine titania resulting in a decrease in the detected amount of gold.
The atomic ratio of oxygen to titania significantly exceeded 2.0 for bare titania (6.9) indicating titania surface enrichment with hydroxyl groups. This was also confirmed by deconvolution of an oxygen peak, which shows an existence of oxygen mainly in the form of free (44%) and bounded (31%) hydroxyl groups (Table 1). The modification of titania resulted in a decrease in the atomic ratio of oxygen to titania to 4.1, 2.7, 2.6 and 1.8 for RuIICP, Au, RuIICP/Au and Au/RuIICP, respectively, which indicates the substitution of surface oxygen by modifiers and/or the decrease in the adsorbed water on the titania surface. After deconvolution of titanium, oxygen and gold peaks, it was found that titanium existed mainly in the Ti4+ form and gold in the Au0 form. In the case of hybrid photocatalysts, change in the deposition sequence resulted in the preparation of samples slightly different in their properties, i.e., the RuIICP/Au sample consisted of only Ti4+ and Au0, while the Au/RuIICP sample possessed also a small amount of Ti3+ (ca. 4%), Auδ− (ca. 9%), and a much smaller amount of oxygen on the surface (especially in the –OH form).
Photocatalytic dehydrogenation of methanol under UV/vis irradiation
As has already been discussed in the preparation of photocatalysts, during gold deposition on different supports (with and without ruthenium(II)), hydrogen was generated from methanol suspensions of titania samples modified with Au/RuIICP, Au/0.5RuIICP and Au. To gain a systematical overview, all the samples were irradiated with UV/vis to compare their photocatalytic activities for methanol dehydrogenation, and the results are shown in Fig. 7. The rates of hydrogen evolution for Au/RuIICP, Au/0.5RuIICP and Au, retained the same trend with the aforementioned results (Fig. 1(a)), but the values changed due to the different conditions used for photoactivity testing than during sample preparations (ca. 5 and 20 times lower amounts of methanol and photocatalysts, respectively).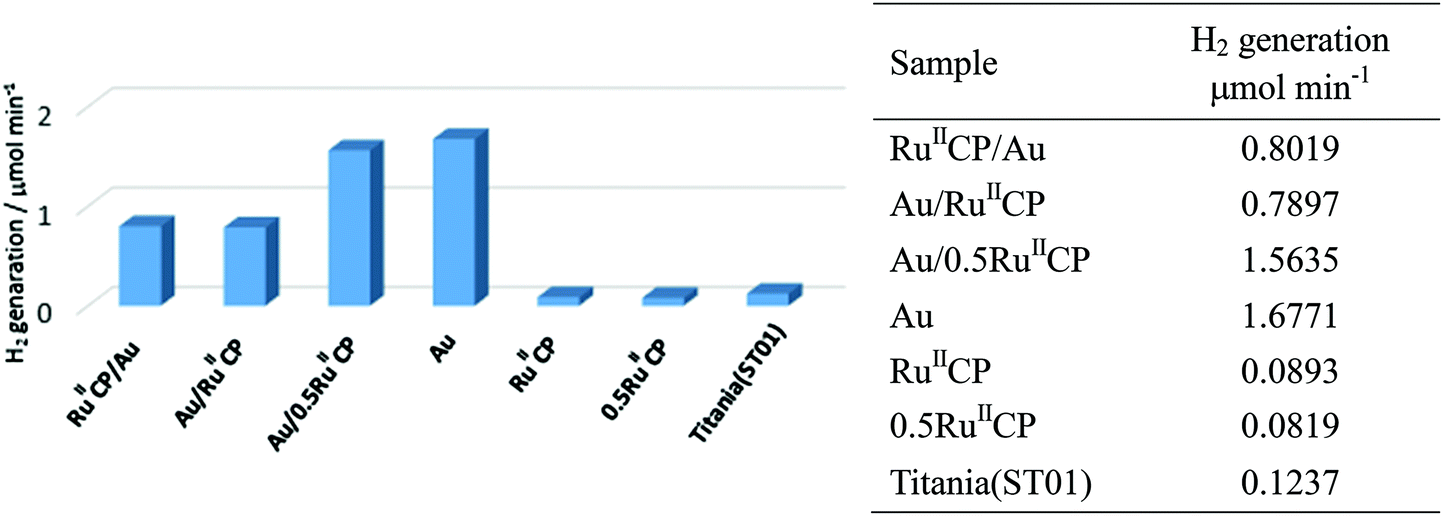 | ||
| Fig. 7 Photocatalytic activity for methanol dehydrogenation under UV/vis irradiation. | ||
Bare titania was practically inactive for alcohol dehydrogenation, attributed to fast recombination of photogenerated charge carriers, and the low driving force for the H+ reduction reaction, resulting in slow H+ reduction kinetics.48 Hydrogenation usually occurs on the co-catalyst site, which collects the photogenerated electrons.49–51 Herein, the presence of Au enhances the photocatalytic activity of titania by ca. 13 times, working as a co-catalyst. However, the samples with RuIICP and 0.5RuIICP inhibit the photocatalytic activity of bare titania ST01. These results are very surprising since our previous report for a similar ruthenium(II) complex showed an enhancement of photocatalytic activity for four different titania samples after adsorption of [Ru(bpy)2(4,4′-(PO3H2)2bpy)]2+,25 which suggested that hydrogen evolution occurred on the Ru site according to the enhancement outcome. The inhibition, observed in the present study, might be caused by different structures of the attached ruthenium(II) complex. It is highly possible that TiO2(e−)–Ru(III) is formed, and the presence of the –CH2– linker slows down the electron transfer, which means that the electrons are faster transferred to titania than are captured by protons. It was observed that during photoirradiation, the suspension with only RuIICP modified photocatalyst turned greenish, which could be owing to Ru(III) formation. Also, under UV/vis irradiation, it could undergo other reactions as we mentioned in the gold deposition section. Therefore, the RuIICP did not result in improvement of the photocatalytic activity in this case. The presence of both modifiers increased the photocatalytic activity when compared to bare titania, but decreased it when compared to the gold modified sample. Moreover, reduced amount of RuIICP resulted in an increase of photocatalytic activity, i.e., titania modified with Au/0.5RuIICP showed ca. two times higher photocatalytic activity than Au/RuIICP. These results are also quite different than those reported for [Ru(bpy)2(4,4′-(PO3H2)2bpy)]2+ for which even a synergetic effect was observed between Au and ruthenium(II) modifiers for a titania photocatalyst of fine NPs (TIO10).25 Thus, it is suggested that the methylene group between the ruthenium coordinated bipyridine and phosphonate groups strongly hindered the electron transfer between titania and ruthenium. To clarify the mechanism of hydrogen evolution the action spectra experiments are presently under study.
Notably, Au/RuIICP and RuIICP/Au showed nearly the same rate of dehydrogenation with about six-fold enhancement to bare titania, which means that the preparation sequences do not affect the photocatalytic activities under UV/vis irradiation conditions. Moreover, it could suggest that even if ruthenium(II) attached partly to the metallic surface (Au) instead of the titania surface, as was suggested in our previous paper25 and confirmed here by the XPS data, this attachment is not crucial for the overall activity.
Therefore, the conclusion that gold improves, but ruthenium(II) inhibits the photocatalytic activity could be drawn in the case of fine anatase titania and the [Ru(bpy)2(4,4′-(CH2PO3H2)2bpy)]2+ complex for methanol dehydrogenation under UV/vis irradiation.
Photocatalytic decomposition of acetic acid under UV/vis irradiation
Photocatalysts were also tested in another reaction system, i.e., under aerobic conditions for acetic acid degradation under UV/vis light irradiation, and the results are shown in Fig. 8.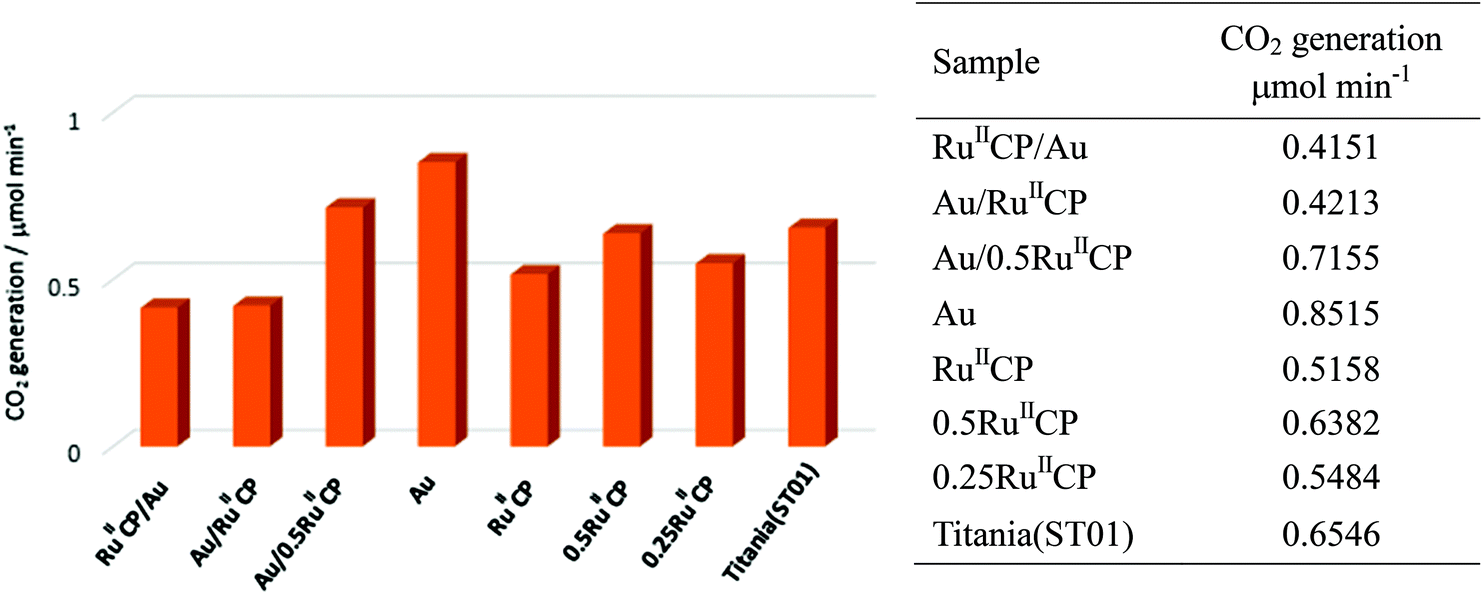 | ||
| Fig. 8 Photocatalytic activity for acetic acid degradation under UV/vis irradiation. | ||
It was confirmed that single modification of titania with gold resulted in an increase in photocatalytic activity, ca. 1.3 times. The introduction of ruthenium(II) inhibited the photocatalytic reactions and the photocatalytic rates for the samples with only ruthenium(II) modifier were slightly lower. Furthermore, for the co-modified titania, a lower amount of RuIICP resulted in a higher level of photocatalytic activity, i.e., titania modified with Au/0.5RuIICP was more active than Au/RuIICP. The change of deposition sequences (Au and RuIICP) did not affect the photocatalytic activity confirming that both modifiers mainly deposited individually and there were no direct interactions between them. These results were consistent with those for methanol dehydrogenation and are discussed above. Au primarily serves as an electron trap which leads to the higher activity of acetic acid degradation. While for RuIICP, its adsorption on titania may reduce the surface bond hydroxyl groups as also proved by the XPS data in Table 2, which under photoreaction would produce surface bond hydroxyl radicals (the reaction between photogenerated holes and surface bond hydroxyl), as was proposed by W. Choi for phosphonate modified titania.22 Hence, mobile hydroxyl radicals (the production of holes with adsorbed water), which are strong oxidant species and may break the ruthenium by oxidizing its –CH2– spacer would be preferred in the presence of RuIICP.28 However, for the [Ru(bpy)2(4,4′-(PO3H2)2bpy)]2+ complex contradictory results were obtained, i.e., (i) enhancement of activity after titania modification with ruthenium(II) for all four anatase titania samples, and only decrease of activity for rutile titania, (ii) enhancement of activity for all hybrid photocatalysts containing Au and ruthenium(II) in comparison with single modification with ruthenium(II) or Au (decrease only for the rutile sample). Considering this, it is proposed that the previously investigated ruthenium phosphonate complex was not decomposed under similar conditions, due to the lack of the methylene group, where enhancement of photocatalytic behaviour was observed. Surely, the kind of titania should also be considered, since it influences the electron injection efficiency as well.52 These data strongly suggest that a slight change in the structure of the ruthenium(II) complex caused a strong influence on the resultant properties and thus on the photocatalytic activities.
| Sample | Ti 2p (%) | O 1s (%) | Au 4f (%) | |||||
|---|---|---|---|---|---|---|---|---|
| Ti+4 | Ti+3 | –OH | –(OH)2 | TiO2 | Auδ+ | Au0 | Auδ− | |
| Titania(ST01) | 100 | 44.1 | 31.3 | 24.7 | — | — | — | |
| RuIICP | 98.5 | 1.5 | 25.9 | 36.3 | 37.8 | — | — | — |
| Au | 96.8 | 3.2 | 22.3 | 26.1 | 51.7 | 2.9 | 97.1 | — |
| RuIICP/Au | 100 | — | 26.8 | 17.4 | 55.8 | — | 100 | — |
| Au/RuIICP | 96.1 | 3.9 | 4.1 | 19.7 | 76.2 | 90.9 | 9.1 | |
Photocatalytic oxidation of 2-propanol under visible light irradiation
Under visible light irradiation, among all the photocatalysts, titania singly modified with ruthenium(II) displayed the highest photocatalytic activity for oxidation of 2-propanol, i.e., about 150-fold enhancement was achieved in comparison with bare titania, and the results are shown in Fig. 9. It should be pointed out that such high enhancement partly resulted from the experimental conditions in this study, i.e., irradiation with wavelengths longer than 450 nm (Y48 filter) to ensure the elimination of photocatalytic activity of bare titania. In addition, it was found that the reaction rates of samples possessing different contents of ruthenium (RuIICP, 0.5RuIICP, 0.25RuIICP, 0RuIICP and also hybrid samples: Au/RuIICP and Au/0.5RuIICP) decreased with a decreasing amount of attached RuIICP, which indicates that the number of the injected electrons to titania is in a direct correlation with the photosensitizers. It is proposed that upon visible light irradiation, the ruthenium(II) injects electrons into the CB of titania, from where they are taken up by oxygen to form the reactive oxygen species (ROS), and finally ROS oxidize 2-propanol. At the same time, other reactions like back electron transfer processes and side reactions of the decomposition products also proceed. Moreover, oxidation of the methylene spacer could also occur in the presence of ROS, therefore the degradation of the attached ruthenium(II) or its detachment could occur.28 To clarify the mechanism, transient spectroscopy is recommended as the next step to gain further details on the ET kinetics.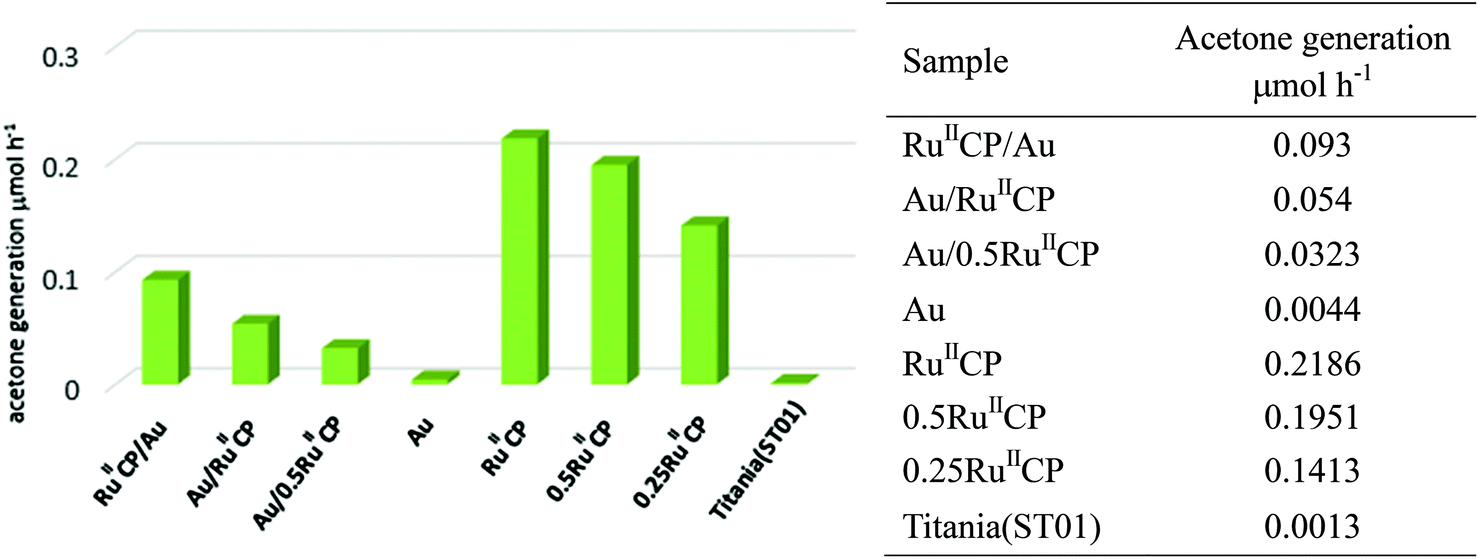 | ||
| Fig. 9 Photocatalytic activity for oxidation of 2-propanol under visible light irradiation. | ||
To compare two slightly different ruthenium(II) complexes used in present and previous reports, i.e., ([Ru(bpy)2(4,4′-(CH2PO3H2)2bpy)]2+ and [Ru(bpy)2(4,4′-(PO3H2)2bpy)]2+, respectively), the photocatalytic activity of the most active samples was tested. It was found that two times higher photocatalytic activity was achieved for titania TIO10 (anatase of fine NPs, similar to ST01) modified with [Ru(bpy)2(4,4′-(PO3H2)2bpy)]2+ than for the most active sample in the present study composed of [Ru(bpy)2(4,4′-(CH2PO3H2)2bpy)]2+. It is suggested that the additional methylene group hinders the electron transfer from ruthenium to the CB of titania or could be oxidized as discussed above.
It is important to mention that Au modified titania (ST01) selected for the present study possessed one of the least photocatalytic activities among other gold modified titanias, e.g., almost eight times lower than that of rutile titania with gold NPs of various sizes and shapes (Au/TiO2(TIO5)).44 This sample was selected to allow a high specific surface area for efficient adsorption of both modifiers (ruthenium(II) and Au) individually on the titania surface. Additionally, ruthenium(II)-modified titania of a high specific surface area exhibited much higher activity than that of a small specific surface area, i.e., 40-times increase in photocatalytic activity was noticed with 33-fold increase in the specific surface area.25 However, it must be pointed out that different behaviours and interactions between gold and RuIICP could be expected for more active gold modified titania of broader LSPR.
For the dual modified titania, since both gold and ruthenium(II) should activate titania under visible light, enhancement of photocatalytic activity was expected. However, different behavior was observed, upon ruthenium(II) introduction to gold modified titania, the photocatalytic activity enhanced, but after introduction of gold to ruthenium(II) modified titania, a decrease in photocatalytic activity was noticed by about 4 and 6 times for RuIICP and 0.5RuIICP samples, respectively. Such a drop effect was also observed previously for other anatase titania samples modified with [Ru(bpy)2(4,4′-(PO3H2)2bpy)]2+.25 Therein, we proposed that the gold NPs serve as an electron sink to trap the electrons (similarly as under UV-activation of titania), which are transferred to the CB of TiO2 from ruthenium(II) sensitizers, therefore hindering the electron scavenging by oxygen. Under current conditions, it is difficult to clarify the function of oxygen, which could lead to different mechanisms.53 The experiments under anaerobic conditions are currently under investigation.
These results strengthened the importance of ruthenium(II) for oxidation of 2-propanol under visible light irradiation, and showed that the presence of gold NPs inhibited the photocatalytic activities of ruthenium(II) modified titania. Interestingly, the samples with co-adsorbed ruthenium(II) and gold NPs, which only differed in the deposition sequence, exhibited very different activities, i.e., RuIICP/Au was more active than Au/RuIICP, in contrast to the results obtained under UV/vis irradiation of methanol (anaerobic) and acetic acid (aerobic) where both the samples exhibited the same activity (Fig. 7 and 8). It is supposed that during the preparation of the latter, the repulsive ruthenium complex induced gold aggregation and thus larger Au NPs were formed (which was confirmed by the DRS data and STEM also). It must be pointed out that the size of plasmonic NPs is important in the visible photocatalytic processes, and generally the larger the size of gold NPs is, the higher is photocatalytic activity under visible light irradiation.51,54 However, in the present system, it seems that the function of gold is different, i.e., an electron trap instead of titania activation, and the larger NPs should exhibit better electron storage properties than the smaller NPs.
It is thought that the present findings could also be helpful for mechanism clarification for plasmonic photocatalysts composed of a wide band-gap semiconductor (usually titania) and plasmonic NPs. Two main mechanisms are suggested under visible light irradiation, i.e., energy55 and electron10 transfers from excited plasmonic NPs to titania. The decrease in activity by co-modification with the ruthenium(II) complex and gold indicates that the electron transfer is more probable than the energy transfer mechanism in the present case. To gain better insight into the mechanism further research studies like dynamic study of charge excitation and recombination are necessary.
Experimental
Materials
Titania ST01 from Ishihara (anatase, specific surface area of ca. 300 m2 g−1) was used as received. RuIICP: [Ru(bpy)2(4,4′-(CH2PO3H2)2bpy)]2+ was synthesized according to an analogue complex in the literature.56 Water for solution preparation and sample washing was from the Milli-Q purification system. Hydrogen tetrachloroaurate(III) tetrahydrate (HAuCl4·4H2O) (Nacalai Tesque and Wako Pure Chemical Industry) was used as received for gold deposition. Methanol, acetic acid, 2-propanol, and acetone (Wako Pure Chemical Industry) were used without further purification.Preparation of photocatalysts
Ruthenium(II) was adsorbed on bare and pre-modified titania with Au (2 wt%) in an aqueous suspension by 3 min sonication, followed by 24-hour stirring in the dark. The mixture was centrifuged, washed three times with water, dried overnight at 100 °C, and ground in an agate mortar. Different amounts of RuIICP corresponding to 0.34 mol% (RuIICP), 0.17 mol% (0.5RuIICP) and 0.085 mol% (0.25RuIICP) to titania, were applied. For the sample of the highest amount of ruthenium(II), the time dependent adsorption (with UV/vis spectroscopy) was performed to ensure that 24 hours is sufficient for the completion of ruthenium(II) attachment.2 wt% of gold, which corresponds to 0.81 mol% to titania, was photodeposited on the surface of bare and pre-modified titania with RuIICP by the photodeposition method from 50 vol% aqueous methanol. The details of photodeposition were shown in our previous reports.16,26
Characterization of photocatalysts
Samples were characterized by diffuse reflectance spectroscopy (DRS), X-ray photoelectron spectroscopy (XPS) and scanning transmission electron microscopy (STEM). DRS measurements were performed on a JASCO V-670 equipped with a PIN-757 integrating sphere. Barium sulfate and bare titania (ST01) were used as references during measurements.The surface composition of samples and oxidation states of elements were measured by XPS on a JEOL JPC-9010MC (MgKα X-ray). 50 scans were carried out for each sample and the average data were taken for determination of titanium, oxygen and carbon. While 500 scans were performed for characterization of gold.
The morphology of samples was observed by STEM on a HITACHI HD2000 at 200-kV accelerating voltage and 30 μA emission current. The droplet of titania suspension (in ethanol) was deposited on carbon-covered copper microgrid, which was dried overnight under vacuum at room temperature. STEM images were acquired as secondary electron (SE), Z-contrast (ZC) and bright-field (TE) modes. For calculation of distribution of gold NP sizes, 449, 460 and 361 NPs of gold were measured for titania samples modified with Au, RuIICP/Au and Au/RuIICP, respectively.
Tests of photocatalytic activity
The photocatalytic activities were examined under UV/vis irradiation (400 W, high-pressure mercury lamp) for methanol dehydrogenation (50 vol% methanol under argon) and acetic acid oxidation (5 vol% acetic acid in air). Visible light photocatalytic activity was examined for 2-propanol oxidation (5 vol% 2-propanol in air) at wavelengths longer than 450 nm (300 W xenon lamp, IR filter, quartz mirror, Y48 cut-off filter). Characterization of set-ups used for irradiation is described in detail in a previous publication.44Conclusions
Ruthenium(II) dye and gold NPs were immobilized on the surface of anatase titania with fine NPs. The photocatalysts were stable since the complex did not desorb even after long-lasting UV/vis irradiation. It was found that under UV/vis irradiation the photocatalysts modified with gold exhibited the highest photocatalytic activities for both tested systems, i.e., anaerobic methanol dehydrogenation and aerobic acetic acid degradation, and introduction of RuIICP decreased the photocatalytic activities. On the contrary, under visible irradiation, gold hindered the photocatalytic activity of ruthenium(II).The DRS, XPS and STEM characterization suggested that the pre-adsorbed ruthenium(II) complex on the titania surface affected the gold deposition, causing aggregation and a broader distribution of gold NP sizes. Also, the introduction of ruthenium(II) induced the change of gold NP sizes, which needs further exploration. This might be utilized to control the metal particle sizes, which can be useful for catalytic (dark) and photocatalytic reactions (it is known that gold size is a key-factor of many reactions).
It was found that the sequence of deposition (ruthenium(II) complex and gold NPs) did not significantly influence the photocatalytic activity under UV/vis irradiation. However, under visible light irradiation formation of slightly larger gold NPs resulted in stronger inhibition of ruthenium(II) activity. It is proposed that larger gold NPs store electrons from the CB of titania (sensitized by ruthenium(II)) hindering their transfer to oxygen.
In addition, it must be pointed out that a small change in the ruthenium(II) complex structure results in a complete change of photocatalytic performance. It is proposed that direct bonding of the bipyridine motif with phosphonate groups allows a better electronic contact and electron transfer between titania and the ruthenium(II) complex. Besides, the presence of the methylene group would also risk the self-oxidization reaction causing dye degradation. In order to achieve the desirable synergetic effects when designing the hybrid photocatalysts, durable ruthenium(II) dyes under photocatalytic conditions with stable attachment properties to allow efficient electron transfer with titania and/or co-deposited metal particles should be considered. The new elongated phosphonate bipyridines, which utilize phenylene and triazole moieties instead of a flexible linker, seem interesting, since they provide π-conjugated systems between ruthenium(II) and titania,29 and also may avoid the oxidation reaction on alkylene linkers.
Detailed investigation on the charge transfer process under specified irradiation ranges (e.g., action spectrum analysis) is still required to clarify the mechanism. It is also recommended to examine other titania supports since titania properties should also influence the mechanism pathways, e.g., by electron storage, recombination of charge carriers and/or preferential self-co-adsorption of ruthenium(II) dyes and gold.
Acknowledgements
This research was funded by a grant from the CONCERT-Japan Joint Call.References
- S. M. Gupta and M. Tripathi, Chin. Sci. Bull., 2011, 56, 1639–1657 CrossRef CAS.
- M. Pelaez, N. T. Nolan, S. C. Pillai, M. K. Seery, P. Falaras, A. G. Kontos, P. S. M. Dunlop, J. W. J. Hamilton, J. A. Byrne, K. O′Shea, M. H. Entezari and D. D. Dionysiou, Appl. Catal., B, 2012, 125, 331–349 CrossRef CAS.
- M. Ni, M. K. H. Leung, D. Y. C. Leung and K. Sumathy, Renewable Sustainable Energy Rev., 2007, 11, 401–425 CrossRef CAS.
- P. Pichat, J. Adv. Oxid. Technol., 2010, 13, 238–246 CAS.
- H. Park, Y. Park, W. Kim and W. Choi, J. Photochem. Photobiol., C, 2013, 15, 1–20 CrossRef CAS.
- P. Chen, L. Wang, P. Wang, A. Kostka, M. Wark, M. Muhler and R. Beranek, Catalysts, 2015, 5, 270–285 CrossRef CAS.
- W. Macyk, G. Burgeth and H. Kisch, Photochem. Photobiol. Sci., 2003, 2, 322–328 CAS.
- D. Mitoraj and H. Kisch, Angew. Chem., Int. Ed., 2008, 47, 9975–9978 CrossRef CAS PubMed.
- C. Wang and D. Astruc, Chem. Soc. Rev., 2014, 43, 7188–7216 RSC.
- Y. Tian and T. Tatsuma, J. Am. Chem. Soc., 2005, 127, 7632–7637 CrossRef CAS PubMed.
- K. Hirano, E. Suzuki, A. Ishikawa, T. Moroi, H. Shiroishi and M. Kaneko, J. Photochem. Photobiol., A, 2000, 136, 157–161 CrossRef CAS.
- M. Grätzel, J. Photochem. Photobiol., C, 2003, 4, 145–153 CrossRef.
- E. Taboada, I. Angurell and J. Llorca, J. Catal., 2014, 309, 460–467 CrossRef CAS.
- M. Hu, J. Chen, Z.-Y. Li, L. Au, G. V. Hartland, X. Li, M. Marquez and Y. Xia, Chem. Soc. Rev., 2006, 35, 1084–1094 RSC.
- M. Serra, J. Albero and H. García, ChemPhysChem, 2015, 16, 1842–1845 CrossRef CAS PubMed.
- E. Kowalska, M. Janczarek, L. Rosa, S. Juodkazis and B. Ohtani, Catal. Today, 2014, 230, 131–137 CrossRef.
- P. A. DeSario, J. J. Pietron, D. E. DeVantier, T. H. Brintlinger, R. M. Stroud and D. R. Rolison, Nanoscale, 2013, 5, 8073–8083 RSC.
- T.-H. Meen, J.-K. Tsai, S.-M. Chao, Y.-C. Lin, T.-C. Wu, T.-Y. Chang, L.-W. Ji, W. Water, W.-R. Chen, I.-T. Tang and C.-J. Huang, Nanoscale Res. Lett., 2013, 8, 450 CrossRef PubMed.
- T. Bora, H. H. Kyaw, S. Sarkar, S. K. Pal and J. Dutta, Beilstein J. Nanotechnol., 2011, 2, 681–690 CrossRef CAS PubMed.
- H. Choi, W. T. Chen and P. V. Kamat, ACS Nano, 2012, 6, 4418–4427 CrossRef CAS PubMed.
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef.
- J. Kim and W. Choi, Appl. Catal., B, 2011, 106, 39–45 CAS.
- J. Kim, J. Lee and W. Choi, Chem. Commun., 2008, 756–758 RSC.
- J. Kim and W. Choi, Energy Environ. Sci., 2010, 3, 1042–1045 CAS.
- E. Kowalska, K. Yoshiiri, Z. Wei, S. Zheng, E. Kastl, H. Remita, B. Ohtani and S. Rau, Appl. Catal., B, 2015, 178, 133–143 CrossRef CAS.
- E. Kowalska, S. Rau and B. Ohtani, J. Nanotechnol., 2012, 2012, 1–11 CrossRef.
- D. L. Ashford, W. Song, J. J. Concepcion, C. R. K. Glasson, M. K. Brennaman, M. R. Norris, Z. Fang, J. L. Templeton and T. J. Meyer, J. Am. Chem. Soc., 2012, 134, 19189–19198 CrossRef PubMed.
- K. Hanson, M. K. Brennaman, A. Ito, H. Luo, W. Song, K. A. Parker, R. Ghosh, M. R. Norris, C. R. K. Glasson, J. J. Concepcion, R. Lopez and T. J. Meyer, J. Phys. Chem. C, 2012, 116, 14837–14847 CAS.
- M. Braumüller, D. Sorsche, M. Wunderlin and S. Rau, Eur. J. Org. Chem., 2015, 27, 5987–5994 CrossRef.
- K. Hanson, M. K. Brennaman, A. Ito, H. Luo, W. Song, K. A. Parker, R. Ghosh, M. R. Norris, C. R. K. Glasson, J. J. Concepcion, R. Lopez and T. J. Meyer, J. Phys. Chem. C, 2012, 116, 14837–14847 CAS.
- K. Nonomura, Y. Xu, T. Marinado, D. P. Hagberg, R. Zhang, G. Boschloo, L. Sun and A. Hagfeldt, Int. J. Photoenergy, 2009, 2009, 1–9 CrossRef.
- N. A. Anderson and T. Lian, Annu. Rev. Phys. Chem., 2005, 56, 491–519 CrossRef CAS PubMed.
- E. Galoppini, Coord. Chem. Rev., 2004, 248, 1283–1297 CrossRef CAS.
- A. S. Polo, M. K. Itokazu and N. Y. Murakami Iha, Coord. Chem. Rev., 2004, 248, 1343–1361 CrossRef CAS.
- H. Zabri, I. Gillaizeau, C. A. Bignozzi, S. Caramori, M.-F. Charlot, J. Cano-Boquera and F. Odobel, Inorg. Chem., 2003, 42, 6655–6666 CrossRef CAS PubMed.
- I. Gillaizeau-Gauthier, F. Odobel, M. Alebbi, R. Argazzi, E. Costa, C. A. Bignozzi, P. Qu and G. J. Meyer, Inorg. Chem., 2001, 40, 6073–6079 CrossRef CAS PubMed.
- E. Bae, W. Choi, J. Park, H. S. Shin, S. Bin Kim and J. S. Lee, J. Phys. Chem. B, 2004, 108, 14093–14101 CrossRef CAS.
- E. Bae and W. Choi, J. Phys. Chem. B, 2006, 110, 14792–14799 CrossRef CAS PubMed.
- H. Park, E. Bae, J.-J. Lee, J. Park and W. Choi, J. Phys. Chem. B, 2006, 110, 8740–8749 CrossRef CAS PubMed.
- K. Hanson, M. K. Brennaman, H. Luo, C. R. K. Glasson, J. J. Concepcion, W. Song and T. J. Meyer, Appl. Mater. Interfaces, 2012, 4, 1462–1469 CrossRef CAS PubMed.
- Y. Xia and N. J. Halas, MRS Bull., 2005, 30, 338–348 CrossRef CAS.
- H. Pan, S. Low, N. Weerasuriya and Y.-S. Shon, ACS Appl. Mater. Interfaces, 2015, 7, 3406–3413 CAS.
- M. Iqbal and G. Tae, J. Nanosci. Nanotechnol., 2006, 6, 3355–3359 CrossRef CAS PubMed.
- E. Kowalska, O. O. P. Mahaney, R. Abe and B. Ohtani, Phys. Chem. Chem. Phys., 2010, 12, 2344–2355 RSC.
- D. A. Panayotov, P. A. DeSario, J. J. Pietron, T. H. Brintlinger, L. C. Szymczak, D. R. Rolison and J. R. Morris, J. Phys. Chem. C, 2013, 117, 15035–15049 CAS.
- W.-T. Chen, A. Chan, Z. H. N. Al-Azri, A. G. Dosado, M. A. Nadeem, D. Sun-Waterhouse, H. Idriss and G. I. N. Waterhouse, J. Catal., 2015, 329, 499–513 CrossRef CAS.
- A. Markowska-Szczupak, K. Wang, P. Rokicka, M. Endo, Z. Wei, B. Ohtani, A. W. Morawski and E. Kowalska, J. Photochem. Photobiol., B, 2015, 151, 54–62 CrossRef CAS PubMed.
- A. Reynal, F. Lakadamyali, M. A. Gross, E. Reisner and J. R. Durrant, Energy Environ. Sci., 2013, 6, 3291–3300 CAS.
- A. J. Bard, J. Photochem., 1979, 10, 59–75 CrossRef CAS.
- A. Dawson and P. V. Kamat, J. Phys. Chem. B, 2001, 105, 960–966 CrossRef CAS.
- E. Kowalska, R. Abe and B. Ohtani, Chem. Commun., 2009, 241–243 RSC.
- R. Katoh, A. Huijser, K. Hara, T. J. Savenije and L. D. A. Siebbeles, J. Phys. Chem. C, 2007, 111, 10741–10746 CAS.
- D. Pei and J. Luan, Int. J. Photoenergy, 2012, 2012, 262831 CrossRef.
- L. Du, A. Furube, K. Hara, R. Katoh and M. Tachiya, Thin Solid Films, 2009, 518, 861–864 CrossRef CAS.
- D. B. Ingram, P. Christopher, J. L. Bauer and S. Linic, ACS Catal., 2011, 1, 1441–1447 CrossRef CAS.
- M. Braumüller, M. Schulz, D. Sorsche, M. Pfeffer, M. Schaub, J. Popp, B.-W. Park, A. Hagfeldt, B. Dietzek and S. Rau, Dalton Trans., 2015, 44, 5577–5586 RSC.
Footnote | ||||||||||||||||||||||||||||||||||||||||||
† Sample abbreviations44
| ||||||||||||||||||||||||||||||||||||||||||
| This journal is © The Royal Society of Chemistry and Owner Societies 2016 |