
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The impact of cationic substituents in phenalen-1-one photosensitizers on antimicrobial photodynamic efficacy†
Isabelle
Tabenski
a,
Fabian
Cieplik
a,
Laura
Tabenski
a,
Johannes
Regensburger
b,
Karl-Anton
Hiller
a,
Wolfgang
Buchalla
a,
Tim
Maisch‡
*b and
Andreas
Späth‡
*c
aDepartment of Conservative Dentistry and Periodontology, University Medical Center Regensburg, Regensburg, Germany
bDepartment of Dermatology, University Medical Center Regensburg, Regensburg, Germany. E-mail: tim.maisch@ukr.de
cDepartment of Organic Chemistry, University of Regensburg, Regensburg, Germany. E-mail: andreas.spaeth@chemie.uni-regensburg.de
First published on 2nd December 2015
Abstract
Light-mediated killing of pathogens by cationic photosensitizers (PS) is a promising antimicrobial approach avoiding resistance as being present upon the use of antibiotics. In this study we focused on the impact of the substituents in phenalen-1-one PS. Photodynamic efficacy depending on positively charged moieties including a primary aliphatic, quaternary aliphatic, aromatic ammonium and a guanidinium cation was investigated against Gram-positive and Gram-negative pathogens. Considering the altered steric demand and lipophilicity of these functional groups we deduced a structure–activity relationship. SAGUA was the most potent PS in this series reaching a maximum efficacy of ≥6log10 steps of bacteria killing at a concentration of 10 μM upon irradiation with blue light (20 mW cm−2) for 60 s (1.2 J cm−2) without exhibiting inherent dark toxicity. Its guanidinium moiety may be able to form strong bidentate and directional hydrogen bonds to carboxylate groups of bacterial surfaces in addition to ionic charge attraction. This may supplement fast and effective antimicrobial activity.
Introduction
The discovery of antibiotics can be seen as one of the most important breakthroughs in medical history. Antibiotics have revolutionized the way patients with bacterial infections are treated and have contributed to the reduction of the mortality and morbidity from bacterial diseases. However, unconsidered and abundant use in human as well as in veterinary medicine and animal fattening finally contributed to the emergence of antibiotic-resistant strains, e.g. methicillin-resistant Staphylococcus aureus (MRSA) or vancomycin-resistant Enterococci (VRE) strains.1 Furthermore, resistance against antiseptic and antimicrobial agents, which are clinically used, e.g. chlorhexidine2 and triclosan,3 is arising. The worldwide spread of resistance among pathogens represents an immense problem for public health.1Consequently, the demand for developing alternative approaches for successful inactivation of pathogens is becoming more and more crucial. These alternative approaches should operate – different from antibiotics – not towards one specific target according to the so-called key-hole-principle, but as multi-target processes, in order to avoid development of resistance in microorganisms.4,5
A promising approach meeting these requirements may be the antimicrobial photodynamic therapy (aPDT), which kills bacteria via an oxidative burst by causing damage to cellular structures and biomolecules.6,7 The concept of aPDT is based on the principle that positively charged photosensitizers (PS) attach to the negatively charged cell walls of pathogens with subsequent killing upon activation of the PS by light of an appropriate wavelength and reactive oxygen species photogeneration.8 The absorption of light by the PS leads to a transition of the excited PS molecule into its triplet excited state, from which there are two general reaction mechanisms for letting the PS regain its ground state: in type I mechanism, charge is transferred to a substrate or molecular oxygen with the emergence of reactive oxygen species (ROS) like hydrogen peroxide (H2O2) or oxygen radicals like superoxide ions (O2˙−) and free hydroxyl radicals (HO˙), which are formed via Fenton-like reactions involving H2O2. In contrast, in type II mechanism, energy – but no charge – is transferred directly to molecular oxygen with the formation of the highly reactive singlet oxygen (1O2).9,10 Due to its topical application form, aPDT may be a superior antimicrobial approach for the treatment of localized infections on mucosal or dermal surfaces and consequently may be used in dentistry and dermatology.
Recently, our group introduced SAPYR (1) as a new class of PS based on a phenalen-1-one structure (Fig. 1), which exhibits a 1O2 quantum yield ΦΔ close to unity (ΦΔ = 0.99) and showed pronounced antimicrobial efficacy against planktonic bacteria11 as well as against monospecies and polyspecies biofilms.12,13
 | ||
| Fig. 1 Chemical structures. Reference-PS SAPYR (1), novel phenalen-1-one derivatives (2), (3), (4) and SAGUA (5) and basic structure phenalen-1-one (6). | ||
In the present study, we evaluate a set of derivatives (Fig. 1) based on a phenalen-1-one structure (6), whereby the effect of the nature of the cationic substituents in the phenalen-1-one derivatives on the antimicrobial photodynamic efficacy is studied.
Different determinants are crucial for effective interactions with the negative charges on a bacterial cell surface such as bulk, geometry and pKa-value. In addition, the binding may be supplemented by H-bonding or π–π-interaction. We wanted to have a closer look on such effects in phenalen-1-one derivatives.
Therefore, we compared SAPYR (1), which is equipped with a pyridinium moiety and serves as a reference PS, to compounds carrying a primary ammonium (2) or a secondary ammonium group (3), respectively. In addition, a cyclohexyl-substituted analog (4) for covering more lipophilic quaternary cations and a permanently positively charged guanidinium derivative (5; SAGUA) are examined.
As oral and dermal infections represent a superior field for clinical applications of aPDT, due to their localized nature, the antimicrobial photodynamic efficacy of this new set of compounds is tested against planktonic cultures of oral and dermal key pathogens: Gram-positive Streptococcus mutans (SM), Enterococcus faecalis (EF), Actinomyces naeslundii (AN), Staphylococcus aureus (SA) and Gram-negative Escherichia coli (EC).
The aim of this study is (i) to evaluate this new set of compounds based on a phenalen-1-one structure regarding their antimicrobial photodynamic efficacy, (ii) to deduce a structure–activity relationship concerning the effect of the nature of their cationic substituents on the antimicrobial efficacy and (iii) to study the efficacy under low light dose conditions using an optimized illumination device.
Materials and methods
General materials and methods
Commercial reagents and starting materials were purchased from Acros Organics (U.S.), TCI Germany (Germany), Fluka (Germany), Merck (Germany), Frontier Scientific (U.S.) or Sigma-Aldrich (Switzerland) and used without further purification. The dry solvents acetone, dichloromethane, dimethylsulfoxide and dimethylformamide were purchased from Roth, Germany (RotiDry Sept) or Sigma-Aldrich, Switzerland (puriss., absolute), stored over molecular sieves under nitrogen and were used as received.Thin layer chromatography (TLC) analyses were performed on silica gel 60 F-254 with 0.2 mm layer thickness and detection via UV light at 254 nm/366 nm or through staining with ninhydrin in ethanol. Flash column chromatography was performed on Merck silica gel (Si 60 40–63 μm) either manually or on a Biotage® solera™ flash purification system. Column chromatography was performed on silica gel (70–230 mesh) from Merck.
Melting points were measured on a SRS melting point apparatus (MPA100 Opti Melt) and are not corrected.
Nuclear Magnetic Resonance (NMR) spectra were recorded on Bruker Avance 300 (1H 300.13 MHz, 13C 75.47 MHz, T = 300 K), Bruker Avance 400 (1H 400.13 MHz, 13C 100.61 MHz, T = 300 K), Bruker Avance 600 (1H 600.13 MHz, 13C 150.92 MHz, T = 300 K) and Bruker Avance III 600 Kryo (1H 600.25 MHz, 13C 150.95 MHz, T = 300 K) instruments. The chemical shifts are reported in δ [ppm] relative to external standards (solvent residual peak). The spectra were analyzed by first order, the coupling constants J are given in hertz [Hz]. Characterization of the signals: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, bs = broad singlet, psq = pseudo quintet, dd = double doublet, dt = doublet of triplets, ddd = double double doublet. Integration is determined as the relative number of atoms. Assignment of signals in 13C-spectra was determined by 2D-spectroscopy (COSY, HSQC and HMBC) or DEPT technique (pulse angle: 135°) and given as (+) for CH3 or CH, (−) for CH2 and (Cq) for quaternary Cq. Error of reported values: chemical shift 0.01 ppm (1H NMR) and 0.1 ppm (13C NMR), coupling constant J 0.1 Hz. The solvents used for the measurements are reported for each spectrum.
Infrared spectroscopy was performed for each compound. IR spectra were recorded with a Bio-Rad FT-IR-FTS 155 spectrometer. Fluorescence spectra were recorded on a ‘Cary Eclipse’ fluorescence spectrophotometer and absorption spectra were recorded on a “Cary BIO 50” UV/VIS/NIR spectrometer from Varian. All measurements were performed in 1 cm quartz cuvettes (Hellma) and UV-grade solvents (Baker or Merck) at 25 °C.
Mass spectroscopy was performed to detect the mass spectra (MS) of all compounds. MS were recorded on a Varian CH-5 (EI), Finnigan MAT95 (EI-, CI- and FAB-MS), Agilent Q-TOF 6540 UHD (ESI-MS, APCI-MS), Finnigan MAT SSQ 710 A (EI-MS, CI-MS) or Thermo Quest Finnigan TSQ 7000 (ES-MS, APCI-MS) spectrometer. Xenon serves as the ionization gas for FAB.
Synthesis and purification of the compounds
Substances 1 and 7 were prepared from 6 based on known literature procedures (Fig. 2 & ref. 11). The purity of all the synthesized compounds was determined by NMR spectroscopic methods (Bruker Avance 300, DMSO-d6) and HPLC-MS confirming a purity of >97%. | ||
| Fig. 2 Scheme of the synthesis of phenalen-1-one PS. (A) Synthesis of phenalene-1-one derivatives 1–4. Conditions: (a) HCl(aq), HOAc, H3PO4, HCHO, 120 °C, overnight, 36%; (b) pyridine, DMF (N,N-dimethylformamide), room temperature, overnight, 50 °C, 5 h, 83%; (c) MeOH, amine, room temperature, overnight, 50 °C, 2–10 h, 60–75%, (d) DMF, N-methyl-piperidine, room temperature, overnight, 50 °C, 2–10 h, 78%. (B) Synthesis of phenalen-1-one derivative SAGUA (5). Conditions: (a) N,N′-di-(tert-butoxycarbonyl)-N′′-triflylguanidine, DCM (dichloromethane) (adapted from ref. 14), triethylamine, 0 °C, then room temperature for 4 h, 88%; (b) HCl in Et2O, DCM, room temperature, 6 h, 50 °C, 5 h, 96%. | ||
In a dry flask with moisture protection dried methanol (20 mL) was stirred in an ice bath. A steady stream of ammonia gas was added into the flask and bubbled through the solution for 30 min. 2-Chloromethyl-1H-phenalen-1-one (7) (228 mg, 1 mmol) in methanol/DMF (dry, 6 mL, 1

![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was added dropwise to the ice-cold solution over a period of 10 min. The introduction of ammonia was continued as a slow stream bubbling through the vigorously stirring solution for 3 h, whilst reaching room temperature (a yellow precipitate begins to form, TLC control shows complete conversion of the starting material). Stirring at room temperature was continued for 10 h, and the solvent and excess ammonia were removed in a stream of nitrogen (fume hood! The N2-purged ammonia vapor was absorbed in a separate flask by diluted sulfuric acid). The residue was dissolved in a minimum amount of DMF and precipitated by the addition of diethylether. The product was settled with the aid of a centrifuge (60 min, 4400 rpm, 0 °C) and the supernatant was discarded. The precipitate was re-suspended in diethylether, settled again and the supernatant was decanted off. The residue was dissolved in a minimum amount of dichloromethane/ethanol 8:1 and precipitated with diethylether again. The product was settled with the aid of a centrifuge (60 min, 4400 rpm, 0 °C) and the supernatant was discarded. The precipitate was re-suspended in diethylether, settled again and the supernatant was decanted off. This washing step was repeated twice. Yellow powder, 46% of theoretical yield.
:1) was added dropwise to the ice-cold solution over a period of 10 min. The introduction of ammonia was continued as a slow stream bubbling through the vigorously stirring solution for 3 h, whilst reaching room temperature (a yellow precipitate begins to form, TLC control shows complete conversion of the starting material). Stirring at room temperature was continued for 10 h, and the solvent and excess ammonia were removed in a stream of nitrogen (fume hood! The N2-purged ammonia vapor was absorbed in a separate flask by diluted sulfuric acid). The residue was dissolved in a minimum amount of DMF and precipitated by the addition of diethylether. The product was settled with the aid of a centrifuge (60 min, 4400 rpm, 0 °C) and the supernatant was discarded. The precipitate was re-suspended in diethylether, settled again and the supernatant was decanted off. The residue was dissolved in a minimum amount of dichloromethane/ethanol 8:1 and precipitated with diethylether again. The product was settled with the aid of a centrifuge (60 min, 4400 rpm, 0 °C) and the supernatant was discarded. The precipitate was re-suspended in diethylether, settled again and the supernatant was decanted off. This washing step was repeated twice. Yellow powder, 46% of theoretical yield.
1 H-NMR (300 MHz, DMSO-d6): δ[ppm] = 8.58 (d, J = 6.7 Hz, 1H), 8.56 (d, J = 6.6 Hz, 1H), 8.52 (m, 3H), 8.32 (d, J = 7.6 Hz, 1H), 8.21 (s, 1H), 8.09 (d, J = 8.4 Hz, 1H), 7.95 (t, J = 7.6 Hz, 1H), 7.79 (t, J = 7.7 Hz, 1H), 3.97 (s, 2H). 13C-NMR (75 MHz, DMSO-d6): δ[ppm] = 183.08 (q), 141.69 (+), 135.93 (+), 133.00 (+), 131.72 (+), 130.33 (q), 127.87 (q), 127.59 (q), 127.38 (+), 126.25 (q), 126.05 (q), 37.51 (−); MS (ESI-MS, CH2Cl2/MeOH + 10 mmol NH4OAc): m/z (%) = 210.1 (MH+, 100%), 193.1 (M+ − NH3, 31%); IR (neat): ν[cm−1] = 3123, 3037, 2809, 2018, 1754, 1638, 1597, 1566, 1399, 1260, 910, 783, 682; MW (molecular weight) = 210.26 + 35.45 g mol−1; MF (molecular formula) = C14H12NOCl.
2-Chloromethyl-1H-phenalen-1-one (7) (113 mg, 0.5 mmol) in methanol (10 mL) was added dropwise to an ice-cold solution of methylamine in methanol (40 mL, 10%) over a period of 1 h. After 30 h of vigorous stirring at room temperature, the solvent and excess methylamine were removed in a stream of nitrogen (fume hood; the N2-purged methylamine vapor was absorbed in a separate flask by diluted sulfuric acid).
The residue was dissolved in a minimum amount of DCM/ethanol 4:1 and precipitated by the addition of diethylether. The product was settled with the aid of a centrifuge (60 min, 4400 rpm, 0 °C) and the supernatant was discarded. This step was repeated one more time.
The precipitate was re-suspended in diethylether, settled again and the supernatant was decanted off. This washing step was repeated twice. Yellow-brownish powder, 78% of theoretical yield (101 mg, 0.39 mmol).
1 H-NMR (300 MHz, DMSO-d6): δ[ppm] = 9.01 (m, app. bs, 1H), 8.57 (d, J = 7.4 Hz, 1H), 8.52 (d, J = 7.6 Hz, 1H), 8.33 (d, J = 8.2 Hz, 1H), 8.29 (s, 1H), 8.09 (d, J = 8.2 Hz, 1H), 7.95 (t, J = 7.5 Hz, 1H), 7.79 (t, J = 7.6 Hz, 1H), 4.06 (s, 2H), 2.60 (s, 3H). 13C-NMR (75 MHz, DMSO-d6): δ[ppm] = 183.17 (q), 143.30 (+), 135.99 (+), 133.30 (+), 133.02 (+), 131.65 (q), 130.43 (+), 130.08 (q), 127.86 (q), 127.61 (+), 127.38 (+), 126.36 (q), 126.00 (q), 46.39 (−), 32.47 (+); MS (ESI-MS, CH2Cl2/MeOH + 10 mmol NH4OAc): m/z (%) = 224.1 (MH+, 100%); IR (neat): ν[cm−1] = 3384, 2989, 2953, 2742, 2669, 2569, 2481, 2435, 1621, 1567, 1509, 1464, 1404, 1375, 1349, 1257, 1230, 1197, 1116, 1020, 952, 906, 849, 778, 597, 571; MW = 224.28 + 35.45 g mol−1; MF = C15H14NOCl.
In an oven-dried, 25 mL round-bottomed flask N,N′-di-Boc-N′′-triflylguanidine (0.41 g, 1.05 mmol) was dissolved in DCM (10 mL). Triethylamine (0.3 g, 0.39 mL, 3 mmol) was added slowly at 2–5 °C under moisture protection. Compound 3 (130 mg, 0.5 mmol) was added in one portion. After 5 h stirring at room temperature, the mixture was diluted with dichloromethane (30 mL) and transferred to a separatory funnel. The organic layer was washed with aqueous potassium hydrogen sulfate (10 mL, 5%), saturated sodium bicarbonate solution (10 mL) and brine (20 mL), and dried over MgSO4, filtered and concentrated under reduced pressure. The raw material was purified by column chromatography with acetone/petroleum ether 1
 :2 to afford the product as bright yellow powder in nearly quantitative yield (0.21 g). For further purification, the material was dissolved in acetone (1 mL) and precipitated with petroleum ether (14 mL). The precipitate was filtered off and the filter cake was washed with petroleum ether.
:2 to afford the product as bright yellow powder in nearly quantitative yield (0.21 g). For further purification, the material was dissolved in acetone (1 mL) and precipitated with petroleum ether (14 mL). The precipitate was filtered off and the filter cake was washed with petroleum ether.
1 H-NMR (300 MHz, DMSO-d6): δ[ppm] = 9.70 (bs, 1H), 8.57 (d, J = 7.3 Hz, 1H), 8.50 (d, J = 7.8 Hz, 1H), 8.29 (d, J = 8.1 Hz, 1H), 8.02–7.96 (m, 1H), 7.94 (t, J = 7.6 Hz, 1H), 7.86 (s, 1H), 7.77 (t, J = 7.6 Hz, 1H), 4.50 (s, 2H), 2.97 (s, 3H), 1.39 (s, 9H), 1.35 (s, 9H). 13C-NMR (75 MHz, CDCl3): δ[ppm] = 185.15 (q), 135.45 (+), 133.66 (q), 132.25 (+), 131.93 (q), 131.18 (+), 128.96 (q), 127.30 (+), 127.07 (q), 126.87 (+), 80.40 (q), 50.00 (−), 31.22 (+), 28.24 (+), 27.98 (+); MS (ESI-MS, CH2Cl2/MeOH + 10 mmol NH4OAc): m/z (%) = 466.1 (MH+, 100%); IR (neat): ν[cm−1] = 3160, 2971, 2927, 1746, 1677, 1588, 1511, 1452, 1391, 1364, 1300, 1250, 1221, 1141, 1067, 1024, 974, 951, 903, 843, 786, 743, 683, 621, 538; MW = 465.53 g mol−1; MF = C26H31N3O5.
Compound 8 (200 mg, 0.45 mmol) was dissolved in dichloromethane (20 mL, dried over CaCl2). A saturated solution of hydrogen chloride in diethylether (2 mL) was added dropwise. After stirring for 4 h at room temperature under moisture protection, the suspension was partitioned between four blue caps and then diethylether was added to a total volume of 15 mL per cap. The product was settled with the aid of a centrifuge (60 min, 4400 rpm, 0 °C) and the supernatant was discarded. The precipitate was re-suspended in diethylether, settled again and the supernatant was decanted off. This washing step was repeated once using diethylether. Afterwards the product was dried at reduced pressure to give 130 mg of yellow powder.

1 H-NMR (300 MHz, DMSO-d6): δ[ppm] = 8.60–8.47 (m, 2H), 8.29 (d, J = 7.4 Hz, 1H), 8.13 (d, J = 7.4 Hz, 1H), 7.94 (t, J = 8.0 Hz, 1H), 7.84–7.73 (m, 2H), 7.50 (m, app. bs, 4H), 4.49 (s, 2H), 3.06 (s, 3H). 13C-NMR (75 MHz, DMSO-d6): δ[ppm] = 183.66 (q), 157.14 (q), 137.68 (+), 135.75 (+), 132.64 (+), 132.19 (+), 131.60 (q), 130.02 (+), 128.19 (q), 127.44 (+), 127.28 (+), 126.48 (q), 126.10 (q), 49.39 (−), 36.22 (+); MS (ESI-MS, CH2Cl2/MeOH + 10 mmol NH4OAc): m/z (%) = 266.1 (MH+, 100%); IR (neat): ν[cm−1] = 3303, 3203, 3125, 1631, 1592, 1563, 1528, 1454, 1402, 1363, 1259, 1199, 1114, 1020, 967, 812, 783, 646, 603; MW = 266.3 + 35.45 = 301.75 g mol−1; MF = C16H16N3OCl.
2-Chloromethyl-1H-phenalen-1-one (7) (230 mg, 1 mmol) was dissolved in DMF (4 mL). N-Methylpiperidin (1.98 g, 2.42 mL, 20 mmol) was added and the solution was stirred for 30 h at room temperature. After cooling to room temperature, diethylether (45 mL) was added, the precipitate was settled with the aid of a centrifuge and the supernatant was discarded. The product was dissolved in methanol (2 mL), precipitated with diethylether (28 mL) and settled again. The precipitate was re-suspended in and washed with diethylether several times.

Afterwards the product was dried at reduced pressure. If necessary, the product is purified by column chromatography with silica gel using chloroform/methanol 6:1 → 5:1 as the eluent. The product is a dark yellow-greenish powder, 166 mg (51%).
1 H-NMR (300 MHz, DMSO-d6): δ[ppm] = 8.60–8.51 (m, 2H), 8.47 (s, 1H), 8.41–8.35 (m, 1H), 8.18 (dd, J = 7.1, 0.7 Hz, 1H), 7.98–7.91 (m, 1H), 7.82 (dd, J = 8.2, 7.1 Hz, 1H), 4.59 (s, 2H), 3.47 (m, 4H), 3.08 (s, 3H), 1.95–1.79 (m, 4H), 1.68–1.46 (m, 2H). 13C-NMR (75 MHz, DMSO-d6): δ[ppm] = 183.31 (q), 149.34 (+), 135.68 (+), 134.01 (+), 133.53 (+), 131.51 (q), 130.71 (+), 128.21 (q), 127.50 (+), 127.22 (+), 126.72 (q), 126.59 (q), 125.99 (q), 60.58 (−), 60.00 (−), 46.18 (+), 20.50 (−), 19.48 (−); MS (ESI-MS, CH2Cl2/MeOH + 10 mmol NH4OAc): m/z (%) = 292.2 (M+, 100%); IR (neat): ν[cm−1] = 3016, 2944, 2869, 1634, 1579, 1510, 1469, 1403, 1360, 1255, 1227, 1191, 1126, 1078, 1030, 938, 875, 831, 774; MW = 292.4 + 35.45 = 327.85 g mol−1; MF = C20H22NOCl.
Photophysical characterization
The 1O2 quantum yields ΦΔ of all compounds were determined using SAPYR (1) with a ΦΔ = 0.99 as a reference PS (synthesized as described earlier11 and according to patent no. WO/2012/11386015 in the Department of Organic Chemistry, University of Regensburg, Germany; purity: 98%) employing the methodology described elsewhere.11,12 Briefly, singlet oxygen luminescence signals were recorded at 1270 nm with a high sensitive photomultiplier system (R5509-42, Hamamatsu Photonics, Hamamatsu, Japan) and an ultrafast multiscaler (P7889, FAST ComTec, Oberhaching, Germany). For spectral resolution, luminescence was detected at wavelengths from 1150 to 1350 nm by using interference filters in front of the photomultiplier. All compounds were excited at a wavelength of 405 nm generated by a tunable laser system (NT 242-SH/SFG, Ekspla, Vilnius, Lithuania). For the determination of the 1O2 quantum yields of each compound the absorbed energies of all the compounds were compared with their emitted singlet oxygen luminescence at 1270 nm with regard to the reference photosensitizer SAPYR, as described recently (Table 1).11,12
| Compound |
|
λ max Absorptiona [nm] | λ max Emission [nm] | Singlet oxygen quantum yield ΦΔb | Octanol/water coefficient logDc,d |
|---|---|---|---|---|---|
| Conditions: at 25 °C, in Millipore water.a All compounds show a broad absorption maximum in this region, which cannot be resolved in distinct maxima; the spectra were collected in air-equilibrated solution.b The 1O2 quantum yield ΦΔ was determined using SAPYR (1) with a ΦΔ = 0.99 as a reference PS (synthesized as described earlier;11 purity: 98%).c Values measured by partition between 10 mM acetate buffer at pH 4.4 and n-octanol (pKb = 4.8 for (2) and 4.4 for (3)).d For compounds 1, 4 and 5 log D = logP, as only one species is present; at the given pH values the guanidinium group in (5) is protonated (pKa = 12). |
|||||
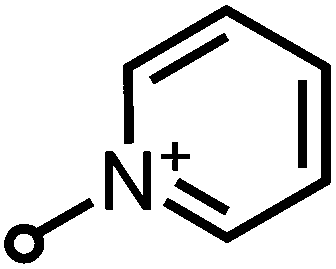
| SAPYR (1) |
|
363–410 (ref. 11) | 489 ± 5 (ref. 11) | 0.99 ± 0.05 (ref. 11) | −1.3 (ref. 11) |
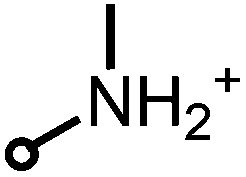
| (2) |

|
360–412 | 486 ± 5 | 0.89 ± 0.05 | −0.3/−1.1c |
| (3) |
|
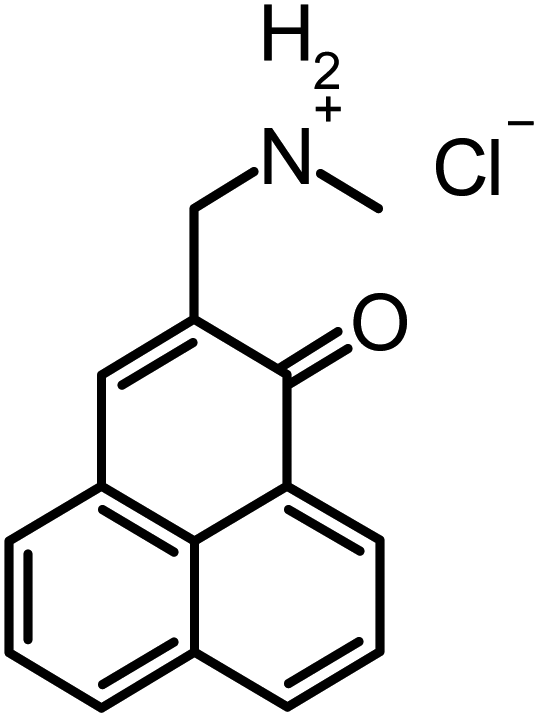
362–415 | 487 ± 5 | 0.98 ± 0.05 | −0.1/−1.0c |
| (4) |

|
360–418 | 492 ± 5 | 0.92 ± 0.05 | −1.2 |
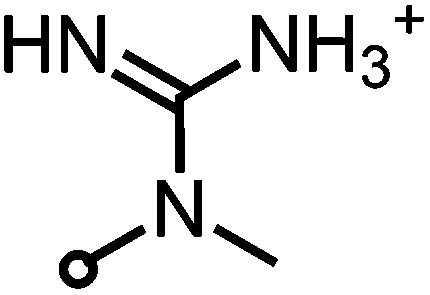
| SAGUA (5) |
|
361–414 | 488 ± 5 | 0.86 ± 0.05 | −1.1 |

For the determination of the photostability of the tested compounds, solutions (100 μM, 2 mL) of 1–5 were irradiated for 10 min with 10 mW at 400 nm, applying a total irradiation energy of 6 J. The solutions were magnetically stirred during irradiation and their transmissions were measured before and after irradiation.
The absorption spectra for the determination of pH-stability and estimation of the octanol–water-partition coefficients were recorded on a Varian Cary BIO 50 UV/vis/NIR spectrometer with temperature control using 1 cm quartz cuvettes (Hellma) and Uvasol solvents (Merck, Baker, or Acros) or Millipore water (18 MΩ). Fluorescence measurements were performed with UV-grade solvents (Baker or Merck) in 1 cm quartz cuvettes (Hellma) and recorded on a Varian “Cary Eclipse” fluorescence spectrophotometer with temperature control.
Light source
Illumination of all the samples was performed for 60 s using a blue light emitting prototype containing a neon tube (BlueV, Medizintechnik Herbert Waldmann GmbH & Co. KG, Villingen-Schwenningen, Germany). An intensity of 20 mW cm−2 at the level of the samples was measured and a total light dose of 1.2 J cm−2 was applied. Intensity was measured with a low-power thermal sensor (Nova 30 A-P-SH, Ophir-Spiricon, North Logan, UT) and the emission spectrum of the light source was recorded by means of a monochromator with a CCD detection system (SPEX 232, HORIBA Jobin Yvon, Longjumeau Cedex, France). The well plates containing the samples were illuminated from below with direct contact to the bottom of the wells, wherefore diffusion of light due to surface tensions in the samples could be excluded.Bacterial culture
Five reference strains, Streptococcus mutans (SM; ATCC 25175), Enterococcus faecalis (EF; ATCC 29212), Actinomyces naeslundii (AN; T14 V), Staphylococcus aureus (SA; ATCC 25923) and Escherichia coli (EC; ATCC 25922) were used in this study.The respective culture media were Brain Heart Infusion Broth (BHI broth; Sigma-Aldrich, St Louis, MO) for SM, EF and AN as well as Mueller Hinton Broth (Sigma-Aldrich, St Louis, MO) for SA and EC. All strains were grown overnight at 37 °C in an orbital shaker under aerobic conditions in order to obtain bacteria in the stationary growth phase. After that, bacteria were harvested by centrifugation (3000 rpm, 10 min; Megafuge 1.0, Heraeus Sepatech, Osterode, Germany) and re-suspended in sterile water. Consequently the samples were diluted to yield an optical density (OD) of 0.6 measured at 600 nm by means of a photospectrometer (DU® 640, Beckman-Coulter, Krefeld, Germany), which corresponds to a number of approximately 107 bacteria per ml. These suspensions were used for aPDT-experiments.
Photodynamic inactivation of bacteria
Bacterial cell suspensions with OD = 0.6 were mixed in 96-well flat-bottom, colorless microtiter plates (Corning Costar®, Corning, NY) one-to-one either with 100 μL H2O or with 100 μL PS obtaining final PS-concentrations of 0 μM, 1 μM, 5 μM, 10 μM, 16 μM, 25 μM or 100 μM, respectively. Immediately after a 10 s incubation period the samples were irradiated for 60 s with the BlueV Prototype (20 mW cm−2) or maintained in the dark for the same period. Irradiation with the BlueV Prototype ensured consistent irradiation and equal intensity over a whole 96-well microtiter plate. Illumination was performed from the bottom side of the 96-well plates to avoid diffusion of light due to surface tensions in the samples. Serial tenfold dilutions (10−1 to 10−6) were prepared in BHI Broth (SM, EF, AN) or Mueller Hinton Broth (SA, EC) and aliquots (3 × 20 μL) were plated on agar plates, as described earlier.16 Mueller–Hinton-agar plates were used for SM, EF, SA and EC and blood agar plates for AN (provided by the Institute of Medical Microbiology and Hygiene, University Medical Center Regensburg, Germany). The plates were incubated aerobically at 37 °C for 24 h (EF, SA and EC) or 48 h (SM, AN); then the colony forming units (CFU) were counted. Six independent experiments with three sub-samples per experimental group were performed.Data analysis
For all aPDT experiments, the CFU counts of experimental data were related to untreated controls (L−, 0 μM, 100%) and expressed as relative survival (% CFU). The results comprise data from at least six independent experiments with three sub-samples per experimental group, whereby the CFU counts of a sample were calculated as the median CFU counts of the three corresponding sub-samples.These obtained data were plotted as bar charts depicting medians and 25–75% quantiles, where the horizontal and dashed lines represent reductions of 3log10 steps (99.9%; antimicrobial effect) and 5log10 steps (99.999%; disinfecting effect), respectively. A reduction of at least ≥3log10 of viable median numbers of bacteria was declared as biologically relevant with regard to the guidelines of hand hygiene.17
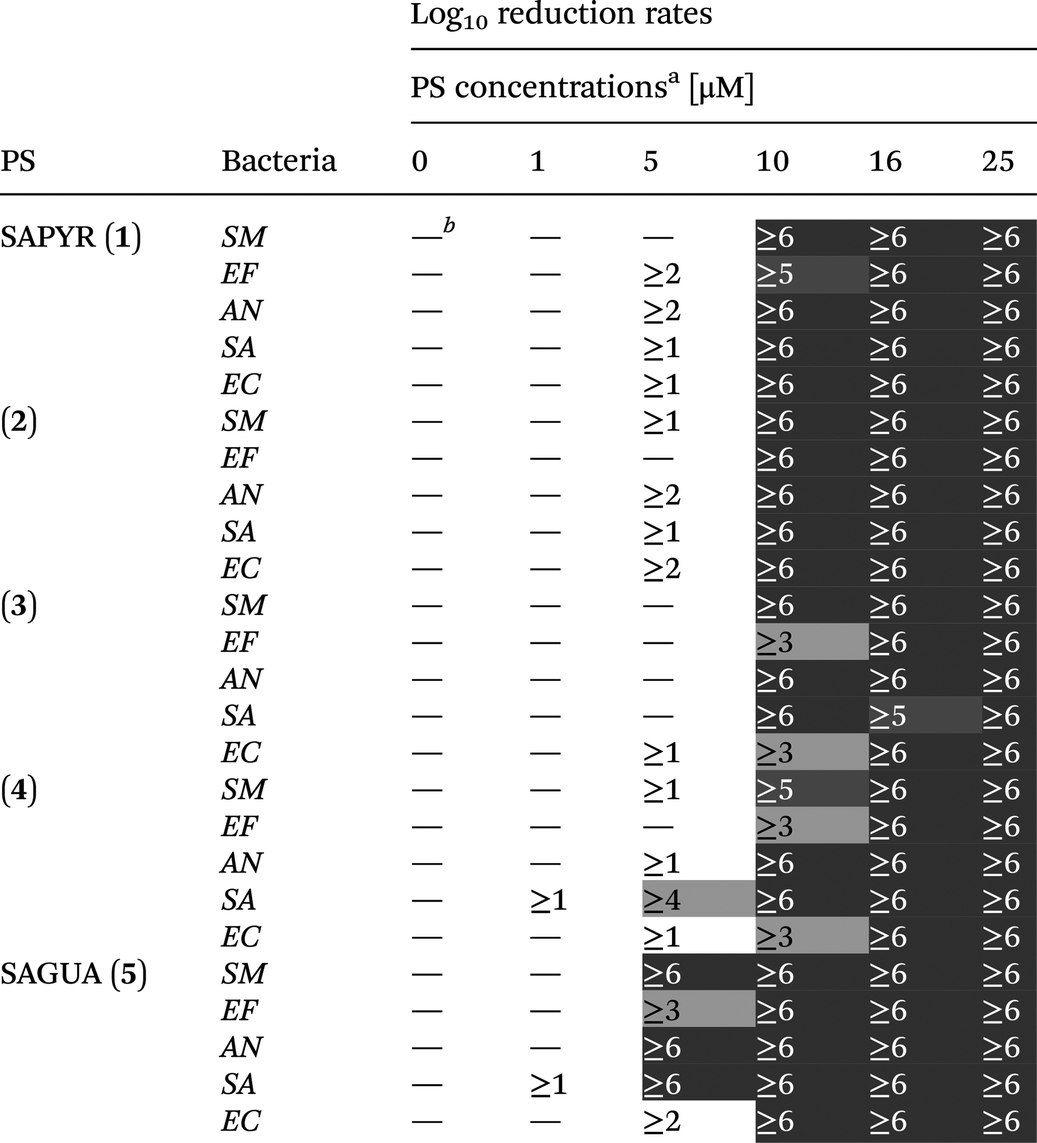
Log10 reduction rates were deduced for each PS from each bacterial species at each PS concentration and are presented in a table (see Table 2). A reduction of less than 1log10 step was defined as virtually no antibacterial efficacy.
| a Photodynamic inactivation rates of compounds 1–5 against all bacterial species investigated in this study upon light activation (1.2 J cm−2). (SM – Streptococcus mutans; EF – Enterococcus faecalis; AN – Actinomyces naeslundii; SA – Staphylococcus aureus; EC – Escherichia coli). b —Indicates <1log10 step reduction of CFU, which was defined as virtually no antibacterial photodynamic efficacy. Thresholds for the evaluation of antibacterial efficacy were inactivation rates of ≥3log10 steps (99.9%; antibacterial effect, light grey) and ≥5log10 steps (99.999%; disinfectant effect, dark grey). |
|---|
|
For each PS and each bacterial species relative survival values were fitted (TableCurve 2D, Systat Software Inc., San Jose, CA, USA) including dose–response-functions. From the resulting four parameter Sigmoid curves (r2 ≥ 0.83) the PS concentrations (including 95% confidence limits) necessary to achieve a reduction rate of 5log10 steps (EC 5log10) (99.999%; disinfecting effect17) were derived and depicted as a bar chart.
For each PS median (25–75% quantiles) of EC 5log10 values for all bacterial species was calculated, depicted as a bar chart, and statistically rated using the Tukey-interval method. Data analysis, except fitting, was performed using SPSS for Windows, version 20 (SPSS Inc., Chicago, IL, USA).
Results
Preparation of photosensitizers
The PS presented in this study are water-soluble dyes based on a phenalen-1-one structure (6). All derivatives were synthesized with a purity of >97%, controlled by NMR spectroscopy and HPLC-MS. Phenalen-1-one (6) was first converted to its chloromethylated analog (7). Derivatives 1–4 are obtained by conversion of 7 in methanol (2, 3) or N,N-dimethylformamide (DMF) (1, 4) with excess amine at slightly elevated temperature in good to excellent yields. Purification was achieved by precipitation with diethylether and re-precipitation from ethanol with diethylether, until the UV/Vis spectra showed constant absorption. The compound with the pyridinium moiety (1, SAPYR) was prepared in a similar manner as described earlier11 (Fig. 2A).Starting from 5 was prepared by reaction with N,N′-di-(tert-butoxycarbonyl)-N′′-triflylguanidine in dichloromethane (DCM) and subsequent deprotection with HCl in diethylether in excellent overall yield (Fig. 2B).
Photophysical characterization
For all compounds 2–5 singlet oxygen quantum yields ΦΔ could be estimated in a similar range compared to the reference PS SAPYR (1) (ΦΔ = 0.99) (Table 1).12 The emission spectrum of the light source (BlueV) and the absorption spectrum of SAPYR have an overlap at wavelengths longer than 400 nm (Fig. 3). | ||
| Fig. 3 Absorption and emission spectra. Absorption coefficient of SAPYR (1) (red dotted line) plotted with the emitted energy of the light source BlueV within 60 s (blue solid line). | ||
Fig. 4 shows the results of the photostability measurements of SAGUA (5). No photobleaching could be observed under the conditions used here (irradiation at 400 nm with 10 mW for 10 min, corresponding to 6 J laser energy). Likewise, when using the same conditions, no photobleaching of compounds 1–4 could be observed (for details please see the ESI†).
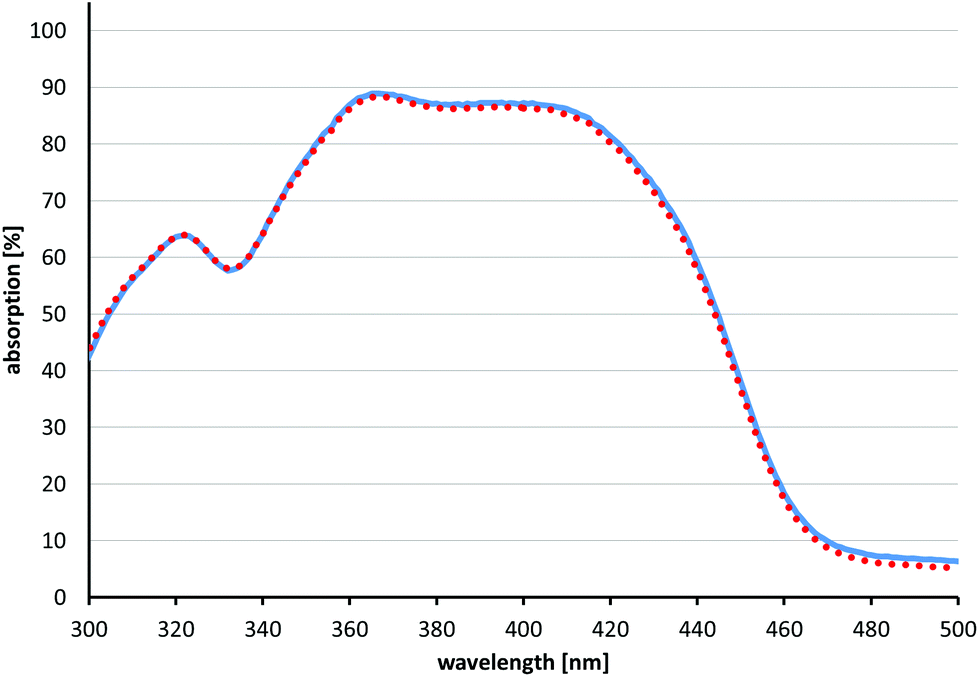 | ||
| Fig. 4 Photostability measurements of SAGUA (5). The solid blue spectrum shows absorption before irradiation, while the red dotted spectrum shows absorption after irradiation at 400 nm with 10 mW for 10 min, corresponding to 6 J laser energy. SAGUA (5) shows no photobleaching under the conditions used. | ||
Physical characterization data are summarized in Table 1. The polarity of compounds 1–5 was estimated by measuring the partition coefficient. Distribution between water and n-octanol of 1 × 10−4 mol of each compound (1–5) was measured by UV/Vis spectroscopy after 20 minutes of stirring at room temperature.
As compounds 2 and 3 can be deprotonated in water, their partition coefficient was also measured using 10 mM acetate buffer at pH 4.4. The pKb values of both compounds were obtained by pH-titration and were found to be 4.8 for (2) and 4.4 for (3), which is similar to the already published data of benzylamine (pKb = 4.65).18
The pH stability of SAGUA (5) was determined by recording UV/Vis spectra in buffered aqueous solutions at different pH values (from pH 2 to pH 12) after incubation for 30 minutes. SAGUA (5) shows excellent stability in acidic medium (down to pH = 2), but decomposes slowly in alkaline solutions with pH > 10 (for UV/Vis spectra analysis please see the ESI†). In comparison with the other phenalen-1-one derivatives, e.g. SAPYR (1), which starts to decompose at pH > 9,11 SAGUA (5) is slightly more stable at high pH values.
The UV/Vis spectra of SAGUA (5) were recorded at different concentrations and showed no differences up to 1 mM concentration (for details please see the ESI†). Thus, aggregation can be neglected below the millimolar range (<1 mM) and has not to be considered for our biological studies.
Photodynamic inactivation of bacteria
aPDT was performed against oral and dermal pathogens Streptococcus mutans (SM), Enterococcus faecalis (EF), Actinomyces naeslundii (AN), Staphylococcus aureus (SA) and Escherichia coli (EC) using the phenalen-1-one compounds 1–5 as PS. Bacterial suspensions were incubated for 10 s with the PS in concentrations ranging from 1 to 100 μM followed by irradiation for 60 s with a blue light emitting prototype (BlueV, Waldmann, Villingen-Schwenningen, Germany; intensity at the sample-level: 20 mW cm−2; light dose: 1.2 J cm−2). Colony forming units (CFUs) were evaluated according to the methodology described by Miles, Misra and Irwin.16Untreated control groups without PS and without light irradiation (L−, 0 μM), which exhibited CFU in a range between 106 and 5 × 107, were set as 100% in order to calculate relative survival rates. In all cases, neither treatment with PS alone nor with light alone led to any decrease of CFU compared to the untreated control groups (L−, 0 μM) (Fig. 5). Fig. 5 exemplarily depicts the photodynamic inactivation rates of compounds 1–5 against Gram-positive EF and Gram-negative EC (results for SM, AN and SA are shown in the ESI†). The medians on or below the black horizontal line represents ≥99.9% efficacy of photodynamic bacteria killing, whereas the black dotted horizontal line represents ≥99.999% killing efficacy, both compared to matching untreated control groups (bacteria only, neither treatment with PS nor with light). In general, a median-reduction of at least 3log10 steps of viable bacteria is stated as biologically relevant with regard to the guidelines of hand hygiene.17 Therefore, the differences seen in the bacterial inactivation of EF and EC is within the experimental accuracy (Fig. 5). All other differences detected are discussed later within the manuscript. For all PS the photodynamic inactivation rates arise with increasing concentrations as dose–response curves.
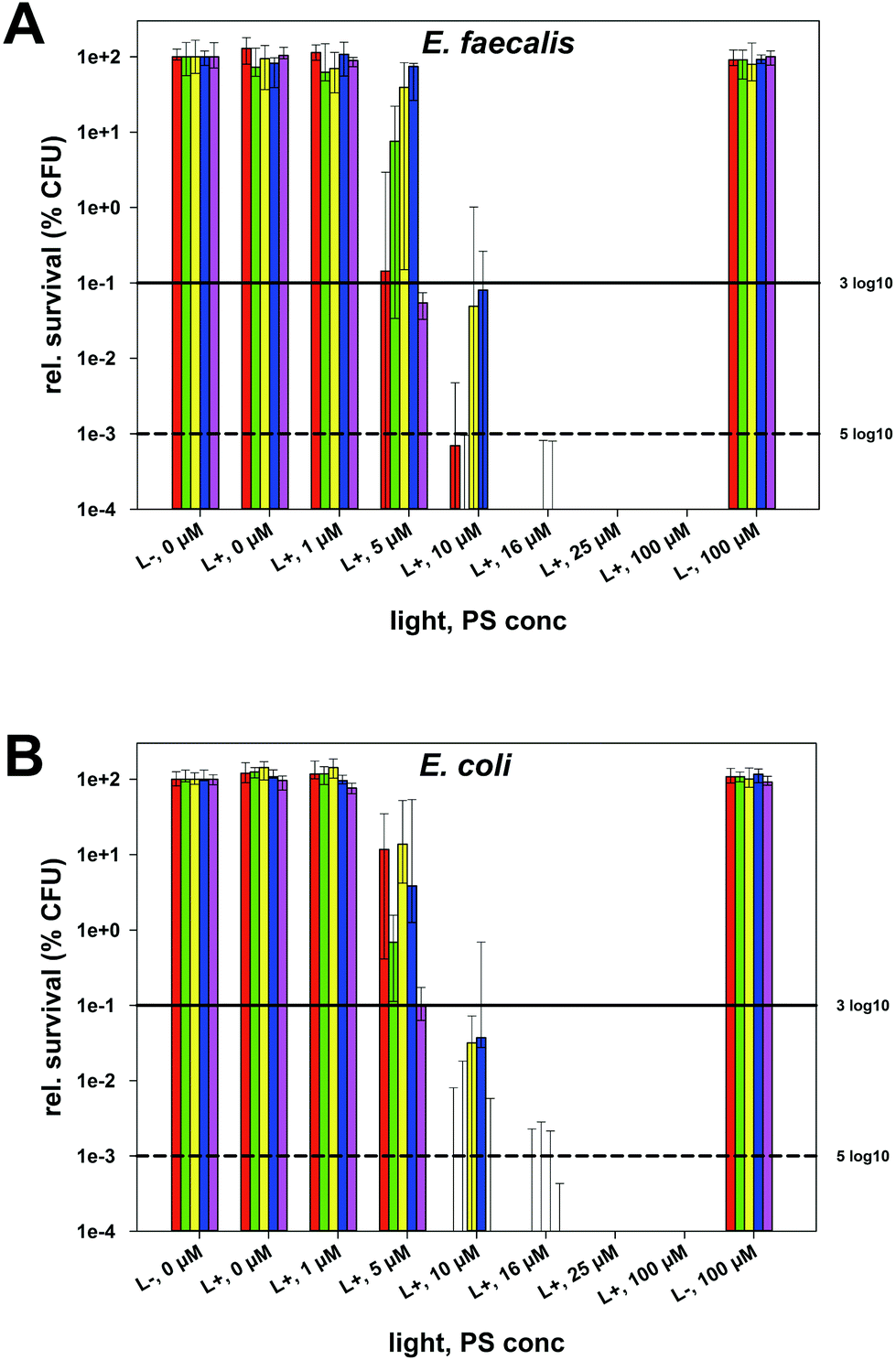 | ||
| Fig. 5 Photodynamic inactivation of Gram-positive E. faecalis (A) and Gram-negative E. coli (B) with phenalen-1-one derivatives 1–5. aPDT results of phenalen-1-one derivatives (1–5) upon light activation (L+; 1.2 J cm−2, samples were irradiated) against Gram-positive E. faecalis and Gram-negative E. coli shown as relative survival rates with untreated controls (L−, 0 μM, without irradiation) being set as 100%. Horizontal and dashed lines depict reductions of 3log10 steps (99.9%; antimicrobial effect) and 5log10 steps (99.999%; disinfectant effect), respectively. (CFU: colony forming units; red: SAPYR (1); green: (2); yellow: (3); blue: (4); pink: SAGUA (5)). | ||
At a concentration of 16 μM all PS showed reductions of ≥5log10 steps following light activation regardless of the respective bacterial species (Table 2). At 10 μM all PS exhibited reductions of ≥3log10 steps. 2 and SAGUA (5) even reached inactivation rates of ≥6log10 steps at a concentration of 10 μM after light activation against all bacterial species, whereby SAGUA (5) even showed reductions of ≥3log10 steps at a concentration of 5 μM against all Gram-positive pathogens (SM, EF, AN and SA).
In order to calculate the minimal effective concentrations (EC 5log10) being necessary for a respective compound to achieve a photodynamic inactivation rate of 5log10 steps (99.999% of bacteria killing, disinfecting effect) (Fig. 6A), CFU-reduction rates of aPDT groups for each bacterial species were fitted. For facilitating the comparison of compounds 2–5 with the reference PS SAPYR (1), medians of EC 5log10 concentrations of each compound over all bacteria were calculated (Fig. 6B).
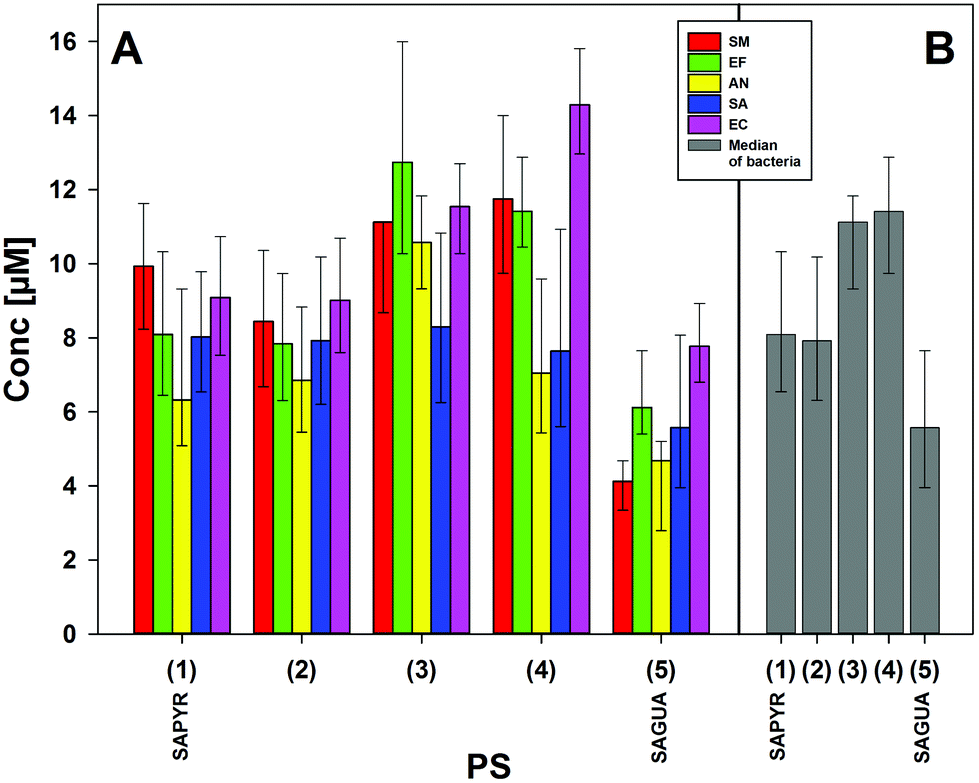 | ||
| Fig. 6 Effective concentrations for 5log10 inactivation. (A) Effective concentrations (95% confidence limits) of compounds 1–5 against every bacteria tested, which were necessary to reach inactivation rates of 5log10 steps of CFU (99.999%; disinfectant effect) upon light activation (1.2 J cm−2) (red: SM – Streptococcus mutans; green: EF – Enterococcus faecalis; yellow: AN – Actinomyces naeslundii; blue: SA – Staphylococcus aureus; pink: EC – Escherichia coli). (B) Median values (25–75% quantiles) of effective concentrations of compounds 1–5 of all bacteria tested, which were necessary to reach inactivation rates of 5log10 steps of CFU (99.999%; disinfectant effect) upon light activation (grey: median of all bacteria). | ||
Consequently, the following ranking could be established: compared to SAPYR (1) (8.1 μM), the EC 5log10 of 2 (7.9 μM) was in a similar range, whereas SAGUA (5) (5.6 μM) was distinctly more effective. In contrast, 3 (11.1 μM) and 4 (11.4 μM) were distinctly less effective (Fig. 6B). According to the Tukey-interval method, only SAGUA (5) was found significantly different from 3 and 4, respectively.
Discussion
The aim of this study was to evaluate the impact of alterations of the cationic charged anchoring groups in phenalen-1-one PS on their antimicrobial photodynamic efficacy since it is well known that already small differences either in the structure or in the physicochemical properties of the substituents of PS can change their antimicrobial potency.11 The photodynamic efficacy of phenalen-1-one PS was evaluated against the planktonic cultures of oral and dermal key pathogens SM, EF, AN, SA and EC (with EC being a representative of Gram-negative bacteria).SAPYR (1), which is equipped with a pyridinium-methyl substituent, was used as a reference PS for comparison. For this compound, pronounced inactivation rates against planktonic bacteria11 as well as against monospecies and polyspecies biofilms formed by oral key pathogens12 have already been demonstrated. The newly synthesized derivatives carry as cationic substituents a primary ammonium ion (2), a secondary ammonium ion (3), a piperidinium group (4) or a permanently positively charged guanidinium group (5; SAGUA).
The whole range of steric demand is covered in this series with the primary and secondary ammonium groups in 2 and 3 being small cations and the pyridinium group in SAPYR (1) being planar, concentrating its quaternary positively charged character on the nitrogen atom (Fig. 7A). The quaternary cation in 4 is also concentrated on the nitrogen atom, but the piperidinium ring is a folded group consisting of sp3 carbon centers with a considerably larger steric demand than a pyridinium unit19 (Fig. 7A). Due to the fast tautomerism of the double bond between the nitrogen atoms the guanidinium group in SAGUA (5) represents a planar positively charged disk.
 | ||
| Fig. 7 (A) Illustration of the steric demand of the substituents in phenalen-1-one derivatives SAPYR (1), 4 and SAGUA (5); atom colours: red = oxygen, blue = nitrogen, white = hydrogen, grey = carbon. (B) Illustration of the binding of phenalen-1-one derivatives SAPYR (1), 4 and SAGUA (5) to glutamate on the cell surface of bacteria. SAGUA (5) shows bidentate and directional hydrogen bonds in addition to the cationic charge attraction, exhibited by all positively charged moieties. | ||
As fluctuations of pH values are omnipresent in a cellular environment, permanently charged moieties are very advantageous for the electrostatic attachment of the PS to bacterial cell surfaces. This is featured by the very alkaline guanidinium ion (pKa ∼ 12) in SAGUA (5) and the quarternary aliphatic ammonium in (4) (cyclohexyl substituted analog of (1)) as well the pyridinium cations in SAPYR (1).
Compound SAGUA (5) was found to be the most effective derivative. The guanidinium group is planar and a permanently charged cation (pKa ∼ 12). The nature of this positively charged group may improve the attachment of the PS to the cell walls of pathogens. No alkyl or aryl substituents shield the strong and permanent charge. In addition, guanidinium groups can establish hydrogen bonds with phosphate and carboxylate groups,20,21 which may improve the binding to target groups, e.g. to bacterial cell walls (Fig. 7B). Furthermore, the combination of the lipophilic chromophore and the guanidinium group may improve cell penetration, as it is known for example for arginine rich peptides.22 In contrast, too hydrophilic molecules may be “washed away” more easily by the surrounding water, thus never reaching a critical amount of PS for good photodynamic efficacy.
When comparing these compounds regarding the antibacterial photodynamic efficacy, the reference PS SAPYR (1) was found to be less effective than SAGUA (5). The median threshold concentration needed for achieving an inactivation rate of ≥5log10 was 8.1 μM for SAPYR (1) vs. 5.6 μM for SAGUA (5) (Fig. 5B).
In contrast compound 4, which can be seen as a structural analog to SAPYR (1) with a completely hydrogenated substituent, was the least effective derivative. Compound 4 is similar in its lipophilicity to SAPYR (1); its minor efficacy however may be attributed to the substantially higher steric demand of the piperidinium residue. This bulky group may hinder the charge to establish a close and strong electrostatic interaction with the negatively charged bacterial cell surface.
Looking at compounds 2 and 3, which are similar to each other regarding the nature of their charged moieties, their steric demand and their lipophilicity, these two derivatives demonstrated different efficacy rates: considering compound 3, the median threshold concentration being necessary for a ≥5log10 inactivation was similar to 4 (11.1 μM compared to 11.4 μM), whereas for compound 2 similar or even lower concentrations than for SAPYR (1) were sufficient to achieve a ≥5log10 reduction (7.9 μM). This may be explained by the protonation equilibria of these charged moieties in the rather complex environment present at the bacterial surface. The conditions that are existent in an aqueous solution are not analogous to the conditions prevailing at the surface of bacterial cells or in their cell membranes. Thus, even small differences in the pKa value may have drastic effects, resulting in a lower local concentration of 3 and a reduced antibacterial photodynamic efficacy. On the surface of a bacterial cell, negative charges predominate over positive, which results in an increased effective pKa value. Inside the membrane different conditions dominate: in this milieu of low dielectric character charged molecules are in a state of higher energy. In particular, small ions like hydroxonium are particularly affected by this, whereas the effect is less drastic for the larger PS molecules. Therefore, the effective pKa value is reduced and the hydroxonium concentration must be far higher compared to the exterior to protonate the phenalen-1-one derivative in the membrane. It is conceivable that both compounds 2 and 3 constantly switch between the bacterial membrane and the outside.
Recently we demonstrated the antibacterial photodynamic properties of SAPYR (1) against planktonic cultures of Gram-positive SM, EF, AN and Gram-negative Aggregatibacter actinomycetemcomitans using a dental light curing unit (Bluephase® C8, Ivoclar-vivadent, Schaan, Liechtenstein) as a light source.11 Although the overlap of the emission of this light source with the absorption of SAPYR (1) was only about 5%, pronounced photodynamic inactivation rates could be achieved due to the high power of the light source (output power: 1360 ± 50 mW cm−2; power at the level of the samples: 1260 ± 50 mW cm−2); thereby, an irradiation for 120 s (light dose: 150 J cm−2) of SAPYR (1) at a concentration of 5 μM was adequate to reduce the CFU of EF and SM by ≥6log10. For achieving the same results for AN with SAPYR (1) at 5 μM an irradiation period of 40 s (light dose: 50 J cm−2) was sufficient.11 However, for Aggregatibacter actinomycetemcomitans aPDT results were distorted by a strong light toxicity of this species towards irradiation with blue light,11 which was substantiated in a subsequent study by the presence of endogenous porphyrines and flavins in Aggregatibacter actinomycetemcomitans, which can act as PS.23
In the present study we employed a light source with an optimized emission spectrum in order to be able to reduce the illumination energy, but to conserve the pronounced efficacy. This is an important point since it is well known that blue light (in particular, at wavelengths shorter than 450 nm) can be detrimental towards human skin cells at high light doses.24 Furthermore, it is well known that high-power dental curing lamps can cause damage to oral soft tissue due to heat generation.25 In contrast, the prototype blue light source (BlueV) reaches a power at the sample level of 20 mW cm−2, which leads to a light dose of 1.2 J cm−2 while irradiating for 60 s only. The match (integral of the convolution) of the absorption spectra of SAPYR (1) and emission spectra of the BlueV (Fig. 3) can be seen as fourfold compared to that found with the Bluephase® C8.11 Considering inactivation of SM with 10 μM of SAPYR (1), in the former study a ≥6log10 inactivation could only be reached upon irradiation for 40 s applying a light dose of 50 J cm−2, whereas in the present study we were able to achieve the same efficacy in a virtually equivalent time of 60 s with a light dose of only 1.2 J cm−2. Consequently, due to the better adaptation of the light source to the absorption spectrum of SAPYR (1), we were able to reduce the light dose in a pronounced manner (up to 60-fold), while high efficacy rates for photodynamic inactivation were observed.
All in all, in this study a structure–activity relationship of phenalen-1-one derivatives 1–5 concerning the effect of the nature of their cationic substituents on the antimicrobial efficacy could be successfully deduced. Due to optimization of the light source the efficacy of aPDT with phenalen-1-ones could be improved.
Conclusion
The structure–activity relationship deduced in the present study shows that a permanently positively-charged moiety in combination with a slightly lipophilic character (logP < 1) as a substituent of a phenalen-1-one PS seems to be a criterion for good photodynamic efficacy. The combination of a permanently positively-charged moiety of a PS and the possibility of a PS to the formation of strong, directional hydrogen bonds towards the cell walls of pathogens seems to be even more beneficial for the antimicrobial action.
The results of this study clearly demonstrate that the functional substituent of guanidinium enhances the photodynamic antimicrobial efficacy substantially (≥6log10; disinfecting effect), inactivating both dental and dermal key pathogens within 60 s of applying a light dose of only 1.2 J cm−2. The use of an optimized light source enables a fast and effective eradication of pathogens at low light doses (up to 60-fold less as demonstrated earlier14) and ensures protection of surrounding tissue.
With its singlet oxygen quantum yield close to unity and the high photostability, SAGUA (5) encourages further research on optimizing handling modalities of aPDT with phenalen-1-one derivatives, e.g. designing an appropriate formulation for clinical application of SAGUA (5) in order to reach pathogens on dermal or mucosal surfaces. In addition, SAGUA (5) may be useful as a promising tool for industrial and clinical purposes where repeatable treatments are necessary and savings in time are critical to achieve efficient inactivation of bacteria. Consequently, SAGUA (5) may also serve as a lead candidate for the development of even more potent phenalen-1-one PS in the future.
Conflict of interest
All authors declare that they have no conflict of interest.Author contributions
Conceived and designed the experiments: TM, AS, KAH, FC and WB. Performed the experiments: IT, JR and AS. Analyzed the data: KAH, IT, FC, JR, TM and AS. Contributed reagents/materials/analysis tools: AS, JR and KAH. Wrote the paper: IT, FC, AS, TM, KAH, LT and WB.Acknowledgements
Ewa Kowalewski and Jana Schiller are gratefully acknowledged for excellent technical assistance. Fabian Cieplik is funded by the University Medical Center Regensburg within the ReForM A program. Johannes Regensburger is funded by a grant of the German Research Foundation (DFG-RE-3323/2-1).References
- G. M. Rossolini and E. Mantengoli, Clin. Microbiol. Infect., 2008, 14(Suppl 6), 2–8 CrossRef PubMed.
- T. Yamamoto, Y. Tamura and T. Yokota, Antimicrob. Agents Chemother., 1988, 32, 932–935 CrossRef CAS PubMed.
- S. P. Yazdankhah, A. A. Scheie, E. A. Høiby, B.-T. Lunestad, E. Heir, T. Ø. Fotland, K. Naterstad and H. Kruse, Microb. Drug Resist., 2006, 12, 83–90 CrossRef CAS PubMed.
- G. Jori and G. Roncucci, Adv. Clin. Exp. Med., 2006, 15, 421–426 Search PubMed.
- E. Alves, M. A. Faustino, M. G. Neves, A. Cunha, J. Tome and A. Almeida, Future Med. Chem., 2014, 6, 141–164 CrossRef CAS PubMed.
- T. Maisch, Lasers Med. Sci., 2007, 22, 83–91 CrossRef PubMed.
- T. Maisch, S. Hackbarth, J. Regensburger, A. Felgenträger, W. Bäumler, M. Landthaler and B. Röder, J. Dtsch. Dermatol. Ges., 2011, 9, 360–366 Search PubMed.
- E. Alves, L. Costa, C. M. Carvalho, J. P. Tomé, M. A. Faustino, M. G. Neves, A. C. Tomé, J. A. Cavaleiro, A. Cunha and A. Almeida, BMC Microbiol., 2009, 9, 70 CrossRef PubMed.
- M. Wainwright, J. Antimicrob. Chemother., 1998, 42, 13–28 CrossRef CAS PubMed.
- T. Maisch, J. Baier, B. Franz, M. Maier, M. Landthaler, R.-M. Szeimies and W. Bäumler, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 7223–7228 CrossRef CAS PubMed.
- A. Späth, C. Leibl, F. Cieplik, K. Lehner, J. Regensburger, K.-A. Hiller, W. Bäumler, G. Schmalz and T. Maisch, J. Med. Chem., 2014, 57, 5157–5168 CrossRef PubMed.
- F. Cieplik, A. Späth, J. Regensburger, A. Gollmer, L. Tabenski, K.-A. Hiller, W. Bäumler, T. Maisch and G. Schmalz, Free Radical Biol. Med., 2013, 65, 477–487 CrossRef CAS PubMed.
- F. Cieplik, A. Pummer, J. Regensburger, K. A. Hiller, A. Spath, L. Tabenski, W. Buchalla and T. Maisch, Front. Microbiol., 2015, 6, 706 Search PubMed.
- T. J. Baker, M. Tomioka and M. Goodman, Org. Synth., 2002, 78, 91 CrossRef CAS.
- W. Bäumler, A. Felgenträger, K. Lehner, T. Maisch, J. Regensburger, F. Santarelli and A. Späth, WO PatentWO/2012/113860, 2012 Search PubMed.
- A. A. Miles, S. S. Misra and J. O. Irwin, J. Hyg., 1938, 38, 732–749 CrossRef CAS PubMed.
- J. M. Boyce and D. Pittet, Infect. Control Hosp. Epidemiol., 2002, 23, 3–40 Search PubMed.
- Z. Rappoport and M. Frankel, CRC handbook of tables for organic compound identification, 1967 Search PubMed.
- S. E. Boiadjiev and D. A. Lightner, Chirality, 2000, 12, 204–215 CrossRef CAS PubMed.
- A. Späth and B. König, Tetrahedron, 2010, 66, 1859–1873 CrossRef.
- A. Späth and B. König, Tetrahedron, 2010, 66, 6019–6025 CrossRef.
- O. M. New and D. Dolphin, Eur. J. Org. Chem., 2009, 2675–2686 CrossRef CAS.
- F. Cieplik, A. Späth, C. Leibl, A. Gollmer, J. Regensburger, L. Tabenski, K.-A. Hiller, T. Maisch and G. Schmalz, Clin. Oral Invest., 2014, 18, 1763–1769 CrossRef PubMed.
- J. Liebmann, M. Born and V. Kolb-Bachofen, J. Invest. Dermatol., 2010, 130, 259–269 CrossRef CAS PubMed.
- T. J. Spranley, M. Winkler, J. Dagate, D. Oncale and E. Strother, Gen. Dent., 2012, 60, e210–e214 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, materials and methods relating to synthesis and characterization, selected NMR spectra, UV-Vis data concerning aggregation and stability and relative survival rates against all bacteria. See DOI: 10.1039/c5pp00262a |
| ‡ These authors share senior-authorship. |
| This journal is © The Royal Society of Chemistry and Owner Societies 2016 |