
Linker proteins enable ultrafast excitation energy transfer in the phycobilisome antenna system of Thermosynechococcus vulcanus†
C.
Nganou
ab,
L.
David
c,
N.
Adir
c and
M.
Mkandawire
*a
aVerschuren Centre for Sustainability in Energy and the Environment, Cape Breton University, P.O. Box 5300, 1250 Grand Lake Road, Sydney, Nova Scotia B1P 6L2, Canada. E-mail: martin_mkandawire@cbu.ca
bDepartment of Chemistry, University of Rochester, RC Box 270216, Rochester, NY 14627-0216, USA
cSchulich Faculty of Chemistry, Technion-Israel Institute of Technology, Haifa, 32000, Israel
First published on 26th October 2015
Abstract
We applied a femtosecond flash method, using induced transient absorption changes, to obtain a time-resolved view of excitation energy transfer in intact phycobilisomes of Thermosynechococcus vulcanus at room temperature. Our measurement of an excitation energy transfer rate of 888 fs in phycobilisomes shows the existence of ultrafast kinetics along the phycocyanin rod subcomplex to the allophycocyanin core that is faster than expected for previous excitation energy transfer based on Förster theory in phycobilisomes. Allophycocyanin in the core further transfers energy to the terminal emitter(s) in 17 ps. In the phycobilisome, rod doublets composed of hexameric phycocyanin discs and internal linker proteins are arranged in a parallel fashion, facilitating direct rod–rod interactions. Excitonic splitting likely drives rod absorption at 635 nm as a result of strong coupling between β84 chromophores (20 ± 1 Å) in adjacent hexamers. In comparison to the absorbance of the phycobilisome antenna system of the cyanobacterium Acaryochloris marina, which possesses a single rod structure, the linkers in T. vulcanus rods induce a 17 nm red shift in the absorbance spectrum. Furthermore, the kinetics of 888 fs indicates that the presence of the linker protein induces ultrafast excitation energy transfer between phycocyanin and allophycocyanin inside the phycobilisome, which is faster than all previous excitation energy transfer in phycobilisome subunits or sub-complexes reported to date.
Introduction
Changes in the ecosystem have induced adaptation of some phototrophic oxygenic species to harsh and changing environments, such as low light intensity by developing specific architecture for efficient harvesting of sunlight energy for carbon fixation and ATP synthesis. Some such oxygenic species include cyanobacteria and red algae. The main light-harvesting complex in cyanobacteria and red algae is the phycobilisome (PBS) complex, which is attached to the thylakoid membrane and whose molecular weight can range between 3 and 7 MDa. The PBS is assembled from pigmented proteins, referred to as phycobiliproteins (PBPs), which covalently bind bilin cofactors and un-pigmented proteins referred to as linker proteins. The PBS absorbs energy from the sun, which is then funnelled by a relatively slow excitation energy transfer (EET) process of between 70 and 200 ps to the photosynthetic reaction centre of photosystem II (PSII).1,2 In PSII, the energy drives the electron transfer processes. A recent report suggests that the PBS can supply the harvesting excitation energy to both photosystem I (PSI) and PSII.3The PBS consists of two sub-complexes, cores and rods, and researchers typically depict its architecture as 6–8 rods surrounding 2–5 core cylinders (see Scheme 1). Based on micrographs obtained using electron microscopy (EM), researchers have suggested two main models for the arrangement of the rods around the core, namely: (a) a radiating arrangement, in which the rods are arranged at regular angles in a radial manner around the core,4 and (b) a parallel rod pair model, in which the rods are arranged as two rod doublets that are parallel to the thylakoid membrane and one or two doublets perpendicular to the membrane.5–7 Researchers verified the latter model using cryo EM micrographs,8 which indicate a structure identical with the crystal structures of phycocyanin (PC). PC is one of the PBP components found in all rods. The PBS of the thermophilic cyanobacterium T. vulcanus is likely to consist of 5 core cylinders surrounded by 8 rod pairs arranged around the core (Scheme 1).7 However, a tricylindrical PBS type may also exist as researchers have suggested for Thermosynechococcus elongatus.9
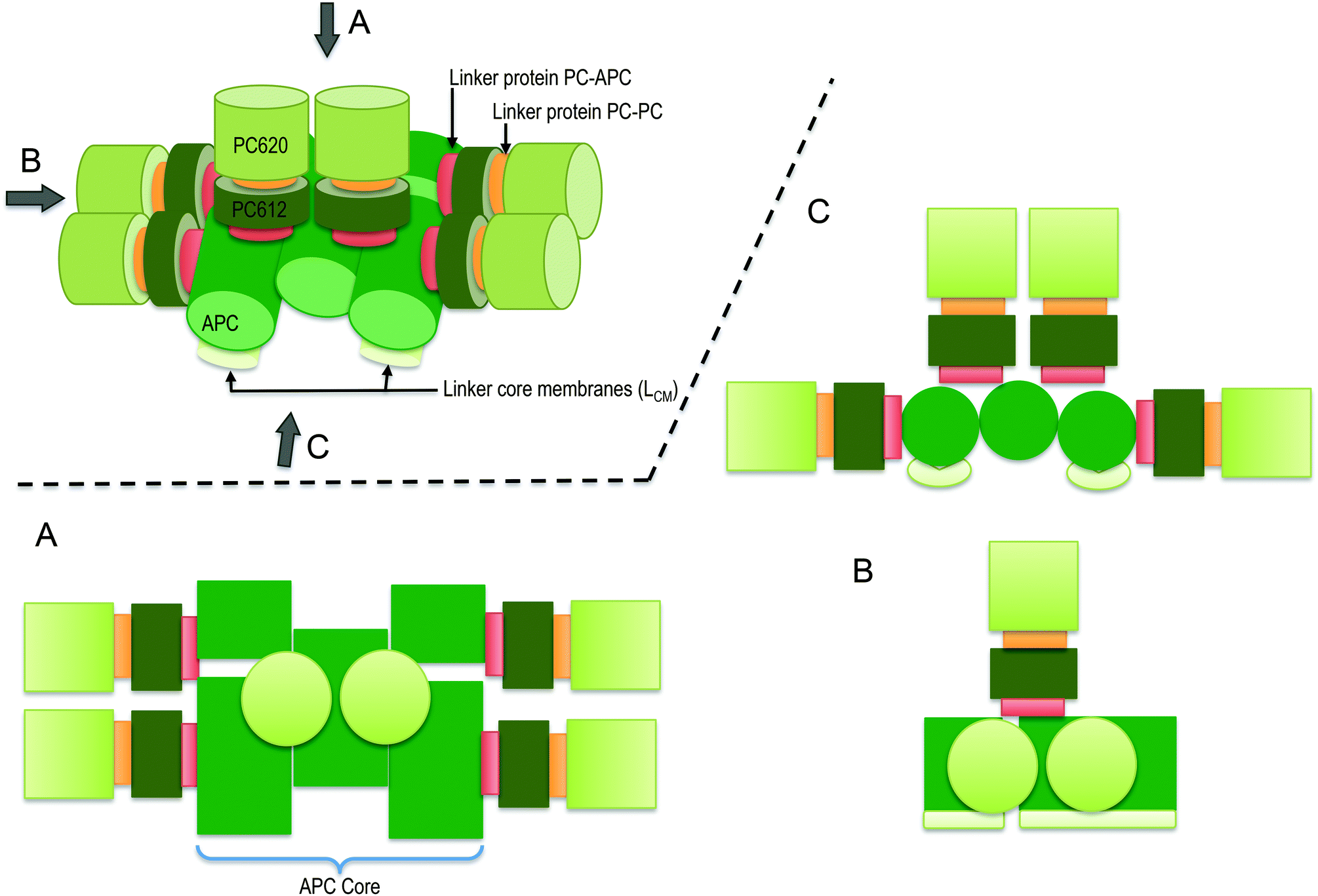 | ||
| Scheme 1 Schematic presentation of the intact-PBS of T. vulcanus showing the PC rod arrayed around the APC core in a parallel disposition. The presentation is shown in three dimension with its orthographic project of plan (A), side (B) and front (C) to show the relevant rod-core interactions based on work of David et al.7 | ||
The biliprotein antenna system of the cyanobacterium Acaryochloris marina is the simplest PBS antenna complex known to date with a molecular weight of about 1.2 MDa. The A. marina PBS is organized as single rod-shaped assembly of PBPs.10,11 Although A. marina and T. vulcanus present PBSs with different structural organizations, they are similar in the efficiency of their EET to the reaction centre (RC) of PS II.12 The building blocks of each PBS are α and β polypeptide subunits that combine to form a monomer, each subunit covalently binding at least one bilin cofactor. PC monomers contain three phycocyanobilins (PCBs), two bound to the conserved cysteines at position 84 (on each subunit) and one to a cysteine at position 155 of the β subunit. Monomers of allophycocyanin (APC), the PBP found in the PBS core, contain only the two cofactors at position 84.4
The absorption of the bilin cofactors in the PC monomer assembly (β155, β84, α84) displays an inhomogeneous broad spectrum at 77 K, with two main resonant absorption maxima between 600–602 nm and 628–630 nm.13 Using a PC monomer devoid of β155 extracted from a mutant strain of Synechococcus sp. PC 7002, Debreczeny et al.13 showed that the β84 cofactor's maximum absorption falls between 628 and 630 nm, while that of the β155 cofactor falls between 600 and 602 nm. They demonstrated that these absorption maxima of the β155 and β84 cofactors at 77 K are consistent with the individual spectra of the β155 and β84 cofactors at room temperature.13 Using a slight modification of the procedure Swanson and Glazer described,14 Debreczeny et al. further separated the α84 and β84 cofactors using high-performance liquid chromatography (HPLC).13 They showed that at room temperature the cofactor α84's absorption maximum is 624 nm. Debreczeny et al. also obtained the steady state absorption at room temperature of the monomeric PC (β155, β84, α84) when using the isolated PC (β155, β84, α84) from the strain of Synechococcus sp. 7002 containing all cofactors. Their results show a peak for a monomer at 617 nm. Based on knowledge of 77 K absorbance,13 one could propose that the cofactor β84 absorbs at the lowest energy level of the PC monomer. However, additional levels of assembly may have significant effects on the absorption characteristics of specific cofactors, as researchers have shown for APC.15
Recently, David et al. showed that the crystal structure of a PBS rod is composed of an assembly of two PC hexamers and three embedded linkers: LR,7,8 LR, and LRC.24 They did not observe the linker proteins’ penetrating out from the rod, and indeed the core discs may penetrate slightly into the last hexamer of the rod, as they previously suggested.24 To date, researchers have proposed different mechanisms of energy transfer between PBS sub-complexes. These can be grouped into either weak or strong-coupled pigments, which are driven by the relative distance and the dipole moment orientations between nearby cofactors in PBS, as Förster resonance energy transfer (FRET) theory describes. The weak coupling between electronic energy levels of neighbouring excited state cofactors matches the explained mechanism of EET in the monomeric PC assembly.13 Sauer,16 showed the limitations of FRET theory in terms of its ability to describe the complete mechanism of EET in the trimeric PC assembly.16 They suggested a fast 370 fs component that they attributed to the relaxation between two excitonic states formed as the result of the excitonic interaction between cofactors in adjacent subunits (from two different monomers) of the PC trimers, specifically α84 and β84. Furthermore, Edington et al. used two-colour time-resolved femtosecond pump–probe spectroscopy to describe the mechanism of excitonic coupling in the APC trimer.17 They reported the presence of two components with excited state lifetimes of 280 fs and 1000 fs, which were consistent with the time resolution of 200 fs. Recent works have focused on the role of the vibronic exciton structure in PC18 and APC.19 Womick et al. suggested that the vibronic exciton structure is consistent with an additional kinetic component found in anisotropy decay.20 Womick et al. found a kinetic component better than 100 fs in APC trimers19 and 970 fs in PC hexamers.18 However, they measured the kinetics on PC hexamers from the commercially purchased Spirulina sp., which was most likely lacking the linker proteins. We recently addressed the EET route in the PC rod of T. vulcanus. Our observations fit well with the activation and deactivation of HOOP wagging, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching, and the N–H rocking in-plane vibration.21 We also observed strong localized vibronic coupling between β84 in adjacent PC trimers in the PC rod of A. marina.9
C stretching, and the N–H rocking in-plane vibration.21 We also observed strong localized vibronic coupling between β84 in adjacent PC trimers in the PC rod of A. marina.9
In the hexameric PC assembly, Womick et al. recently showed that the fastest EET component is 970 fs,18 while, in a fragment of PC bound to APC, Sandström et al. found 17 ps and 50 ps kinetic components.22 Emphasizing the estimation of the EET component in PBS, Zhang,23 used time-resolved fluorescence on an isolated fragment of one PC hexamer connected to the APC core by a LRC. They found an 18 ps component suggested to be an EET from three β84 in a terminal PC trimer to the α84 in the APC core.23 They presented no further information about the presence of the LRC.23 Despite the extensive investigation of EET in PBS components, the estimation of EET in the intact PBS has remained an open issue. Detailed mapping of potential EET pathways in the intact PBS is critical for understanding how this complex functions. Therefore, the aim of the study reported in this paper was to ascertain if studying extracted PBS of represented close to real-life dynamics of EET transfer in T. vulcanus and A. marina. Thus, we compared the EET transfer dynamics in extracted and intact PBS of T. vulcanus and A. marina.
Results and discussion
Absorbance
We measured the room temperature absorbance spectra of PC rods, APC cores, and PBS in A. marina and T. vulcanus. Our results show that the inhomogeneous absorption of PBS rods in A. marina displays a maximum absorption at 618 nm, while its second derivative is indicative of PC component absorption at 618 nm and APC component absorption at 640 nm (Fig. 1). The PC rods and APC cores of T. vulcanus show absorption maxima at 635 and 650 nm, respectively. Prior to the PBS steady state absorption we measured (Fig. 1), the known maximum absorption of PBS sub-compounds in high phosphate fell between 620 and 635 nm for PC trimers and the intact PBS, respectively.24 The presence of the three different linkers in the PBS and the hexamer formation of T. vulcanus induce a 15 nm spectral red shift. The consequence of the latter change may be the shortening of the distance between β84 cofactors in adjacent hexamers. The mutual orientation and close proximity of adjacent PCB cofactors drive excitonic splitting. One can observe this from α84 and β84 in adjacent monomers in APC trimers, as MacColl25 and McGregor et al. discussed.15 Therefore, the distance change between β84 pigments in adjacent hexamers may drive the excitonic splitting from 630 to 635 nm. Consequently, one observes a bathochromic shift in PC rods when compared to PC trimers. An additional observation is the broad minimum in the second derivative of the PBS absorption spectra of T. vulcanus between 600 and 615 nm likely due to the β155 cofactor. Regarding the parallel disposition of PC rods around the core of T. vulcanus, strong coupling may occur between β155 in adjacent rods. Elsewhere, we will describe our analysis of the rates of this coupling and the aforementioned excitonic splitting.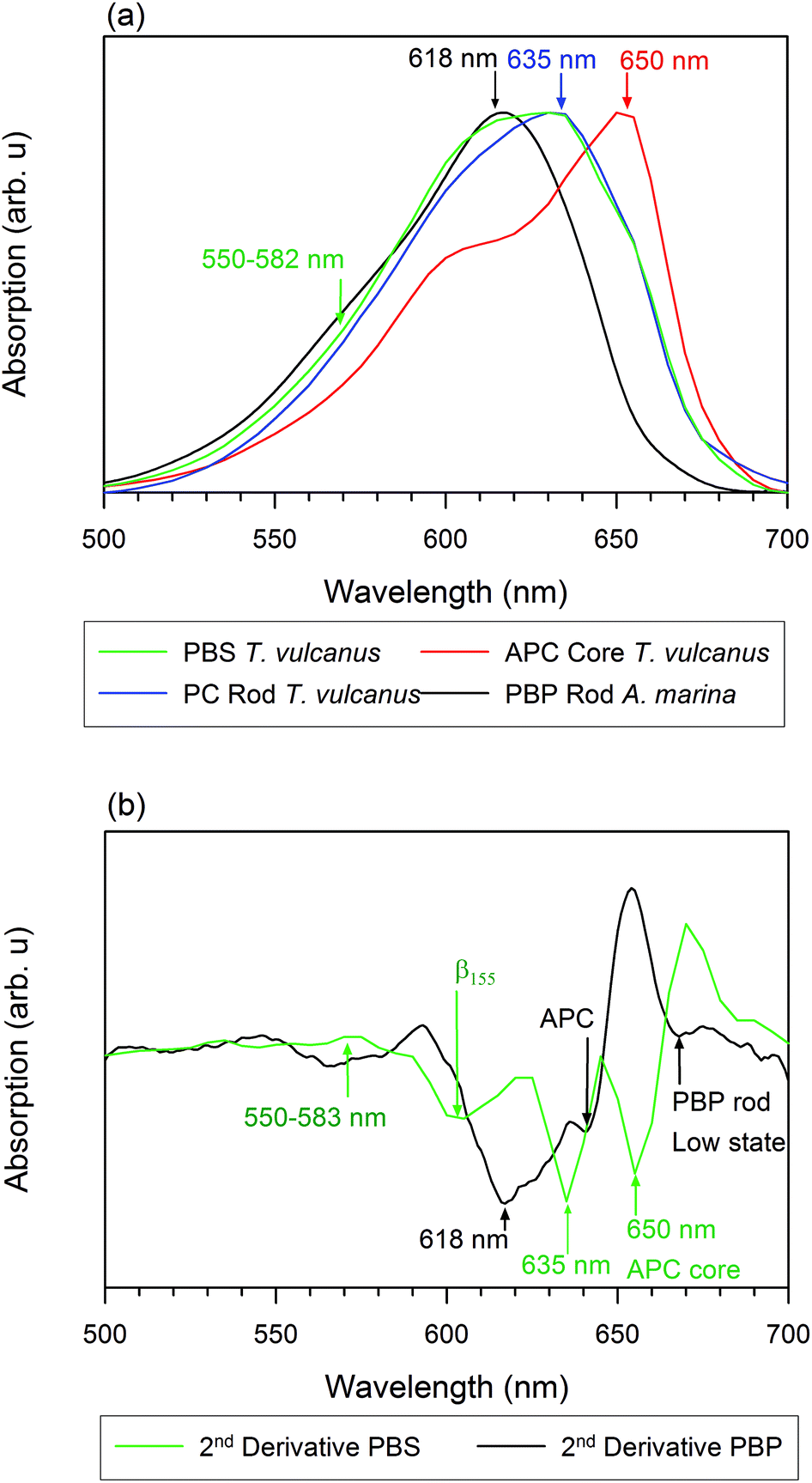 | ||
| Fig. 1 Absorption spectra of phycobilisomes: (a) absorbance of an intact PBS antenna system of T. vulcanus (dark green) in comparison to the absorbance of an isolated rod in T. vulcanus (blue), a core of T. vulcanus (red), and the PBP rod antenna system of A. marina (black) in the presence of high phosphate; (b) the second derivative spectra of the aforementioned antenna systems. | ||
Photo-induced absorption changes
To estimate the EET components in the PBS antenna system of T. vulcanus, in comparison to those in the PBP antenna system of A. marina, we excited samples at 620 nm to monitor the time-resolved absorption changes at different probe wavelengths. The choice of the excitation wavelength was based on the maximum of absorption peak of the PBS antenna system of A. marina for comparison purpose. The femtosecond transient absorption kinetic trace (ΔA spectrum) is different from the kinetic components, because the kinetic components is derived from fitting transient absorption kinetic trace into the kinetic model, eqn (1).21,26 In the section immediately below, we present the result of transient absorption traces (Fig. 2), while the section that follows after is devoted to the analysis of kinetic component. The reference therein gives more informative insight into the kinetic trace and its component analysis in transient absorption spectroscopy.26,27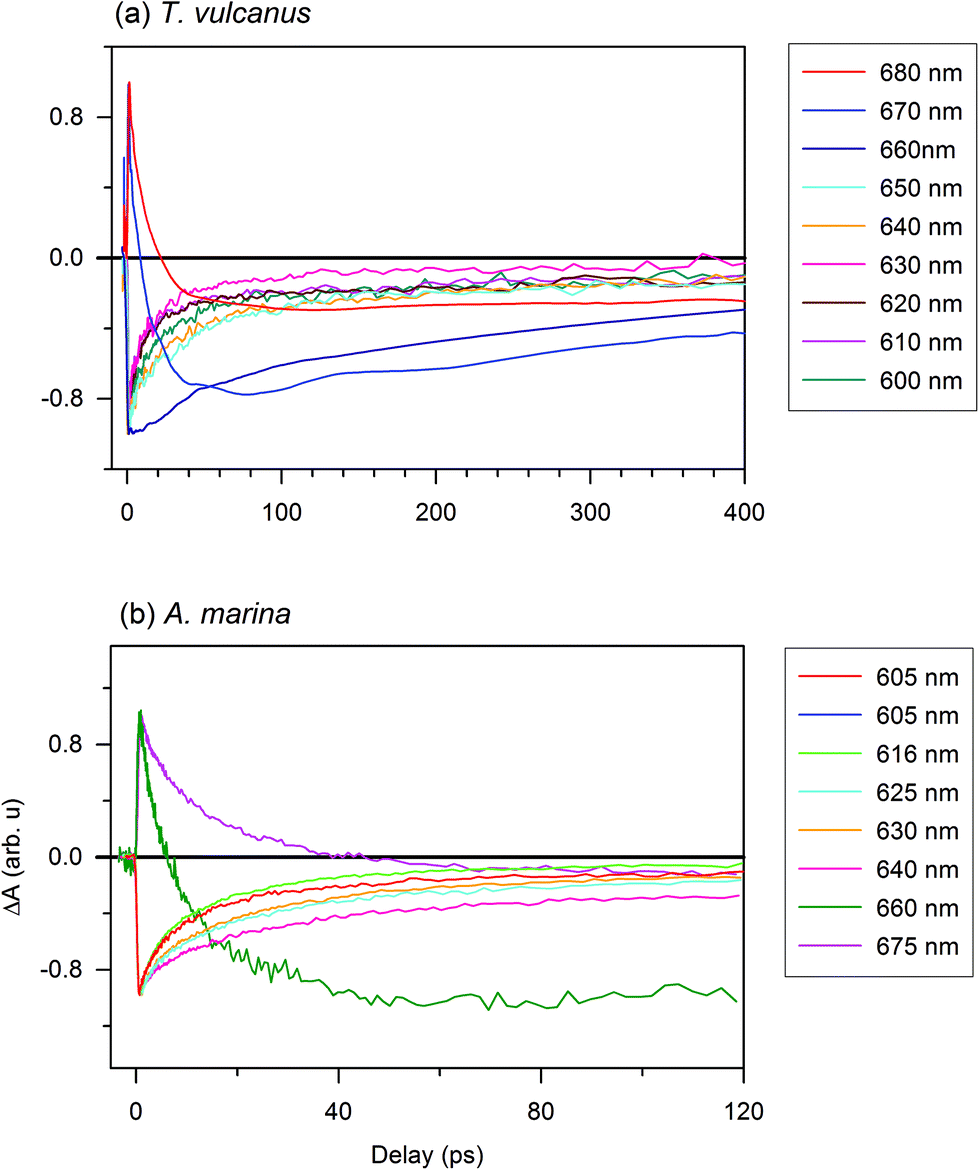 | ||
| Fig. 2 Time resolved absorption of PBS and PBP: (a) photo-induced absorption change of the PBS antenna system of T. vulcanus after excitation at 620 nm; and (b) the PBP rod antenna system of A. marina after excitation at 620 nm. | ||
An additional interesting observation is the evolution of the 630 nm component, which displays faster kinetics in the absorption change trace for PC rods. Based on the analysis in the previous section, the β84 with a maximum absorption at 630 nm is strongly coupled to its homologue in adjacent hexamers through the effects of the LR. The trace at 635 nm has also been shown to display the same behaviour as that at 630 nm.26 The 640 nm component shows the same recovery component as that of the APC core. The excited state decay of this pigment is likely coupled in a different way from that of other pigments in the PC upper states. Prior to this observation, Adir and Lerner suggested that PC612, unmethylated PC with an absorbance maximum at 612 nm (in its isolated trimeric form), is functional near the core.29 Furthermore, the presence of the LRC close to this spectral position might induce strong coupling between PC612 and the APC core. This component could serve as the real low energy state for PC rods.21
The bleaching observed in PC at 620 nm is the electronic transition of the PC620 inside the PC rod. This is the high energy level of PC Rod, which is induced by the direct excitation at 620 nm. The bleaching observed at different low energy levels of the intact PBS are the consequence of excitation energy transfer from PC620 to the sub-light harvesting complex, where PC620 does not absorb. Therefore, the current results revealing the time-resolved absorption changes shows that the presence of the different linkers optimizes the EET between adjacent PC rings of the PBS in T. vulcanus. These linkers connect and stabilize the two PC hexamers in the rod. Furthermore, the LRC, which connects the terminal rod hexamer to the APC core, also optimizes the EET from PC's low state to the APC core (also see ESI Fig. SI†). In Fig. 2a, the 660, 670 and 680 nm kinetic components appear at the redshifted electronic transition. These observation agree with Kuzminov et al. who explained that such red shift translation are due to coupling of β18 in APC-like trimer to α2 in the adjacent monomer, and also the coupling of LCM cofactors to β2 in the other adjacent monomer.30 The 660 nm transition is slower than all the kinetic traces between 600 and 650 nm. This means that the 660 nm transition is the activation of APC-like trimer, and the energy is transferred to 670 and 680 nm in the terminal emitter. Thus, the very quick and short-lived rise profile of both of 670 and 680 nm is attributed to excited state absorption (ESA) from PC; while the delayed bleaching that follows, is the absorption of the terminal emitter. This has been previously reported too.31 Kuzminov et al. explained that the two terminal emitters can be differentiated through their content of linker encoded apcE and apcD.30 Further, we earlier observed that the absorption peaks within the same region.9 Therefore, we exclude the possibility of stimulated emission (SE) influencing our current measurements (Fig. 2). Further, the two relaxation kinetics at 670 and 680 nm confirms the difference in terminal emitters vis-à-vis one with apcE and the other with apcD.
In order to identify the contribution of PC's and APC's ESA, the amplitude and time constant of the 3 kinetic components were globally fit into eqn (1) (Fig. S5†). Hence, the PC contribution at 670 nm and 680 nm is done by comparing the kinetics of PC ground state bleaching and the ESA profile fit. The obtained amplitude of the kinetic component of PC is an indication of the fraction population of excited PC that contributes to the ESA. Therefore, the second component of 670 and 680 nm can be attributed to a fraction of a photoexcited population that undergoes a slow vibrational cooling. Since the PBS system used in the current study are not connected to the RC, future studies using complete system with PBS connected to RC would give better mapping of the terminal kinetic component than in the isolated system.
We observed a further difference between the PBS in A. marina and T. vulcanus at the lowest state of A. marina, where we detected an absorption change as a result of pigments’ absorbing between 660 and 675 nm. Regarding the absorption of the A. marina PBS in Fig. 1, the observation reveals a weak absorption of APC.
In addition to the one-phonon profile of A. marina, APC fluorescence displays two red shift peak absorption bands at 660 and 673 nm (Fig. 3). Therefore, the regular APC trimer pigments absorbing at 645 nm are not functional at 660 or 673 nm. Regarding the evolution of the absorption change trace, two different states displayed at first glance different evolutions of the absorption change trace at 660 and 675 nm. There are two different states not located in the last hetero-hexamer, which some have suggested is the terminal emitter of the PBS in A. marina.21 Since the APC trimer already forms an excitonic absorption state at 645 nm, there is no more evidence of further localized APC trimer excitonic state. Therefore, the origin of the terminal emitter, or its pigment composition, remains an open issue. Further biochemistry investigation is necessary to identify the pigment composition of these states at 660 and 675 nm. Gindt et al. suggested that these pigments are as different as β18 (absorbing at 660 nm) and LCM (absorbing at 676 nm) in Synechococcus sp. 7002.32
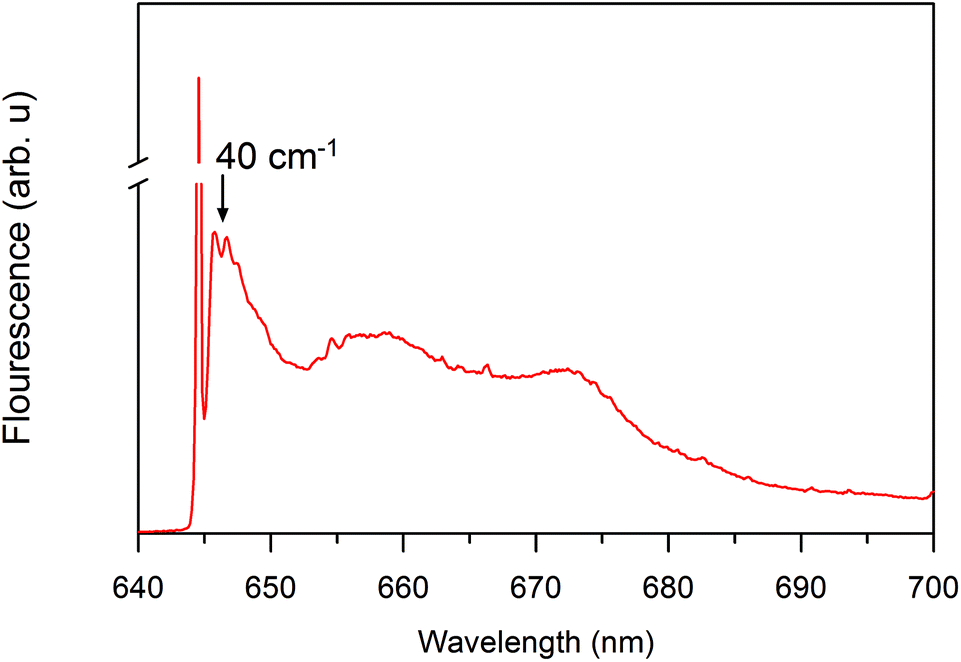 | ||
| Fig. 3 Site selective spectroscopy showing nonlinear narrowed and line-narrowed fluorescence spectrum33,34 of the A. marina PBS after excitation at 645 nm at 4.5 K. The sharp line, which is at the laser excitation, is partially contaminated by the scattered light. It is cut off to allow clear visualization of the non-resonant fluorescence contribution. | ||
Global analysis of the EET's kinetic components
In the PBS, the 888 fs kinetic component of the EET exhibits a negative spectrum between 600 and 650 nm and a positive spectrum between 650 and 690 nm. The negative spectral gap of the 888 fs component exhibits a minimum around 620 nm and a shoulder between 635 and 645 nm, where the maximum absorption is localized and where researchers have suggested the low energy state of the PC rod occurs.21 The zero cross-section of 888 fs is 652 nm, where the APC core absorbs (see Fig. 1). Therefore, after formation of the excited state, a fraction of PC pigments transfer the energy to the APC core pigment in 888 fs.
The positive spectral band of the 888 fs kinetic component in Fig. 4(a) can have different sources. These sources include the ESA of PC or the different behaviour of the increased ground state bleaching for low energy level compound pigments. These compounds can be the APC core or LCM. Despite the broad inhomogeneous absorption of the APC core, its absorption remains low (at 660 nm), where the upper electronic state of the linker membrane is functional. Furthermore, an increase in the ground state bleaching will induce a negative evolution of the kinetic component. At this specific absorption band (660 nm), a kinetic component above 1 ns is an indication of a trapping energy site attributed to a compound at a lower energy level than that of the APC core. Additionally, the ESA decays with three different kinetic components: 888 fs, 17 ps, and the one above 1 ns. The 17 ps is the kinetic component of the EET from the APC core to the LCM. We therefore expect that the positive spectral band of the 888 fs component can be mainly due to PC's ESA. The above 888 fs kinetic component is the fastest ever reported EET component from PC to APC in all previous studies of the PBS antenna system of cyanobacteria (Sandström22 and Zhang23). This 888 fs is more than three times faster than the component of the EET found in A. marina (see the A. marina section). From this section, we can observe the apparent contribution on the 888 fs EET that has the presence of the linker inside the system.
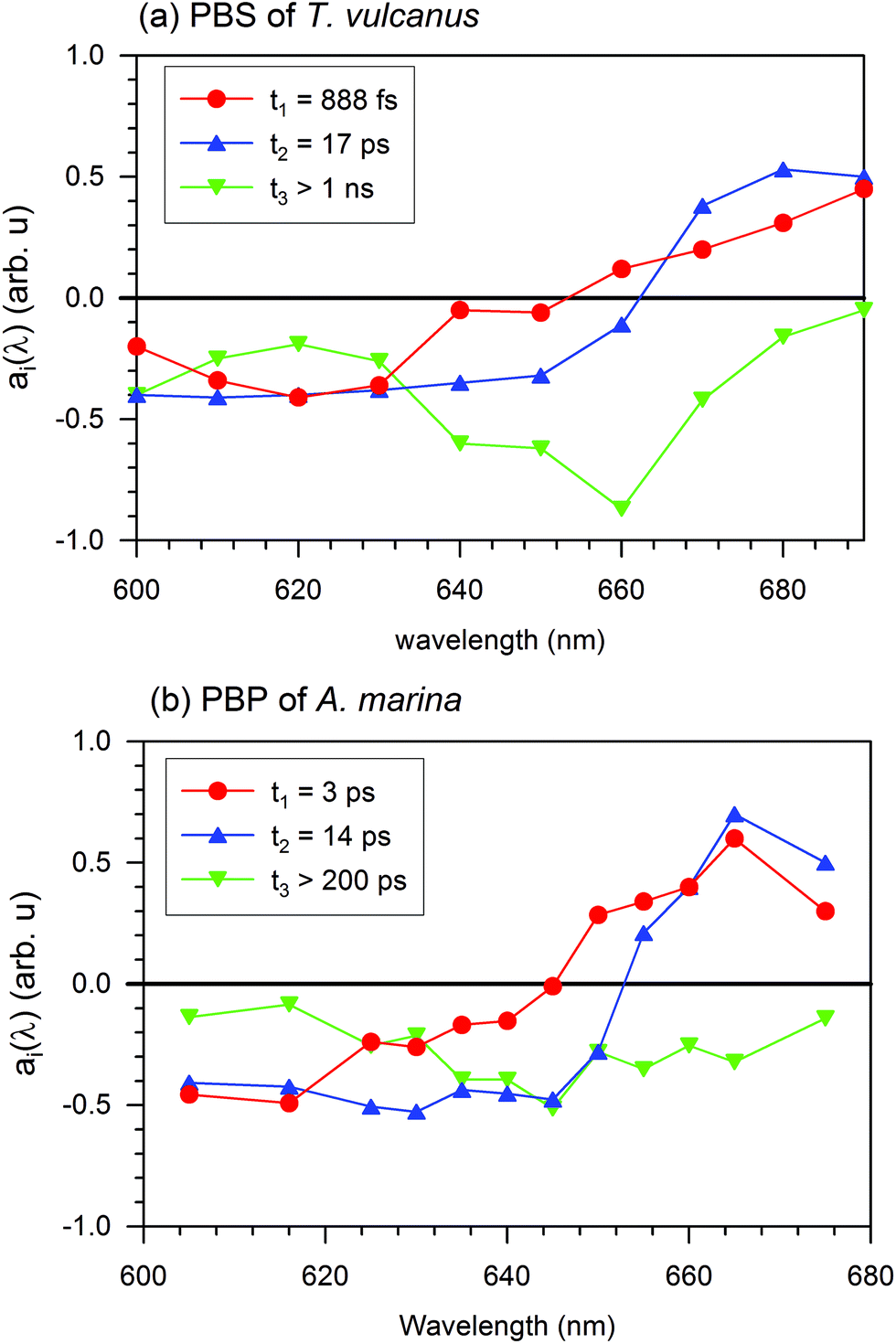 | ||
| Fig. 4 Decay analysis spectra: (a) EET components gathered by global analysis of the photo-induced absorption change in the PBS of T. vulcanus containing linker proteins in high phosphate and (b) the PBP of A. marina in high phosphate. | ||
In Fig. 4(a), the spectra for the 17 ps and 888 fs components of the EET have similar trends of having both negative and positive profiles. The 17 ps component has negative profile between 600 and 660 nm and a positive above 660 nm, while the 888 fs component has positive profile above 650 nm. In the positive profiles, the 17 ps component has higher amplitude, which exhibits a maximum between 670 and 680 nm, than the 888 fs component. The zero cross-section of the 17 ps component is 660 nm of the upper electronic state absorption in the LCM. These characteristics suggest that the 17 ps decay component reflects mainly an EET from a fraction of the high energy level of the APC in the PBS to the LCM, which are the low energy level as well as the terminal emitter component of the PBS. From there, the energy is expected to be transferred to the RC in a system with RC. For a review on PBSs from other cyanobacteria see Liu et al.,3 Lundell and Glazer.3,35,36 Such kinetics is on the same order as the 14 ps kinetic component Holzwarth,37 found. Since we performed the EET for an isolated PBS antenna system, the EET to the PS II and I RC experienced an interruption. This is in fair agreement with the prolongation of the excited state lifetime. This lifetime period exhibits a completely negative spectrum in all spectral ranges with a pronounced minimum at 660 nm, which is the non-resolved lifetime of the pigment trapping the EET at 660 nm. The minimum at 660 nm and the kinetic component above 1 ns suggest that the 660 nm band is consistent with the functionality of the LCM. Hence, the mechanism of the EET in the terminal emitter from the PBS to the RC of PS II and I experiences interruption in an isolated PBS system. The terminal emitter is therefore located at the LCM absorption energy level. The energy flow is trapped, and the kinetics reflects the relaxation dynamics of the LCM, as shown in Fig. 4(a) for the component τ3 > 1 ns. We cannot fully resolve this kinetic component with our experimental setup. However, the broad minimum of the evolution of this component at 660 nm of the zero cross-sections is another indication that the pigment absorbing at 660 nm has a different excited state lifetime than that for the APC core. According to the spectral position where it is functional, and based on Zhao et al.'s work,38 we suggest that it is the APC E membrane linker, which is present in the PBS of T. vulcanus.7 No additional information was obtained with 4 kinetic component of the EET (S4). Therefore, the DAS describes better the EET with 3 components not 4.
The fluorescence spectrum of the intact PBS of T. vulcanus after excitation of its high energy level (PC620) displays its emission on the PC612 electronic absorption spectrum between 640 and 645 nm. We observed the next peak of fluorescence line narrowing spectroscopy (FLN)33,34 at 660 nm, which Gindt et al. previously suggested was the β18 cofactor copy of the linker core membrane distinct from the APCE copy at 680 nm (the PBS's terminal emitter).32 The decay-associated spectra of the ground state bleaching are consistent with the fluorescence peak of different cofactors (see more details are discussed in sections below). The plateau we observed in the 888 fs is apparent between 640 and 645 nm as with the fluorescence peak of PC612. One can describe the absence of the peak between 650 nm and 655 nm as an increase in coupling strength between PC612 and the APC core. Gindt et al. indicated that the emission peak at 660 nm stems from the emission of APC in the APC core.32 This 660 nm peak, known as the electronic transition of the β18 cofactor, is likely to act as an intermediate energy site where the photoexcitation that migrated to the terminal emitter at 680 nm (APCE) may be trapped for regulation. We will investigate this regarding the fact that the cross-linked purification here discussed has been shown to supply the photoexcitation to both PSI and PSII.3
Target and quantitative relaxation dynamics of the PBS of T. vulcanus
To understand how the dynamics of each compound contribute to the EET or exciton in the PBS of T. vulcanus, we investigated the ground state bleaching in PC620 and PC612. The transient absorption (TA) bleach signal we obtained agrees within experimental uncertainty. For PC620, the high-energy transition at 620 nm displayed a relaxation amplitude of 30% and 70%. This transition is mainly located at the α84 cofactor's peak absorption.13 Since α84 and β84 are strongly coupled in the trimeric unit of the PC hexamer for fast EET, we can attribute the 30% of the relaxation dynamic to the deactivation of the S1 state at 620 nm toward the S1 state at the 630 nm activity mode, which is the transition of the β84 cofactor.21 We observed increased amplitude from 30% to 40% and a fast relaxation dynamic from 876 fs to 500 fs of the deactivation transition at 630 nm, which was similar to the transition at 635 nm. This is an apparent indicator of the change in coupling. As demonstrated in our previous communication,21 this coupling takes place between β84 in adjacent trimers as an intermediate state to promote exciton or energy transfer from PC620 to PC612 in 500 fs. We attributed the kinetic component above 1.00 ns to the lifetime dynamic of the transition of the cofactors α84 and β84 at 620 and 630 nm, respectively. Fig. 5 displays the activation and deactivation mechanism from the PC620 to PC612 transition in intact PBS. We detected the activation of the S1 state of the β84 in PC 612 as a result of the ground state bleaching of the TA signal's kinetic component. However, we observed a deactivation kinetic component of 890 fs not seen in the isolated PC rod.21 This further indicated that the coupling in PC612 has changed. Due to directional energy transfer from a high energy to a low energy level, the expected coupling was consistent with that of the APC core. This expectation was confirmed throughout the TA signal of the APC core at 650 nm, which appeared to be indistinguishable from that at 640 nm (also see ESI Fig. SI†). This is additional behaviour of the ground state bleach signal we did not observe in any of the previously investigated PBS pieces containing PC and APC compounds. The coupling between PC612 and APC in the 890 fs kinetic component validates the DAS excitation energy flow from the PC rod to APC core in 888 fs.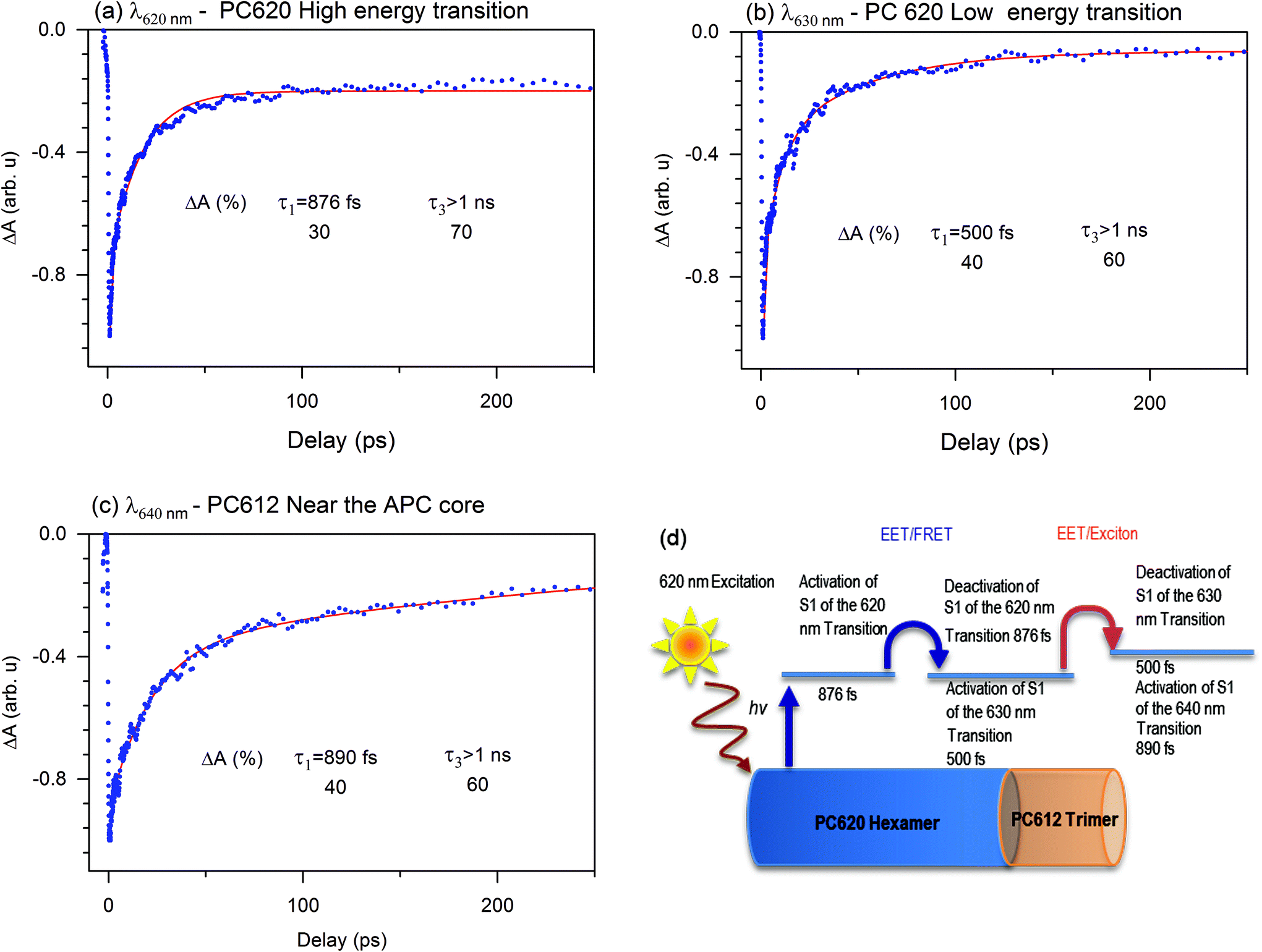 | ||
| Fig. 5 Photo-induced absorption change of the PBS antenna system of T. vulcanus at (a) 620 nm, (b) 630 nm, and (c) 640 nm after excitation at 620 nm. (d) Scheme of the activation and deactivation channel in the PC rod in T. vulcanus (for clarity, the APC core is not shown in the scheme). One hexamer represents PC620 at a high energy level of the PC rod, while the trimeric PC612 is located at the rod's low energy level. | ||
Regarding the coupling of 888 fs in light with previously studied PBS containing fragments of a PC hexamer bound to APC, the best kinetics obtained between PC and APC was 18 ps. In the PBS of A. marina, which researchers have considered until now to have a faster EET from PC to APC, the kinetic component was 3 ps component. Researchers claimed such kinetics to be faster because of the single rod geometry of the PBS of A. marina, where APC is present in the rod forming a hetero-hexamer with a PC trimer.9,31,39 This hypothesis appears to be not consistent in light of the current result where the difference in structure between the PBS of A. marina and that of T. vulcanus does not confer to the PC/APC coupling kinetic component a faster kinetic in the PBS of A. marina than that in the PBS of T. vulcanus.
Furthermore, the early investigations of excited state dynamics in PBS were done in the absence of linker proteins between PC and APC. We suspect the presence of the linker protein might change the coupling between the PC612 and the APC core. This coupling is stronger than that in all previously reported studies in PBS as it involves an isolated fragment of one PC hexamer connected to the APC core by a LRC.23 This coupling is especially strong with regard to the APC trimer fragment bound to PC612 via the linker rod-core (SI). We expected the excitation energy to migrate inside the APC core toward its deactivation in 17 ps, which started the LCM's activation mode (more details in sections below). The decrease in the kinetic component from 888 fs to 17 ps may indicate a weak coupling between the LCM and the APC core. These couplings have been discussed before by Kuzminov et al.30Fig. 5 illustrates the relaxation mechanism between the PC rod and the APC core. Therefore, the excitation that triggers the EET in PBS cannot be well described in absence of some sub-compounds as linker. It is appears that the PBS needs the fast relaxation dynamics from PC to APC core system as an adaptation strategy to environment where light intensity is low.
Influence of the coupling distance in PC rod
Using visual molecular dynamics (VMD), we have reconstructed the rod of the T. vulcanus PBS model (see Fig. 6). The rod was based on a single hexamer of PC620 (pdb 3o2c) and a trimer of PC612 (pdb 1on7) without the three different linker proteins David et al. reported.24 Since these linkers have not yet been crystallized, and isolating them is still a challenge, we obtained the distance between pdb 3o2c and pdb 1on7 with the help of the ruler algorithm in VMD. The model using ruler algorithm predicted that the relative distance between PC620 and PC612 cofactors is as close as 21 ± 1 Å to induce strong dipole–dipole coupling. The π-stacking distance that occurs between the two adjacent β84 of PC620 and PC612 cofactors at their interface was observed at AM1 level of theory, with the ring A frozen to have a fraction of their D ring population below 20 ± 1 Å (result not shown).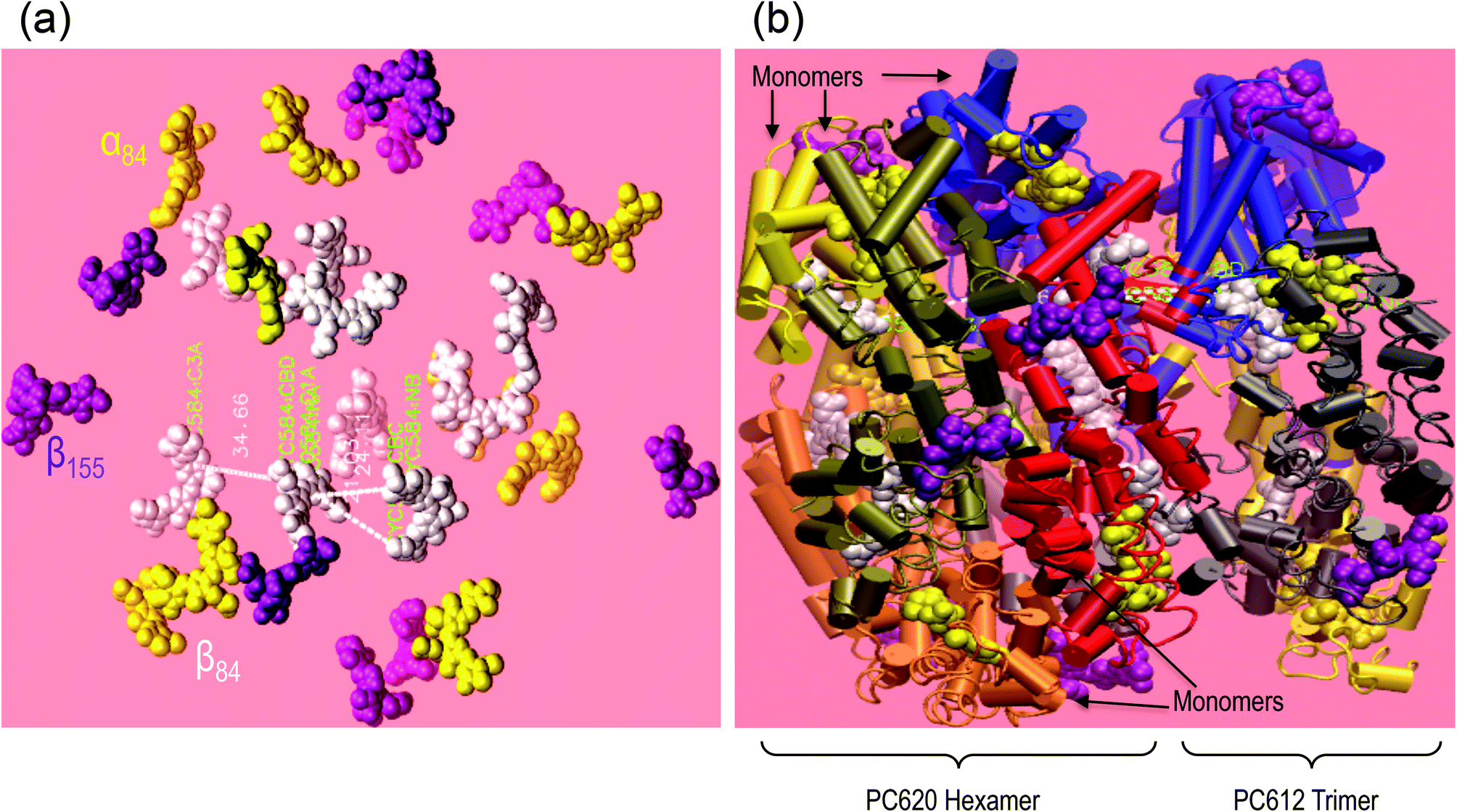 | ||
| Fig. 6 Model of the PC rod form with one APC trimer of T. vulcanus's PBS. The cofactors are shown on the right: The β155, α84, and β84 (PC rod cofactors) are displayed in purple, yellow, and white, respectively, while the APC's α84 and β84 cofactors are displayed in pink and green, respectively. The distance between adjacent β84 cofactors in PC612 and APC is 36.28 Å. | ||
MacColl reviewed this dipole coupling between α84 and β84 in adjacent monomers in the APC trimer.25 With support from the literature and the circular dichroism spectrum of the APC trimer, MacColl suggested that the peak absorption in APC monomer cofactors (α84 and β84) at the 615 nm shift to 650 nm stems from strong dipole–dipole coupling due to excitonic splitting. MacColl suggested excitonic splitting with a relative distance of ∼21 Å between α84 and β84 in adjacent monomers in a specific environment.25 MacColl attributed the shoulder at 620 nm, as observed in Fig. 1a (APC core), to the higher energy state for the split spectrum.25
In the present case of the intact T. vulcanus PBS, the PC rod's peak absorption appears at 635 nm (Fig. 1a, b and 8). This was different from the isolated PC trimer's peak absorption at 620 nm,21,24 which is also the absorption peak for PC620. Debreczeny et al. showed the spectrum shoulder observed at 630 nm to yield the β84 cofactor's absorption peak,13 which was recently observed in the PBS rod of A. marina.9 This would likely have supported the work of Hu et al., who did not find two copies of PC in the PBS of A. marina.39 Furthermore, the low absorption of A. marina PBS does not have this 635 nm peaks9 which suggests that it appears at the interface between PC612 and PC620. Since the PBS of T. vulcanus has two different copies of PC, the PC620 and PC612 Adir et al. found,24,29 the assembly of these PCs may be the result of the peak absorption at 635 nm, if the relative distance between adjacent cofactors in PC620 and PC612 is short enough to induce strong dipole–dipole coupling. From results we obtained from VMD simulation, we observed that the relative distance between adjacent β84 in PC620 and PC612 is about 21 ± 1 Å. This distance between the ring D of the β84 in PC612 and the centre of its homologue β84 in PC620 is apparently in the same order of value from 21 ± 1 to 20 ± 1 Å when we considered the measured distance from ring D (β84 in PC620) to ring D (β84 in PC612). However, the distance from ring A of the β84 in PC612 to the centre of the adjacent β84 in PC620 appears to be 24.11 Å. In our previous study, we found that the ring A has a limited degree of liberty, which contributes less to the conformation promoting the face-to-face coupling between adjacent β84 in adjacent trimers.21 Therefore, the distance between β84 of PC620 and PC612 (Fig. 7) supports that the peak absorption at 635 nm is due to strong coupling between PC620 and PC612 via β84, where the higher energy is at 630 nm and the lower at 635 nm.
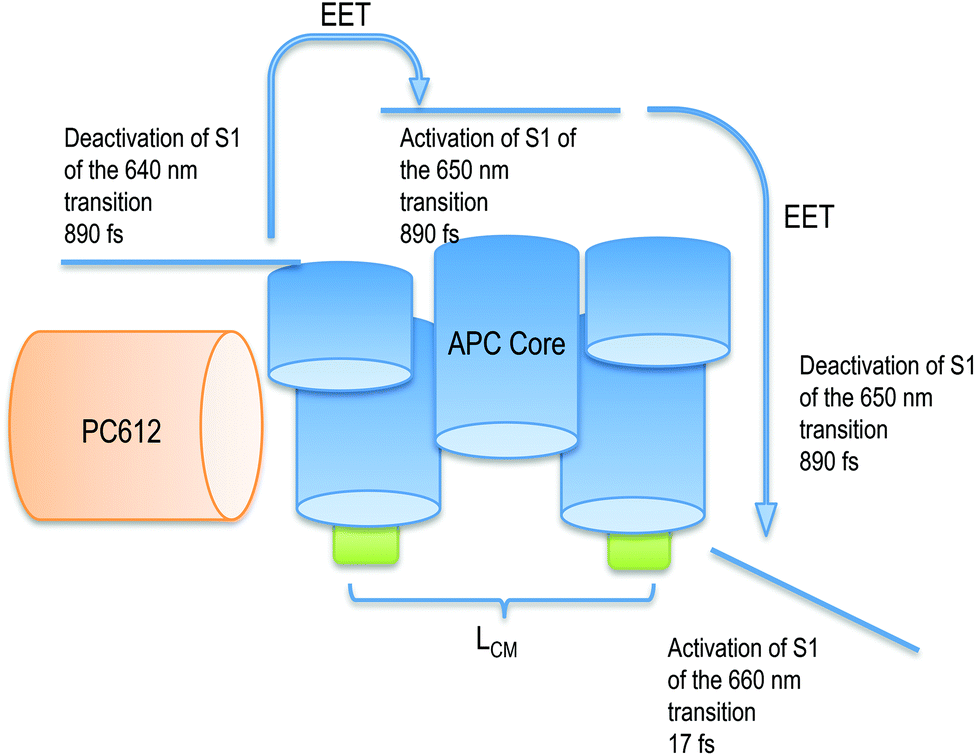 | ||
| Fig. 7 Scheme of the activation and deactivation channel from the PC rod to the APC core after activation of PC612. The deactivation of APC is followed by the activation of LCM. | ||
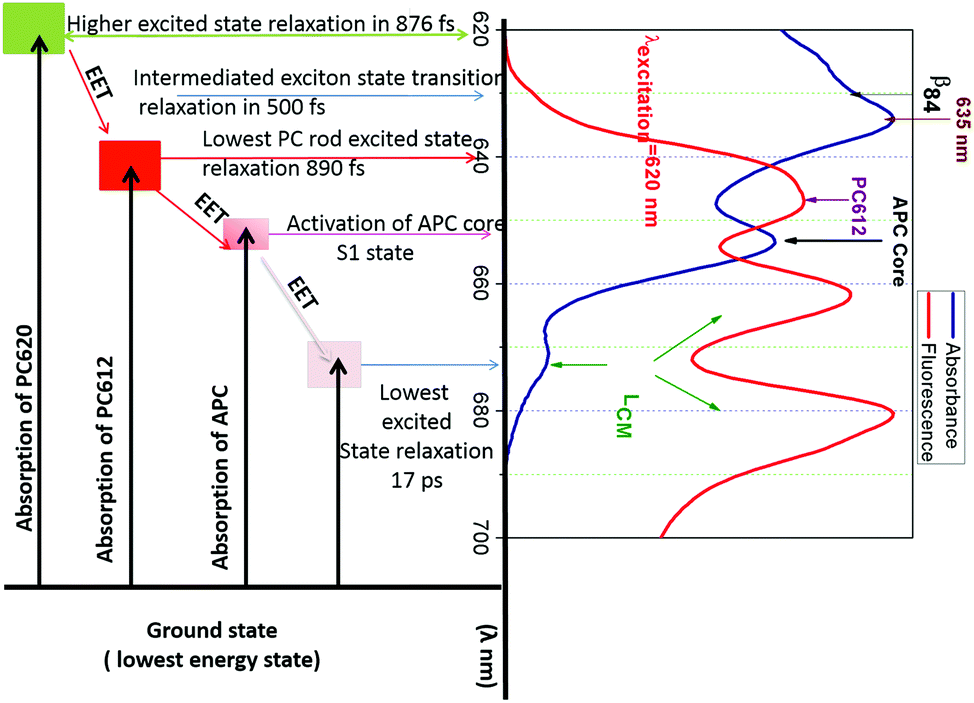 | ||
| Fig. 8 Photoexcitation migration in intact PBS of T. vulcanus. The low temperature absorption shows a distinct peak for the absorption trace of the terminal emitter (LCM), while the one phonon profile of the intact PBS after excitation at 620 nm (PC620) portrays a fine view of two electronic absorption peaks at 660 nm and 680 nm, which is indication of the LCM activity. | ||
The change in the kinetic component from 876 to 500 fs and their kinetic amplitude enhance the role played by the assembly via the different linkers, which maintain the PC620 hexamer and PC612 trimer in a strong coupling conformation. The change of the kinetic amplitude rises with 14.3%. The gain in kinetic from 876 to 500 fs at 635 nm is slower than the 370 fs exciton relaxation in PC calculated by Sauer.16 Despite that we detected an average kinetic trace of the ground state depletion, which is both the EET and the exciton dynamics. Therefore, the 14.3% increase in amplitude of 500 fs is due to the exciton dynamics at 635 nm that contribute to transfer population.21 According to the coulombic interaction, the exciton π-stacking energy, which is an integration over the π-stacking D ring distance (between the adjacent β84), is expected to increase at low interaction distance of the stacking D ring.6,25,40 This may give the exciton the capacity of crossing the thermal fluctuation in weak exciton coupling diffusion. Since this mechanism is for prime importance in nano-sciences and beyond the scope of this work, it will be investigated elsewhere.
The linker connecting PC620 to PC612 is responsible for the assembly of PC620 and PC612 with all the pieces into the PC rod. The linker-PC rod and linker rod-APC core probably act on the distance between PC612 and the APC core for strong coupling toward the deactivation and activation modes in APC with the same conformation as in PC612. We discussed this activation and deactivation process in our previous work in the case of an isolated PC rod.21 Reuter et al. suggested that the linker found on the cavity axis (C3) interacts with two of the β84 cofactors and brings the cofactors close to their adjacent homologues, which induces a change in the spectra.41 We suspect that this change is driven by ring D of these cofactors in close proximity because ring A has less degree of liberty than D.21 Furthermore, Moran et al. investigated the exciton structure that defines the chromophore interaction in APC. They found that exciton states are 96% localized to the individual molecular site, which exist in dimers. However, they were not able to find the same trend of exciton in PC, and we believe that this was due to the absence of linker in their PC system as they used isolated pieces of either PC or APC. They further showed evidence of dominant internal channel activity via hydrogen out-of-plane (HOOP) wagging, which Womick et al. suggested to favour a quick and efficient relaxation of the photoexcitation from the high energy to the lower level exciton.19 However, we earlier found that the complete activation and deactivation mode can only be captured in intact PC rod.21 The high energy level of the exciton was earlier suggested to be at 620 nm,25 the shoulder observed in the APC absorption spectrum, while the low energy exciton was thought to be localized around 650 nm. Prior to the work of Moran et al., it was believed that the exciton was delocalized along the two states.19,25
For PC, we observed a vibronic localized state in the PC rod of A. marina, especially in the interface between two hexamers. Fig. 8 illustrates the route of the photoexcitation migration in intact PBS (also recall influence of the coupling distance in PC rod outlined in sections above). The localized state occurred at 635 nm, which is a 5 nm red shift from the β84 cofactor.9 This state was not apparent at room temperature in the A. marina PBS, but was in the PBS of T. vulcanus. Furthermore, the inhomogeneous broader spectrum at room temperature of the PBS of T. vulcanus does not allow for the discrimination between the high and the low exciton states that may be formed in PC. Decreasing the temperature of the medium to around 4 K, the cofactors overcome the thermal fluctuation that inhibits the observation of the high exciton state at 630 nm (Fig. 8). The low absorption spectrum of the intact PBS of T. vulcanus displays an apparent sharp peak at 635 nm similar to the one for APC at 650 nm. However, the shoulder, which may express the high exciton state, appears at 630 nm. This is in line with the localized exciton state at 635 nm at the A. marina PC hexamer interface. The localized vibronic coupling of 51 cm−1 in PC, as compared to that of APC of 40 cm−1, is weak.9,18,19 But, the difference of 11 cm−1 is an indication of the strength of the coupling that is present in APC and PC. The distances between this cofactor are the same in PC and APC. Earlier, Zhang et al. studied the kinetic trace of components from PC to APC in extracted PBS and reported the fastest component of 18 ps.24 If the linker was not present to enhance the photochemistry that trigger the fast 888 fs component from PC rod to APC core in our current investigation, Zhang et al. would also have detected a kinetic trace with a component faster than 18 ps. Therefore the linker is important not only for the assembly of the PBS, but also for the EET process.
Experimental
Samples
We grew the A. marina strain MBIC11017 in a seawater K medium42 and the cultures at 26 °C with filtered air bubbling and shaking. We ensured continuous illumination using the bias of a white fluorescence tube with a light intensity of 20 μE.11 To carry out the femtosecond pump–probe experiment with a high and low ionic strength, we buffered the PBP antenna system with and without phosphate, respectively.We isolated the PBP antenna complexes of A. marina from the cultures of A. marina using procedures similar to those Hu et al. described previously.39 For the isolation of the PBS of T. vulcanus, we re-suspended 12 litres of harvested cells in a 0.9 M phosphate buffer (pH 7.0) and disrupted them using a French pressure cell treatment followed by centrifugation at 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm. We then re-suspended the pellet with a 0.9 M phosphate buffer, incubated it for 1 hour with 2% Triton X-100 (w/v, sigma), and then performed clarification by centrifugation. We centrifuged the resulting supernatant (Beckman Coulter, optima L-90 K ultracentrifuge, Beckman Type T70.1 rotor) in 10 ml tubes for 2 hours at 35000 rpm. Then we re-suspended the resulting blue pellet with a 0.9 M phosphate buffer, placed it on a 0.8 M sucrose cushion, and centrifuged it for 2 hours at 40000 rpm. We next re-suspended the resulting pellet with a 0.9 M phosphate buffer and placed it on a two-step sucrose gradient constructed with 1 M and 1.2 M sucrose in the presence of a 0.9 M buffer. We centrifuged the gradient sucrose overnight at 35000 rpm. We isolated three different bands from the sucrose gradient and characterized them by absorption (Varian spectrophotometer – Cary Bio 50) and fluorescence spectroscopy (Flourolog, excitation at 580 nm, with slit width of 5 nm for excitation and 1 nm for emission).
000 rpm. We then re-suspended the pellet with a 0.9 M phosphate buffer, incubated it for 1 hour with 2% Triton X-100 (w/v, sigma), and then performed clarification by centrifugation. We centrifuged the resulting supernatant (Beckman Coulter, optima L-90 K ultracentrifuge, Beckman Type T70.1 rotor) in 10 ml tubes for 2 hours at 35000 rpm. Then we re-suspended the resulting blue pellet with a 0.9 M phosphate buffer, placed it on a 0.8 M sucrose cushion, and centrifuged it for 2 hours at 40000 rpm. We next re-suspended the resulting pellet with a 0.9 M phosphate buffer and placed it on a two-step sucrose gradient constructed with 1 M and 1.2 M sucrose in the presence of a 0.9 M buffer. We centrifuged the gradient sucrose overnight at 35000 rpm. We isolated three different bands from the sucrose gradient and characterized them by absorption (Varian spectrophotometer – Cary Bio 50) and fluorescence spectroscopy (Flourolog, excitation at 580 nm, with slit width of 5 nm for excitation and 1 nm for emission).
Low temperature investigations
To achieve a better optical density at 4 K, we added 70% of glycerol to perform a low temperature measurement. We conducted this low temperature experiment (absorption and fluorescence) using the Spectra Physics model 375 dye laser (line width of <0.5 cm−1), which is pumped by an Ar-ion laser (model 171, Spectra Physics, USA). We kept the samples in He-bath cryostat (Utreks, Ukraine), in a plastic cuvette with an 8 mm optical path, and above the level of liquid helium at 4.2 ± 0.2 K. We recorded the presented spectra using a 0.3 m spectrograph (Shamrock SR-303i, Andor Technology, UK), which was combined with an electrically cooled CCD camera (DV420A-OE, Andor Technology, UK).Femtoseconds investigations
The pulses generated by Tsunami resonator (Spectra physics) are stretched before entering a Ti: sapphire RegA amplifier (in Spitfire, Spectra physics), which provides an 800 nm beam in 1 KHz with a pulse energy of 1 J and 100 fs FWHM after re-compression. The output beam from the Spitfire entering an optical parametric amplifier is capable of generating beams in the 475 up to 800 nm region with pulse energy of 1–35 nJ at a repetition rate of 1 kHz and 120 fs FWHM. A fraction of the beam entering the OPA is focused on a sapphire plate to generate a white light continuum (probe beam). The pulse generated from the OPA is sent to a variable delay line and made to overlap with the probe beam at the sample spot (150 μm).We placed the sample of the PBS antenna complex in a 2 mm path silica cuvette and used a magnetic stir bar to homogenize the sample's illumination. We used two photodiodes connected in a difference circuit to detect the probe light. We set the polarization of the pump laser beam to the magic angle (54.7°) with respect to the probe beam. The measured FWHM of the beam was ∼160–200 fs at the sample spot. The fluency was 1.7 × 1013 photons per cm−2. Using the procedure Womick et al. described,18 we estimated an average 0.62 excitations per hexamer. We increased the pulse energy without any observed influence. There was no apparent illumination-induced aggregation formation of the PBS in the buffer. More information on the time resolve and the fluorescence used in the study can be found Nganou et al.21
Excitation and probing the samples
A 10 nm full-width half-maximum (FWHM) was used to excite PC at 620 nm, and we successively probed the system with 10 nm FWHM. It was apparent that APC was excited through excitation energy transfer from PC at 620 nm to APC at 652 nm. The detection signal of APC contained information on the coupling between PC and APC. Hence, the overall system dynamic was monitored by this procedure, and individual bleaching dynamics were recorded. The dynamics of APC were well observed in PBS of T. vulcanus (652 nm) and PBS of A. marina (640 nm).Decay associated spectra of the ground state bleaching
The DAS consist of a global analysis of the available lifetime components found in the sample, enabling clear visualization of the spectral dynamics of the excited states of the system. When the wavelength-dependent form of the amplitude at each time constant rises from negative to positive, it can be interpreted as energy transfer, provided that the wavelength at which the amplitude crosses zero coincides with an absorbing electronic state. For each lifetime, we observe the evolution of the amplitude as a function of wavelength. The following results of global and independent fit were acquired using a multi-exponential decay model: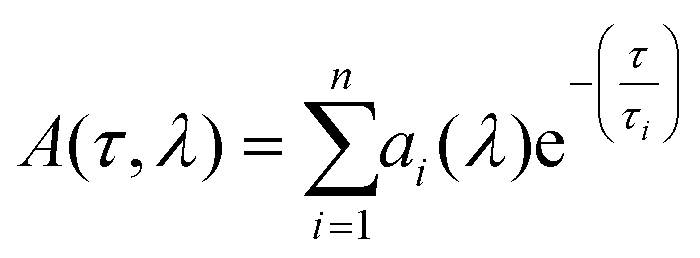 | (1) |
The kinetics at different wavelength intervals were fitted together, with the common values of the lifetimes τi as a linked parameter; the wavelength dependent amplitude factor ai(λ) was set as a non-linked parameter. Due to the considerably low IRF of 280 fs, its influence on the kinetic components of 888 fs is negligible – i.e. it allows the quantitative measurement of 888 fs. The noise contributed a time constant error of 0.18 ps.
Conclusions
In summary, the current work has demonstrated the ultrafast kinetic components of the EET along the PC rod domain containing linker proteins to the APC core domain in the T. vulcanus PBS antenna system, which has never been reported before in an intact PBS system. The EET for the PBS of T. vulcanus is ∼888 fs, in comparison to the kinetics found in A. marina (3 ps component) and in other cyanobacteria species, which all displayed a slower EET component in high phosphate.The prior maximum absorption of the PBS was 635 nm; the linker proteins embedded in the rod changed the absorption spectrum of the pigments in the antenna system. These linkers have also optimized the EET components between sub-compounds in the PBS.
Furthermore, the fast kinetics of the 888 fs is too fast to be explained by the FRET-type mechanism. Rather, such kinetics must stem from an excitonic relaxation along the rod. We therefore indicated that the LR and LRC have not only optimized the EET pathway between PC and APC but also induced strong coupling between low and high state pigments in adjacent sub-compounds located face to face along the symmetry axis. Additionally, the LR linker has induced an excitonic splitting to 635 nm as result of strongly coupling β84 pigments in adjacent hexamers. We will address the rate of this excitonic coupling in the future as well as the coupling between β155 in adjacent PC rods. The 17 ps component is shown to transfer energy from APC in the core to the upper electronic state of the terminal emitter linker membrane between 660 nm and 670 nm. In comparison to the 14 ps component from APC to the terminal emitter in the A. marina PC rod, the 17 ps component of EET is slower. This forms a stark contrast with EET from PC to APC. The latter is 3 ps component in the A. marina PC rod. Although we observed a slow 3 ps component in the PBS of A. marina, this antenna system also contains different linkers in PC. We performed both experiments under the same experimental conditions. Since the purification differs, this may be the key factor needed by the assembly of the PCB cofactor in PBS for a faster kinetics. Thus, it would be recommended to investigate the kinetic components using PBS of A. marina pre-washed with a cross-linked buffer. To fast regulate the EET from the high energy level sub-compound (PC) to the low energy sub-compound in the APC core, the PBS need its entire compounds to be present. This seems to play a crucial role in the adaption of the organism to sustain photosynthesis in a harsh medium. The details are subject of discussion elsewhere.
Acknowledgements
This work was supported by DAAD (fellowship to C.N.), the Dora Grant (to C.N.), the USA-Israel Binational Science Foundation (to N.A.) (2009406), the Israel Science Foundation Grant to N.A. (1576/12), the Technische Universitaet Berlin, the Department of Biophysics at the University of Tartu, the computational facilities at the University of Rochester (USA) and ACENet (Canada), and the ECBC support to the Industrial Research Chair of Mine Water (held by M.M.) at Cape Breton University. The authors are very thankful to Prof. Dr. Christian Thomsen for his support and advice; Prof. Dr. Rienk Van Grondeller for the helpful technical insight; and, Prof. David W. McCamant of Chemistry Department at University of Rochester for his kind support.Notes and references
- Z. Petrasek, F.-J. Schmitt, C. Theiss, J. Huyer, M. Chen, A. Larkum, H. J. Eichler, K. Kemnitz and H.-J. Eckert, Photochem. Photobiol. Sci., 2005, 4, 1016–1022 CAS.
- C. W. Mullineaux, Photosynth. Res., 2008, 95, 175–182 CrossRef CAS PubMed.
- H. Liu, H. Zhang, D. M. Niedzwiedzki, M. Prado, G. He, M. L. Gross and R. E. Blankenship, Science, 2013, 342, 1104–1107 CrossRef CAS PubMed.
- R. MacColl, J. Struct. Biol., 1998, 124, 311–334 CrossRef CAS PubMed.
- A. N. Glazer, Biophys. Biophys. Chem., 1985, 14, 47–77 CrossRef CAS PubMed.
- N. Adir, Photosynth. Res., 2005, 85, 15–32 CrossRef CAS PubMed.
- L. David, M. Prado, A. A. Arteni, D. A. Elmlund, R. E. Blankenship and N. Adir, Biochim. Biophys. Acta, Bioenerg., 2014, 1837, 385–395 CrossRef CAS PubMed.
- Z.-W. Yi, H. Huang, T.-Y. Kuang and S.-F. Sui, FEBS Lett., 2005, 579, 3569 CrossRef CAS PubMed.
- C. Nganou, J. Chem. Phys., 2013, 139, 045101 CrossRef PubMed.
- J. Marquardt, H. Senger, H. Miyashita, S. Miyachi and E. Mörschel, FEBS Lett., 1997, 410, 428 CrossRef CAS PubMed.
- M. Chen, M. Floetenmeyer and T. S. Bibby, FEBS Lett., 2009, 583, 2535–2539 CrossRef CAS PubMed.
- S. Miyachi, K. Strassdat, H. Miyashita and H. Senger, Z. Naturforsch., C: J. Biosci., 1997, 52, 636–638 CAS.
- M. P. Debreczeny, K. Sauer, J. Zhou and D. A. Bryant, J. Phys. Chem., 1993, 97, 9852–9862 CrossRef CAS.
- R. Swanson and A. Glazer, Anal. Biochem., 1990, 188(2), 295–299 CrossRef CAS PubMed.
- A. McGregor, M. Klartag, L. David and N. Adir, J. Mol. Biol., 2008, 384, 406–421 CrossRef CAS PubMed.
- K. Sauer and H. Scheer, Biochim. Biophys. Acta, Bioenerg., 1988, 157–170 CrossRef CAS.
- M. D. Edington, R. E. Riter and W. F. Beck, J. Phys. Chem., 1995, 99, 56995–15704 CrossRef.
- J. M. Womick and A. M. Moran, J. Phys. Chem. B, 2009, 113, 15771 CrossRef CAS PubMed.
- J. M. Womick and A. M. Moran, J. Phys. Chem. B, 2009, 113, 15747–15759 CrossRef CAS PubMed.
- J. M. Womick, S. A. Miller and A. M. Moran, J. Chem. Phys., 2010, 133, 024507 CrossRef PubMed.
- C. Nganou, L. David, R. Meinke, N. Adir, J. Maultzsch, M. Mkandawire, D. Pouhè and C. Thomsen, J. Chem. Phys., 2014, 140, 085101 CrossRef CAS PubMed.
- Å. Sandström, T. Gillbro, V. Sundström, R. Fischer and H. Scheer, Biochim. Biophys. Acta, Bioenerg., 1988, 933, 42–53 CrossRef.
- J.-M. Zhang, J.-Q. Zhao, L.-J. Jiang, X.-G. Zheng, F.-L. Zhao and H.-Z. Wang, Biochim. Biophys. Acta, Bioenerg., 1997, 1320, 285–296 CrossRef CAS.
- L. David, A. Marx and N. Adir, J. Mol. Biol., 2011, 405, 201–213 CrossRef CAS PubMed.
- R. MacColl, Biochim. Biophys. Acta, Bioenerg., 2004, 1657, 73–81 CrossRef CAS PubMed.
- R. Berera, R. V. Grondelle and J. T. M. Kennis, Photosynth. Res., 2009, 101, 105–118 CrossRef CAS PubMed.
- A. C. Nganou, L. David, N. Adir, D. Pouhe, M. J. Deen and M. Mkandawire, Photochem. Photobiol. Sci., 2015, 14, 429–438 CAS.
- V. Mikas, S. L. Delmar, V. Leonas, K. Gerdenis, H. Christian, G. Devens, M. Thomas, M. Ana and G. van Rienk, J. Phys. Chem. B, 2013, 117, 11372–11382 CrossRef PubMed.
- N. Adir and N. Lerner, J. Biol. Chem., 2003, 278, 25926–25932 CrossRef CAS PubMed.
- F. I. Kuzminov, Y. V. Bolychevtseva, I. V. Elanskaya and N. V. Karapetyan, J. Photochem. Photobiol., B, 2014, 133, 153–160 CrossRef CAS PubMed.
- C. Theiss, F.-J. Schmitt, J. Pieper, C. Nganou, M. Grehn, M. Vitali, R. Olliges, H. J. Eichler and H.-J. Eckert, J. Plant Physiol., 2011, 168, 1473–1487 CrossRef CAS PubMed.
- Y. M. Gindt, J. H. Zhou, D. A. Bryant and K. Sauer, Biochim. Biophys. Acta, Bioenerg., 1994, 1186, 153–162 CrossRef CAS.
- R. Jankowiak and G. J. Small, Anal. Chem., 1989, 61, 1023A–1031A CrossRef CAS PubMed.
- V. Zazubovich and R. Jankowiak, in Photonics, John Wiley & Sons, Inc., 2015, pp. 129–164, DOI:10.1002/9781119011804.ch4.
- D. J. Lundell and A. N. Glazer, J. Biol. Chem., 1983, 258, 8708–8713 CAS.
- M. Mimuro, C. Lipschultz and E. Gantt, Biochim. Biophys. Acta, Bioenerg., 1986, 852, 126–132 CrossRef CAS.
- A. R. Holzwarth, E. Bittersmann, W. Reuter and W. Wehrmeyer, Biophys. J., 1990, 57, 133–145 CrossRef CAS PubMed.
- K.-H. Zhao, P. Su, S. Bfhm, B. Song, M. Zhou, C. Bubenzer and H. Scheer, Biochim. Biophys. Acta, 2004, 1706, 81–87 CrossRef PubMed.
- Q. Hu, J. Marquardt, I. Iwasaki, H. Miyashita, N. Kurano, E. Morschel and S. Miyachi, Biochim. Biophys. Acta, Bioenerg., 1999, 1412, 250–261 CrossRef CAS.
- R. G. Fisher, N. E. Woods, H. E. Fuchs and R. M. Sweet, J. Biol. Chem., 1980, 255, 5082–5089 CAS.
- W. Reuter, G. Wiegand, R. Huber and M. E. Than, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 1363–1368 CrossRef CAS.
- H. Miyashita, H. Ikemoto, N. Kurano, K. Adachi, M. Chihara and S. Miyachi, Nature, 1996, 383, 402–402 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Comparison of ground state recovery trace between 640 and 650 nm components in intact PBS. See DOI: 10.1039/c5pp00285k |
| This journal is © The Royal Society of Chemistry and Owner Societies 2016 |