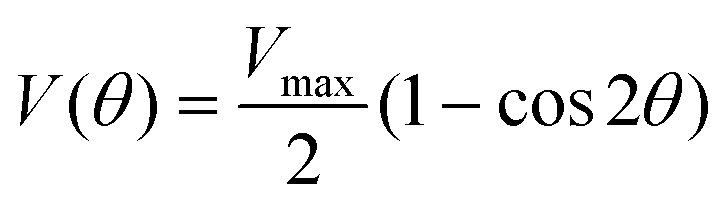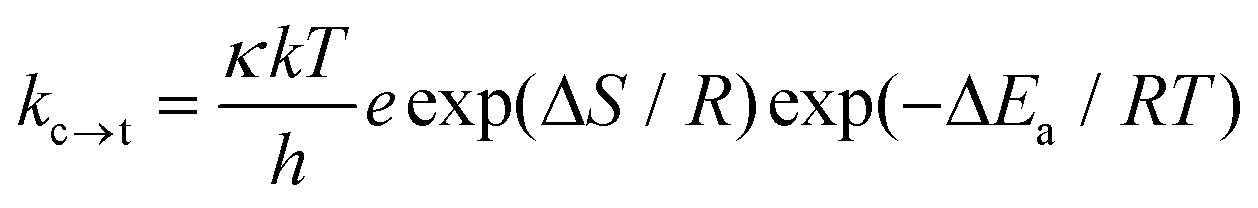Photophysical properties of symmetrically substituted diarylacetylenes and diarylbuta-1,3-diynes†
Received
7th May 2015
, Accepted 7th November 2015
First published on 13th November 2015
Abstract
A series of symmetrically substituted diarylacetylenes and diaryl-1,3-butadiynes were prepared and studied with an emphasis on their spectral and photophysical properties. The photophysical characteristics of these compounds were studied in relation to their structures and the influence of solvent or temperature. The observed spectral and photophysical properties are explained using potential energy maps of the ground and excited states obtained from density functional theory calculations. The structure–property relationships of all of the compounds are discussed and compared with the appropriate diphenylacetylene and diphenyl-1,3-butadiyne derivatives.
Introduction
Asymmetrically substituted acetylene derivatives containing aromatic or heteroaromatic substituents are fluorescent compounds which have been applied to a variety of purposes. They can be used as polarity-sensitive probes,1 for labelling biological molecules2 and intracellular imaging,3 as oligonucleotide analogues,4 as metal or pH sensors,5–8 as fluorescence switches,9,10 as electrochemiluminescent materials11–13 or to synthesize novel fluorescent probes.14–17 Moreover, so-called pull–push chromophores containing electron donors and acceptors connected by a π-electron bridge (D–π–A) show a large nonlinear optical response (NLO) which allows their use in second-order NLO applications.18–20 Poly(aryleneethylene)s find wide application as components of light-emitting diodes and solar cells,21 as well as for the construction of nanoarchitectures22 and molecular machines.23–26 Buta-1,3-diyne derivatives are less applicable than acetylene derivatives, however, substituted buta-1,3-diynes undergo 1,4-addition reactions in the solid state to give polydiacetylene27 which can be used as a label-free fluorogenic chemosensor.28 While the asymmetrically substituted derivatives of acetylene and buta-1,3-diyne are fluorescent, their symmetrically substituted phenyl counterparts show very weak or practically no measurable fluorescence at room temperature and their fluorescence quantum yields show strong dependence on temperature. This applies in particular to diphenylacetylene (DPA) and diphenyl-1,3-butadiyne and their derivatives containing electron-donating substituents in position 4,4′ in the phenyl ring.29–37 These phenomena have been explained by Zgierski and Lim38,39 based on theoretical calculations. The authors found that excited singlet states of DPA demonstrate the existence of a low energy crossing between the initially excited ππ* (B1u) state in the linear D2h symmetry and the “non-fluorescent, dark πσ* state” (11Au) in the bent C2h symmetry, which, as demonstrated Saltiel and Kumar,40 is a weakly fluorescent state. The state switch from the linear ππ* state to the bent πσ* state accounts for all the unusual photophysical and spectroscopic properties of DPA and its derivatives. The experimental confirmation of the presence of the πσ* state was a strong πσ* ← πσ* absorption at about 700 nm in picosecond transient absorption for different DPA derivatives containing electron-donating substituents, and a lack of such absorption for electron-withdrawing substituents38,39 as was predicted by theoretical calculations. Moreover, for 1,4-diphenylbuta-1,3-diyne, similarly to DPA, a dramatic reduction of the fluorescence quantum yield was observed when the photoexcitation energy becomes higher than about 2000 cm−1 above the 11B1u state. This was explained assuming the presence of the close location of the 11A1u state above 11B1u state.35
The big differences between the photophysical properties of DPA and diphenylbuta-1,3-diyne and their non-symmetrical substituted derivatives containing polycyclic aromatic/heteroaromatic groups raises a question about the reason for such differences. To find the answer to this question, symmetrical derivatives of acetylene and buta-1,3-diyne containing polycyclic aromatic substituents were synthesized and their spectral and photophysical properties were examined (Fig. 1). Moreover, there is a lack of systematic studies of the spectral and photophysical properties of symmetrically substituted diarylacetylene and diarylbuta-1,3-diyne derivatives, but at the same time there is considerable interest in systems containing a diyne motif.41,42 On the basis of the obtained results, the similarities and differences between the diarylacetylene and diarylbuta-1,3-diyne derivatives are discussed. Moreover, the properties of diarylacetylene and diaryl-1,3-butadiyne are compared to the previously published properties of DPA and diphenylbuta-1,3-diyne.
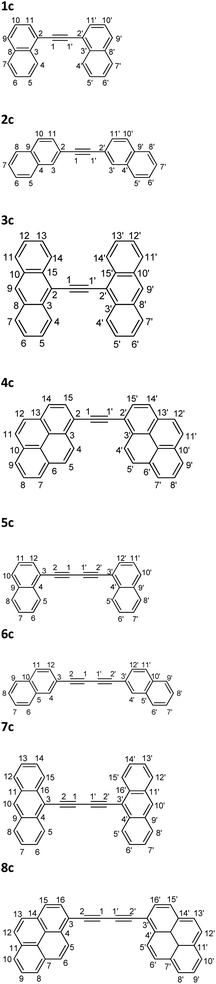 |
| | Fig. 1 Structures and atom numbering of studied compounds. | |
Experimental
Materials and methods
All halogenoarenes: 1-iodonaphtalene (Sigma-Aldrich), 2-bromonaphtalene (Fluka AG), 9-bromoanthracene (Fluka), and 1-bromopyrene (Sigma-Aldrich); and acetylene derivatives: 2-methyl-3-butyn-2-ol (Fluka), trimethylsilylacetylene (Sigma-Aldrich), and 1-ethynylpyrene (Alfa Aesar) were commercially available and used as received. Symmetrical monoacetylene derivatives (1c–4c) were prepared based on the Sonogashira–Hagihara method43,44 using the respective halogenoarenes (procedure A and A′), while symmetrically substituted 1,3-butadiyne derivatives (5c–8c) were made based on the Glaser method45 (procedure C) (see ESI†). The progress of reaction was monitored by means of TLC (Merck plates, Kieselgel 60 F254). The products were isolated by means of column chromatography (Merck, Kieselgel 60 (0.040–0.063 mm) and semi-preparative RP-HPLC (Kromasil column, C-8, 5 μm, 250 mm long, i.d. 20 mm). The identification of products was based on: 1H and 13C NMR spectra recorded on a Bruker AVANCE III (500 MHz) spectrometer in CDCl3, mass spectra recorded on a Bruker Daltonics (HCT ultra) instrument and Raman spectra recorded on a Bruker FRA-106 instrument (see details in ESI†).
Spectroscopy and fluorescence measurements
Absorption spectra were measured using a Perkin-Elmer Lambda-40P spectrophotometer, whereas emission spectra were measured using a FluoroMax-4 (Horiba-Jobin-Yvon) spectrofluorimeter. Low temperature luminescence spectra were obtained using an FL-1013 liquid nitrogen dewar assembly.
Fluorescence quantum yields were calculated using quinine sulphate in 0.5 M H2SO4 (QY = 0.52 ± 0.02) (for 1,2-di(naphtalen-1-yl)ethyne (1Nac), 1,2-di(naphtalen-2-yl)ethyne (2Nac), 1,4-di(naphtalen-1-yl)buta-1,3-diyne (1Nac2), and 1,4-di(naphtalen-1-yl)buta-1,3-diyne (2Nac2)), fluorescein in 0.1 M NaOH (QY = 0.87) (for 1,2-di(anthracen-9-yl)ethyne (Aac), 1,2-di(pyren-1-yl)ethyne (Pac), and 1,4-di(anthracen-9-yl)buta-1,3-diyne (Aac2)) and coumarin 480 in acetonitrile (QY = 0.91) (for 1,4-di(pyren-1-yl)buta-1,3-diyne (Pac2)) as references and were corrected for the different refractive indices of solvents. The fluorescence lifetimes and TRES spectra were measured with a time-correlated single-photon counting fluorescence lifetime spectrometer FT300 (PicoQuant) using a subnanosecond pulsed diode PLS-340 (for 1Nac, 2Nac, 1Nac2, 2Nac2) or a diode laser LHD 420 (for Aac, Pac, Aac2, Pac2).
Theoretical calculations
All calculations were performed using density functional theory (DFT) within the Turbomole v. 6.4 suite programs on a PC cluster, similarly to those published previously.36,37 The structures of all compounds were prepared and initially optimized using the TmoleX program. Energy-minimized structures were searched for their ground and lowest excited states using a B3-LYP hybrid functional with a def2-TZVP basis set of triple-ζ quality. This procedure was considered satisfactory if the energy difference between the optimized cycles was <1 × 10−6 Hartree for ground state structure optimization and <1 × 10−7 Hartree for excited state structure optimization. In both states, a gradient of <1 × 10−3 au was achieved. The low-lying excited states were treated within the adiabatic approximation of time-dependent density functional theory (DFT-RPA) using a B3-LYP functional. The geometry optimization of the excited state was performed using a grid parameter equal to 4. The convergence of all the studied systems was checked by harmonic vibrational analysis. No imaginary frequencies were observed. The rotation energy barriers in the ground and excited states were performed by imposing constraints on the dihedral angle optimizing the energy of molecule. It does not impose additional constraints such as the symmetry of the molecule. In a similar way the energy of the bent conformation of the molecule was calculated by imposing the bent angle value and optimizing the remaining part of the molecule; the same functional and base set were used as in the case of energy optimization.
Results and discussion
Absorption spectra
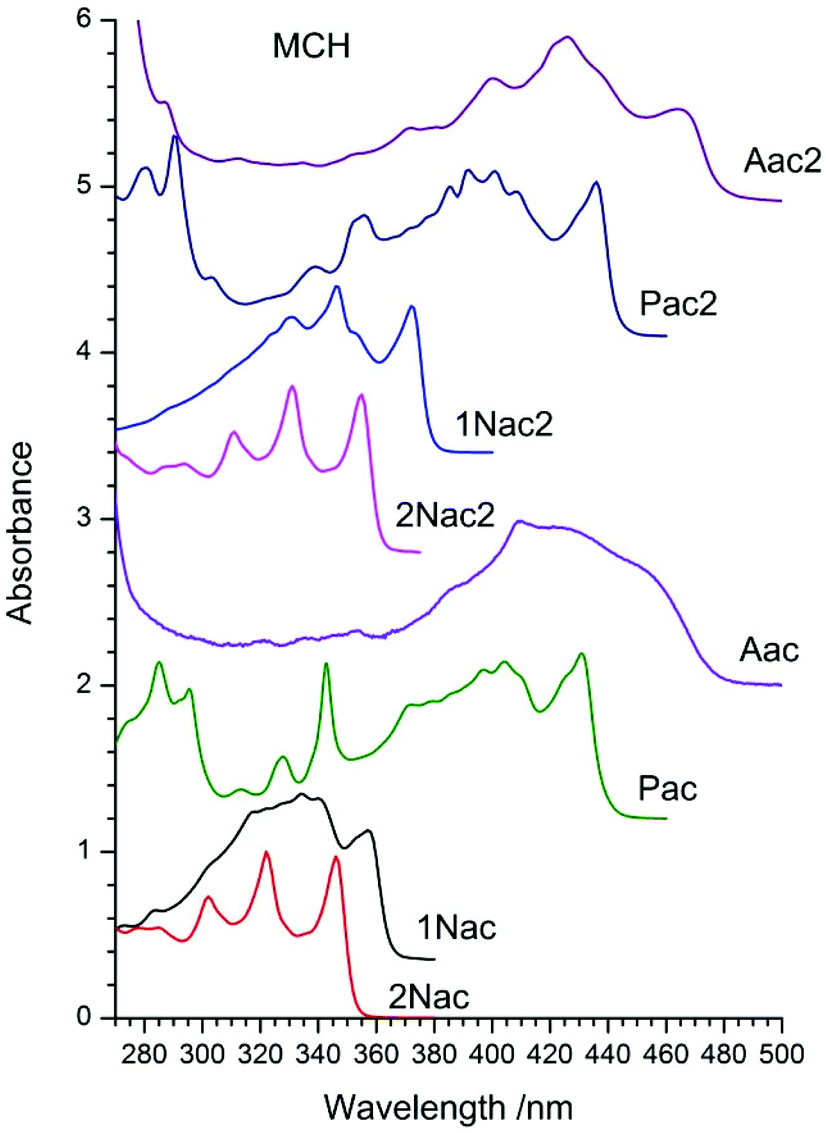
The absorption spectra of the studied compounds measured in methylcyclohexane (MCH) are presented in Fig. 2, whereas those measured in 2-methyltetrahydrofurane (2-MeTHF) and acetonitrile (MeCN) are in Fig. 1–8, ESI.† All the studied compounds, except for 2Nac and 2Nac2, show complicated vibrational structures. Among some of them, the vibrations can be recognized as associated with the vibration of the triple bond (about 2000 cm−1) and the ring breathing mode of an aromatic ring (at about 1400 cm−1). However, for acetylene and buta-1,3-diyne derivatives containing 2-naphthyl substituents (2Nac and 2Nac2 and to a lesser extent in the case of 1Nac2) a clear long progression of C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C stretching mode (ca. 2150 cm−1), which is an indication of the large displacement of the CC bond upon the electronic excitation, is seen. This is similar to the absorption spectra of the simple phenyl derivatives of acetylene and buta-1,3-diyne.30,34–37,46 The positions of long-wavelength absorption bands measured in MCH are presented in Table 1 of the ESI.† The position of the absorption maximum depends on the size of the aromatic substituent. The bigger it is, the more bathochromically shifted the observed spectrum is (2Nac < 1Nac < Pac < Aac). The introduction of the second acetylene unit does not change the position order of the absorption spectra of the studied compounds, albeit it shifts the spectra to longer wavelengths by about 10 nm (15 nm for 1Nac). However, for 1Nac2, substantial changes in the shape of the absorption spectrum compared to that of 1Nac are observed, which points to a larger participation of the triple bonds in the conjugation between the naphthalene substituents. A more polar solvent induces small changes in the position of the absorption bands, however, in the case of anthryl derivatives, the band shape changes (Fig. 1–8, ESI†). In 2-MeTHF a small bathochromic shift of the absorption spectrum is observed, whereas in MeCN a blue shift by a few nanometers occurs. Simultaneously, with the increase of solvent polarity, the vibrating structure is blurred and the ratio of the vibronic band changes. This is particularly evident for anthryl and to a lesser degree for pyrenyl, as well as for di(naphtalen-1-yl) derivatives.
C stretching mode (ca. 2150 cm−1), which is an indication of the large displacement of the CC bond upon the electronic excitation, is seen. This is similar to the absorption spectra of the simple phenyl derivatives of acetylene and buta-1,3-diyne.30,34–37,46 The positions of long-wavelength absorption bands measured in MCH are presented in Table 1 of the ESI.† The position of the absorption maximum depends on the size of the aromatic substituent. The bigger it is, the more bathochromically shifted the observed spectrum is (2Nac < 1Nac < Pac < Aac). The introduction of the second acetylene unit does not change the position order of the absorption spectra of the studied compounds, albeit it shifts the spectra to longer wavelengths by about 10 nm (15 nm for 1Nac). However, for 1Nac2, substantial changes in the shape of the absorption spectrum compared to that of 1Nac are observed, which points to a larger participation of the triple bonds in the conjugation between the naphthalene substituents. A more polar solvent induces small changes in the position of the absorption bands, however, in the case of anthryl derivatives, the band shape changes (Fig. 1–8, ESI†). In 2-MeTHF a small bathochromic shift of the absorption spectrum is observed, whereas in MeCN a blue shift by a few nanometers occurs. Simultaneously, with the increase of solvent polarity, the vibrating structure is blurred and the ratio of the vibronic band changes. This is particularly evident for anthryl and to a lesser degree for pyrenyl, as well as for di(naphtalen-1-yl) derivatives.
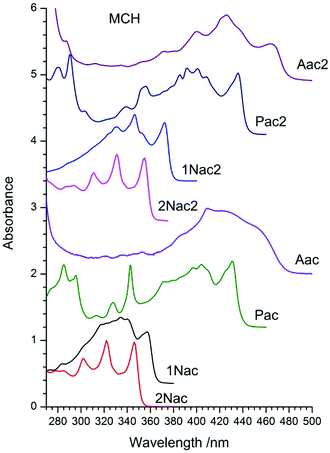 |
| | Fig. 2 Normalized absorption spectra of acetylene and buta-1,3-diyne derivatives in methylcyclohexane (MCH). | |
Room temperature fluorescence
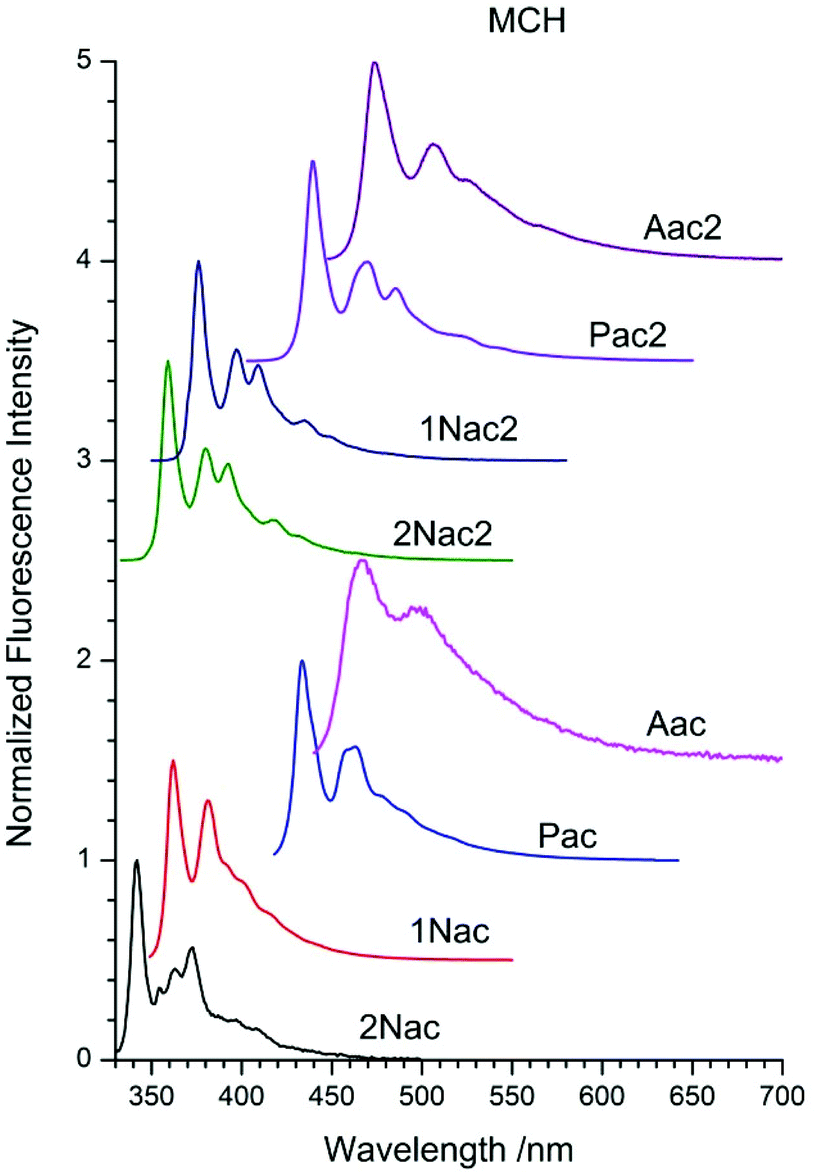
The fluorescence spectra of acetylene and buta-1,3-diyne in MCH are presented in Fig. 3, whereas those in 2-MeTHF and MeCN are in Fig. 9–16, ESI.† These spectra contain all the characteristic features of the emission spectra of the symmetrical derivatives of acetylene and buta-1,3-diyne, as well as phenyleneethylene oligomers.30,34–37,47–51 Contrary to the DPA spectrum in which two emission bands from 11Bu and 11Au were observed,40 in the spectra of the studied compound only one emission band was recorded. The fluorescence spectra are narrow and exhibit clear vibronic bands. Moreover, the optical spectra are characterized by a substantial asymmetry between absorption and fluorescence. These effects have been explained in terms of torsional disorder and quadratic coupling between the ground and first excited states52 or the exciton model developed by Liu et al.53,54 The red shift of the fluorescence maximum depends on the size of the substituent for both acetylene and buta-1,3-diyne derivatives in the following order: 2-naphthyl, 1-naphthtyl, pyrenyl and anthryl (Table 1 ESI†).
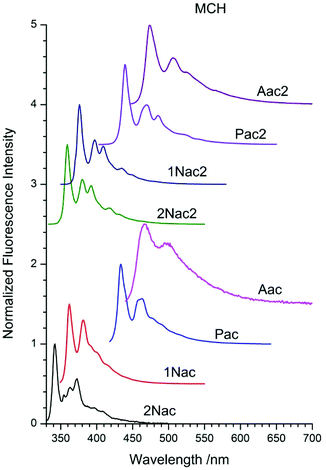 |
| | Fig. 3 Normalized fluorescence spectra of acetylene and buta-1,3-diyne derivatives in methylcyclohexane (MCH). | |
Similarly to the absorption analysis observations, the second acetylenic unit shifts the emission spectrum in the red direction by about a dozen nanometers for naphthyl and by several nm for the anthryl and pyrenyl derivatives (Table 1 ESI†). The emission spectra of the compounds in 2-MeTHF are similar to those in MCH, however, they are shifted by several nm in the red direction. MeCN has a greater impact on the shape and position of the emission band. The vibrational structure is blurred, and for anthryl derivatives it completely disappears. Moreover, the emission bands are broader and the ratio of the intensity of the vibrational peaks changes, in comparison to those of the compounds in 2-MeTHF. Such influence of the polarity of solvent indicates a fairly large contribution of the emission from the charge transfer excited state. This statement is supported by the femtosecond time-resolved studies of the fluorescence spectra of 1,2-bis(9-anthryl)-acetylene in solvents of different polarity.51 The fluorescence quantum yield of all acetylene and buta-1,3-diyne derivatives in MCH, 2-MeTHF and MeCN are collated in Table 1. Moreover, in Table 2 ESI† the fluorescence quantum yields and fluorescence lifetimes measured in five additional non-polar solvents differing in viscosity are presented.
Table 1 The fluorescence quantum yield (ϕ), fluorescence lifetime (τ), pre-exponential factor (α) and quality of fit χR2 of the studied compounds in MCH, 2-MeTHF and MeCN
| Compound |
Solvent |
ϕ
|
τ/ns |
α
|
χ
R
2
|
| 1Nac |
MeCN |
0.745 |
1.82 |
1 |
0.98 |
| MeTHF |
0.841 |
1.56 |
1 |
0.99 |
| MCH |
0.836 |
1.69 |
1 |
0.95 |
| 1Nac2 |
MeCN |
0.083 |
0.24 |
1 |
0.89 |
| MeTHF |
0.165 |
0.27 |
1 |
0.96 |
| MCH |
0.160 |
0.29 |
1 |
0.97 |
| 2Nac |
MeCN |
0.109 |
3.23 |
0.063 |
0.95 |
|
|
|
0.45 |
0.937 |
|
| MeTHF |
0.119 |
4.02 |
0.067 |
0.92 |
|
|
|
0.42 |
0.933 |
|
| MCH |
0.110 |
4.70 |
0.060 |
1.12 |
|
|
|
0.48 |
0.940 |
|
| 2Nac2 |
MeCN |
0.004 |
3.02 |
0.014 |
1.14 |
|
|
|
0.22 |
0.986 |
|
| MeTHF |
0.006 |
0.23 |
1 |
1.15 |
| MCH |
0.010 |
0.23 |
1 |
1.13 |
| Aac |
MeCN |
0.683 |
2.63 |
1 |
1.14 |
| MeTHF |
0.660 |
1.75 |
1 |
0.97 |
| MCH |
0.634 |
1.73 |
1 |
1.07 |
| Aac2 |
MeCN |
0.199 |
5.05 |
0.100 |
0.89 |
|
|
|
1.83 |
0.900 |
|
| MeTHF |
0.200 |
4.45 |
0.118 |
0.98 |
|
|
|
1.44 |
0.882 |
|
| MCH |
0.147 |
3.72 |
0.153 |
0.93 |
|
|
|
0.91 |
0.847 |
|
| Pac |
MeCN |
0.920 |
1.93 |
1 |
0.94 |
| MeTHF |
0.986 |
1.54 |
1 |
0.99 |
| MCH |
0.975 |
1.37 |
1 |
1.06 |
| Pac2 |
MeCN |
0.723 |
1.60 |
1 |
1.00 |
| MeTHF |
0.758 |
1.30 |
1 |
0.98 |
| MCH |
0.779 |
1.14 |
1 |
0.97 |
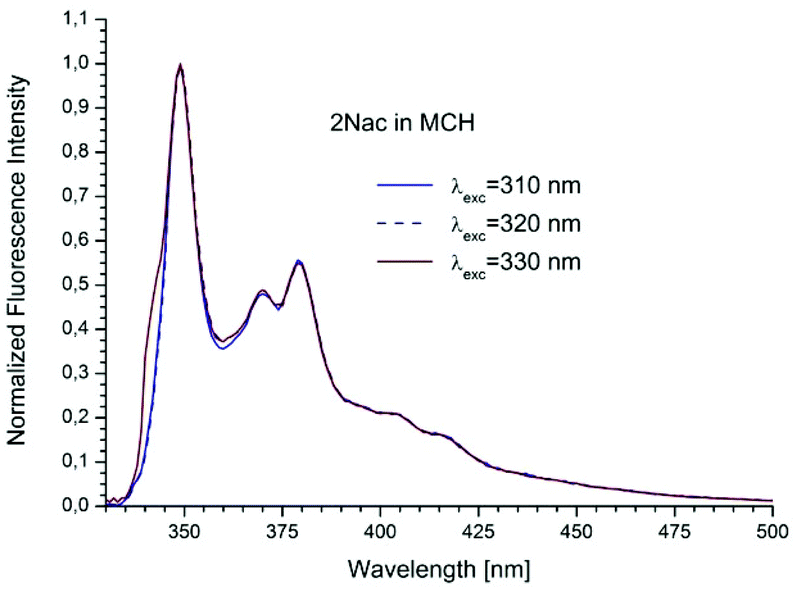
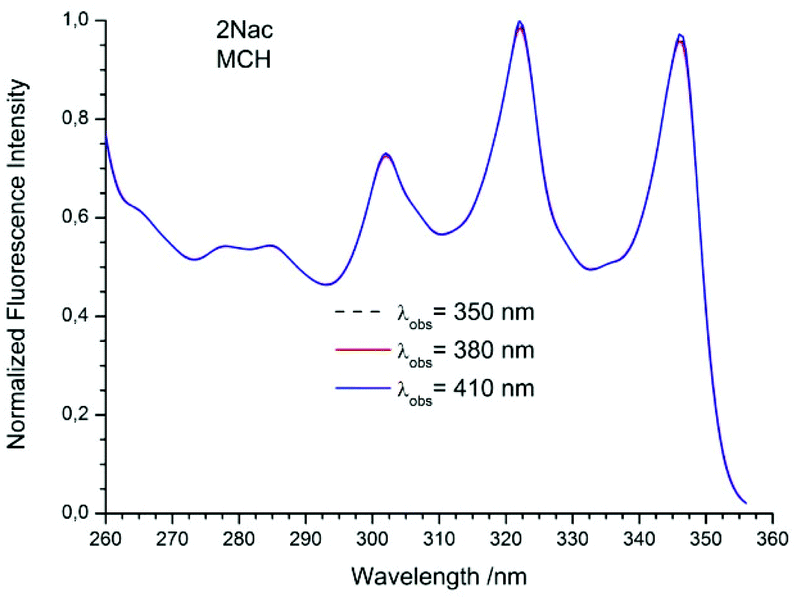
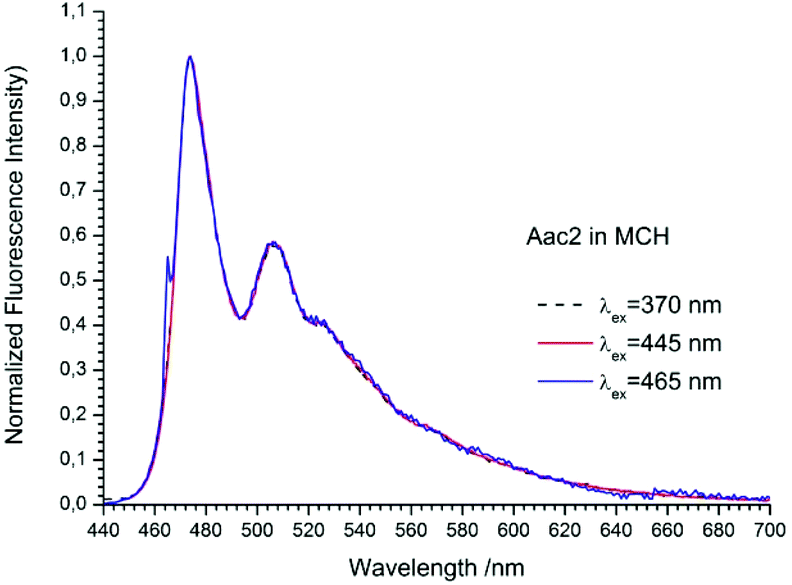
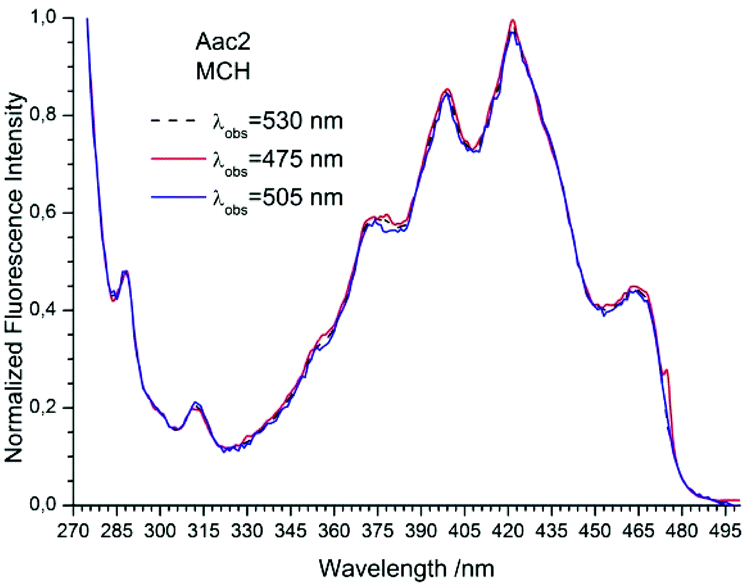
The studied compounds have various values of fluorescence quantum yield which depend primarily on the structure of the compound and the size of the substituent and substitution sites, as well as the amount of triple bonds (Table 1). Thus, a 1-naphthyl derivative of acetylene has a much higher fluorescence quantum yield than a 2-naphthyl derivative, (about one order of magnitude). As can be seen from the absorption spectrum, this seems to be due to the greater involvement of the triple bond in conjugation with the aromatic system for 2-naphthyl than for the 1-naphthyl derivative. The anthryl and pyrenyl derivatives of acetylene have high fluorescence quantum yields, and for the pyrenyl derivative the value is close to 1. However, 1,3-diyne derivatives, except for the pyrenyl one, have much lower fluorescence quantum yields than the acetylene derivatives (Table 1 and Table 2 ESI†). This indicates the participation of an additional deactivation process of the excited state. The polarity of solvent has a minor influence on the fluorescence quantum yield. In acetonitrile the yield is a bit lower than that in 2-MeTHF or MCH (Table 1). Also, the solvent viscosity does not significantly affect the fluorescence quantum yield (Table 2 ESI†). The fluorescence lifetimes of studied compounds are also differentiated as are the fluorescence quantum yields (Table 1 and Table 2 ESI†). The fluorescence intensity decay for 1Nac, 1Nac2, Pac and Pac2 is well described by a mono-exponential function, while for 2Nac and 2Nac2 and Aac in viscous solvents by a bi-exponential function. For 1Nac, the fluorescence lifetime values fall within a range from 1.82 to 1.46 ns whereas those for 1Nac2 are much shorter (about 0.3 ns). The 2-naphthyl derivatives of acetylene and buta-1,3-diyne have bi-exponential fluorescence intensity decay, however, 2Nac2 has a shorter decay time. For 2Nac the longer component has a lifetime of about 5 ns while the shorter one has a value of about 0.5 ns. It is worth emphasizing that the contribution to longer fluorescence lifetimes seems to depend on solvent viscosity. For solvents with a low viscosity, the contribution to the fluorescence intensity decay is about 4–6% while for viscous solvent this increases to several percent (Table 2 ESI†). The major component of the fluorescence lifetimes of 2Nac2 (contribution of about 94%) is in the range from 0.2 to 0.3 ns, while the minor component is about 3 ns. Due to the very low fluorescence quantum yield of this compound, the measured fluorescence lifetimes may be wholly flawed due to Raman scattering and solvent impurities. The fluorescence lifetime of Aac in low viscous solvents is about 2 ns while in viscous solvents the second component (about 0.6 ns) appears with several percent of contribution, whereas Aac2 has two fluorescence lifetimes in all studied solvents. The major component (about 90%) is in a range from 0.9 to 1.4 ns while the minor one is in a range from 5 to 3.6 ns. For the pyrene derivatives of acetylene and buta-1,3-diyne, mono-exponential fluorescence intensity decay is recorded. For Pac, the fluorescence lifetime is about 1.4 ns in non-polar solvents and 1.93 ns in polar acetonitrile, while for Pac2 the fluorescence lifetimes are shorter; about 1.2 ns in non-polar solvents and 1.6 ns in acetonitrile. The photophysical and theoretical studies suggest that phenyleneethynylenes exist in planar and several twisted conformations in their ground state due to the nearly free rotation of the arene rings along the molecular axis. On the other hand, their emission spectra are highly structured as a consequence of the planarization of the molecule in the excited state.47–49,55,56 The emission properties (fluorescence spectra, fluorescence excitation spectra and fluorescence quantum yield) of 2Nac, 2Nac2 and Aac2 were found to be independent of the excitation/observation wavelengths (Fig. 4–7, Fig. 17–24 and Table 3, ESI†). The bi-exponential fluorescence decays can be reasonably explained by the delayed fluorescence caused by thermal repopulation of the second excited state, which was reported for diphenylacethylene derivatives.57–59 However, for 2Nac2 and Aac2, contrary to 2Nac, the energy barrier of rotation in the excited state is relatively low (about 16 kJ mol−1), Table 5 ESI† (for details see the Theoretical calculation section), causing crossing through the energy barrier to occur several times during the lifetime of the excited state. Thus, the recorded fluorescence excitation/emission spectrum is averaged over the emission of different rotamers resulting in a heterogeneous decay of fluorescence intensity.
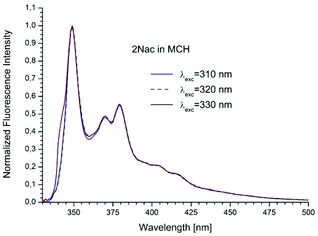 |
| | Fig. 4 The normalized emission spectra of 2Nac in methylcyclohexane recorded for different excitation wavelengths. The shoulder in the spectrum recorded at λex = 330 nm is caused by scattered light. | |
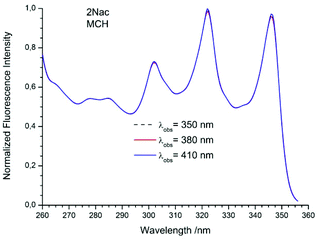 |
| | Fig. 5 The normalized fluorescence excitation spectra of 2Nac in methylcyclohexane recorded for different observation wavelengths. | |
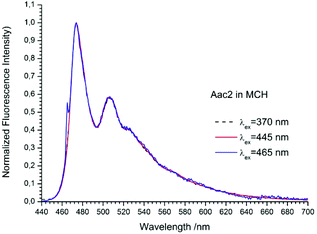 |
| | Fig. 6 The normalized emission spectra of Aac2 in methylcyclohexane recorded for different excitation wavelengths. The shoulder in the spectrum recorded at λex = 465 nm is caused by scattered light. | |
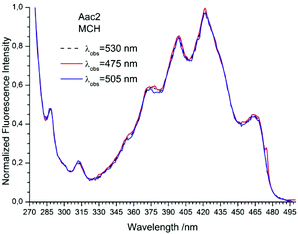 |
| | Fig. 7 The normalized fluorescence excitation spectra of 2Nac in methylcyclohexane recorded for different observation wavelengths. The shoulder in the spectrum recorded at λobs = 475 nm is caused by scattered light. | |
For compounds that have a mono-exponential fluorescence intensity decay, it is possible to calculate the radiative (kr) and non-radiative (knr) rate constants, according to equations: kr = ϕ/τ and knr = (1 − ϕ)/τ, where ϕ is the fluorescence quantum yield and τ is the fluorescence lifetime. For 1Nac, 2Nac2, Aac, Pac and Pac2 the corresponding rate constants for the three solvents used (MeCN, 2-MeTHF and MCH) are summarized in Table 4 of the ESI.† The fluorescence rate constant is very similar for all the above-mentioned derivatives in the non-polar 2-MeTHF and MCH while a little lower in MeCN. In contrast, there is a big difference in the knr rate constant between the acetylene and buta-1,3-diyne derivatives. The non-radiative rate constant for a buta-1,3-diyne is one order higher than that for the corresponding acetylene derivative. This indicates an additional deactivation channel of the excited state, probably related to the greater conformational freedom. Also, values of the fluorescence rate constant in acetonitrile are smaller than those in nonpolar solvents, which seem to confirm that the emission of the anthryl and pyrenyl derivatives of acetylene and 1,3-diyne in this solvent comes from the charge-transfer (CT) state. Fig. 25–28,† of the ESI presents time-resolved emission spectra (TRES) for the anthryl and pyrenyl derivatives showing the bathochromic evolution of the fluorescence in a short time after excitation. Such a process has been observed for 1,2-bis(9-anthryl)acetylene by Gutierez-Arzuluz et al.51 These authors demonstrated, on the basis of femtosecond fluorescence up-conversion experiments, that in acetonitrile, the spectral shift is biphasic, with the first sub-picosecond decay time dictated by the solvent response and a much slower one, about 37 ps, is connected to the relaxation of the excited state structure leading to a change in the relative orientation of the two anthracenic end groups which allows for further stabilization of the CT state. Thus, before the system is able to relax to the full CT state, an initial equilibrium between the local excited state and partial charge-transfer excited state takes place. It seems that the process of solvent-mediated symmetry reduction in the excited state also takes place for the anthryl derivative of buta-1,3-diyne as well as the pyrenyl derivative of acetylene and buta-1,3-diyne and to some extent for the naphthyl derivatives of acetylene. The broad, structure-less fluorescence spectra of the anthryl derivatives in MeCN suggest the possibility of the formation of an orthogonal twisted-internal charge transfer (TICT) state.51 The dependence of fluorescence decay times on the observation wavelengths found for Aac2 (Table 2) suggests relatively slow conformation changes.
Table 2 Fluorescence lifetime (τ), pre-exponential factor (α) and quality of fit (χR2) of Aac and Aac2 in acetonitrile determined for different observation wavelengths
| Observation wavelength/nm |
λ
exc = 420 nm |
|
τ/ns |
α
|
χ
R
2
|
| 495 |
5.05 |
0.10 |
1.10 |
| 1.83 |
0.90 |
|
| 515 |
5.40 |
0.04 |
0.97 |
| 1.88 |
0.94 |
|
| 530 |
6.06 |
0.007 |
0.99 |
| 1.87 |
0.993 |
|
| 560 |
1.87 |
1 |
1.09 |
| 575 |
1.87 |
1 |
1.03 |
| Aac |
| 460 |
2.65 |
1 |
1.11 |
| 475 |
2.62 |
1 |
1.12 |
| 500 |
2.65 |
1 |
1.12 |
| 530 |
2.64 |
1 |
1.03 |
| 570 |
2.63 |
1 |
1.08 |
This may indicate the transformation to an orthogonal TICT state, and all the more, that the energy barrier of rotation decreases with increasing solvent polarity.50 Such a dependence is not determined for Aac (Table 2). However, according to Gutierez-Arzuluz et al.51 the relative orientation of the two anthracenic groups of the Aac molecule occurs rapidly, over time of about 40 ps, shorter than the resolving power of our instrument. Based on measurements of the fluorescence quantum yield, it is difficult to conclude the influence of the solvent viscosity from the photophysical properties, however, the measurements of the fluorescence intensity decay indicate that the 2-naphthyl and 9-anthryl derivatives of acetylene can be used as molecular rotors to study the solvent viscosity (particularly for viscous solvents at lower temperature60).
Low temperature luminescence
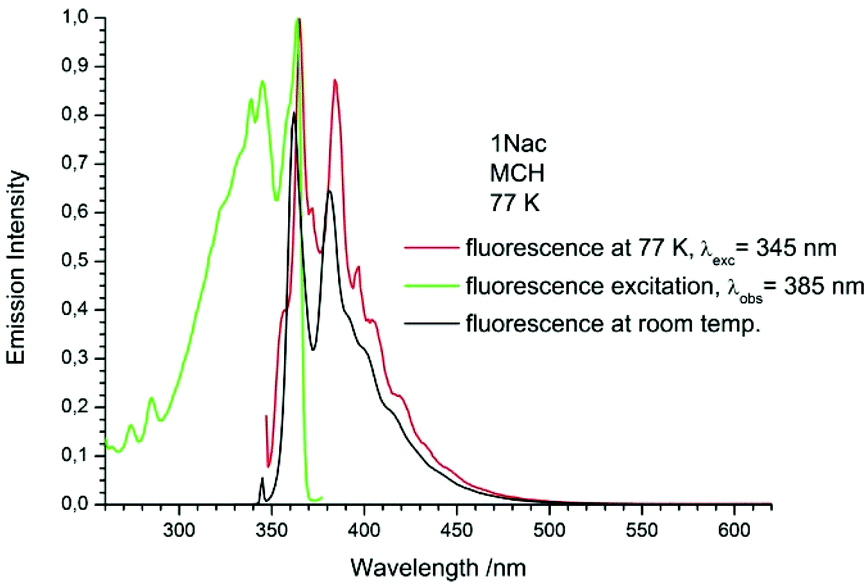
In order to check the influence of environment stiffness on the fluorescence intensity, the luminescence spectra of the studied compounds were measured in the glasses of MCH and 2-MeTHF. As the measurements at the temperature of liquid nitrogen in 2-MeTHF gave the same results as in MCH, they are not presented here. The luminescence, fluorescence excitation and room temperature fluorescence spectra of the 1-naphthyl and 2-naphthyl acetylene and buta-1,3-diyne derivatives in MCH are presented in Fig. 8–11, and the anthryl and pyrenyl derivatives in Fig. 29–32, ESI.†
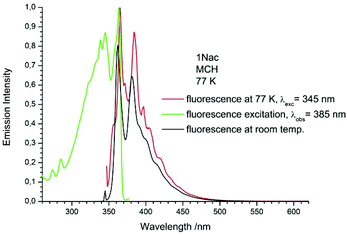 |
| | Fig. 8 The normalized emission (solid red line), and excitation (solid green line) spectrum measured at 77 K and fluorescence spectrum measured at room temperature (black solid line) of 1Nac in methylcyclohexane. | |
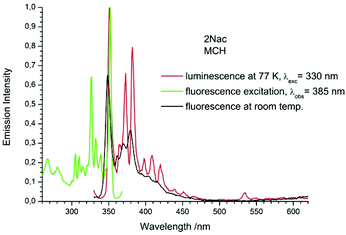 |
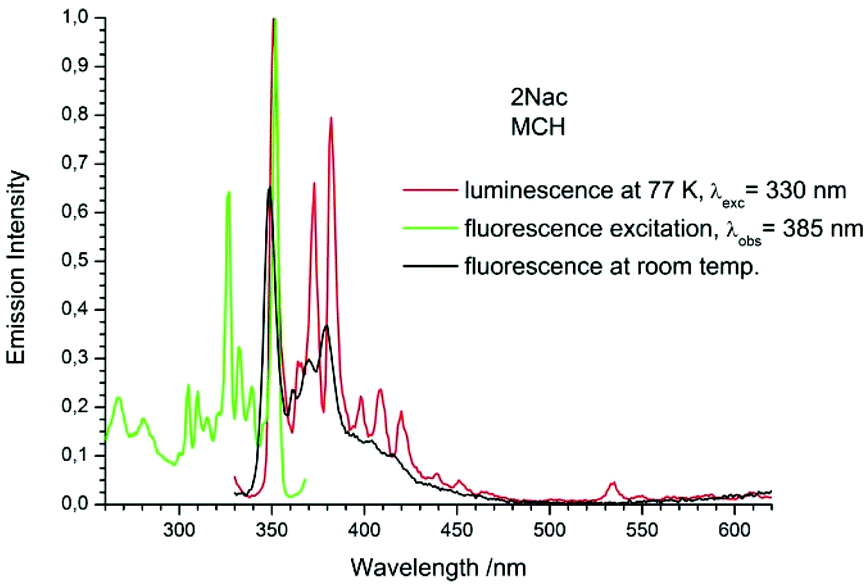
| | Fig. 9 The normalized emission (solid red line), and excitation (solid green line) spectrum measured at 77 K and fluorescence spectrum measured at room temperature (black solid line) of 2Nac in methylcyclohexane. | |
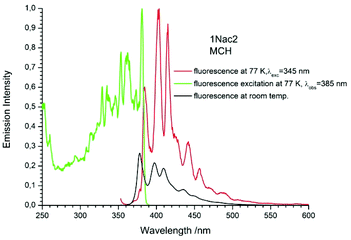 |
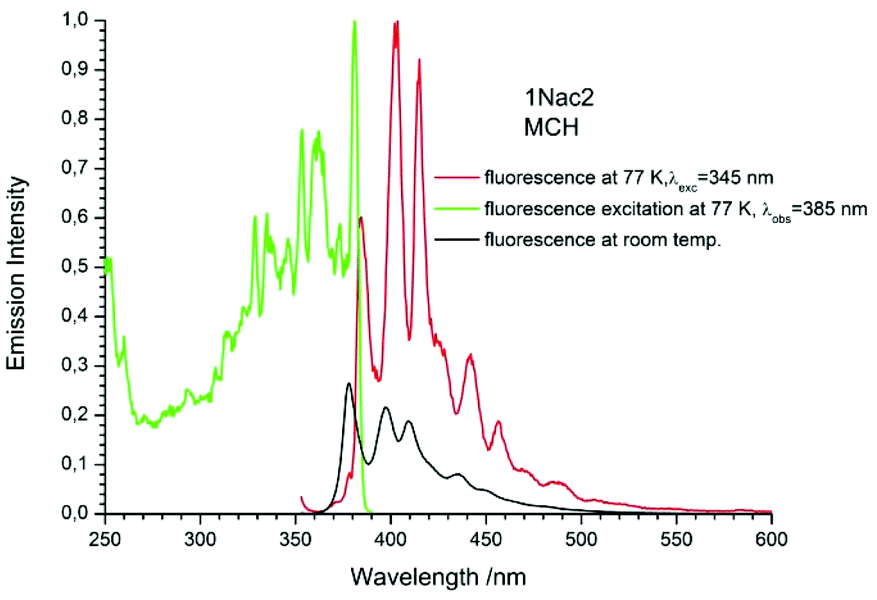
| | Fig. 10 The normalized emission (solid red line), and excitation (solid green line) spectrum measured at 77 K and fluorescence spectrum measured at room temperature (black solid line) of 1Nac2 in methylcyclohexane. | |
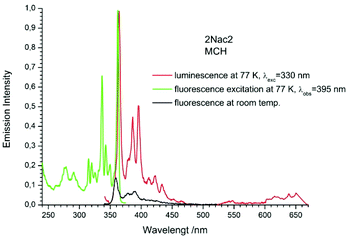 |
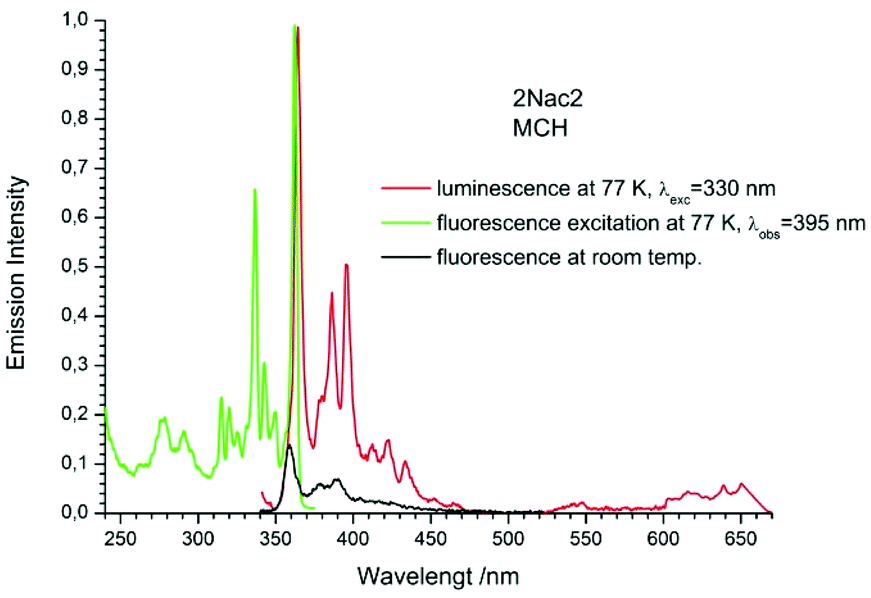
| | Fig. 11 The normalized emission (solid red line), and excitation (solid green line) spectrum measured at 77 K and fluorescence spectrum measured at room temperature (black solid line) of 2Nac2 in methylcyclohexane. | |
The fluorescence spectra measured at 77 K have a richer vibrational structure than those recorded at room temperature and are red-shifted by a few nanometers. Furthermore, they exhibit very small Stokes’ shift. In contrast to the room temperature results, for all studied compounds, except the pyrenyl derivatives, the fluorescence and fluorescence excitation spectra at 77 K are almost mirrored symmetrical images. The lack of mirror symmetry between the fluorescence and fluorescence excitation spectra for the pyrenyl derivative indicates that a long absorption band is a superposition of over at least two absorption transitions. The mirror symmetry indicates hindered ring torsion in the MCH organic glass. It is worth mentioning that for 2-naphtyl derivatives only very weak phosphorescence is detected indicating a low intersystem crossing rate constant in both the acetylene and buta-1,3-diyne derivatives. For all the studied compounds, the room temperature fluorescence intensity is less intense than that recorded at 77 K. The exceptions are the pyrene derivatives (Pac and Pac2, Fig. 31 and 32, ESI†) for which the room temperature fluorescence is much more intense, probably because of their solubility at low temperature. It should be noted that for the compounds with high or moderate values of fluorescence quantum yield (acetylene derivatives), the increase of fluorescence intensity with decreasing temperature is relatively small, contrary to the buta-1,3-diyne derivatives (Fig. 8–11). Thus, the measurements in stiff organic glasses confirm the greater conformational freedom of the buta-1,3-diyne derivatives in comparison to acetylene derivatives.56
Theoretical calculations
To understand the influence of the substituent dimensions on the torsional motions of the acetylene and buta-1,3-diyne derivatives, we studied the torsional potential in both the ground and excited states within the framework of the local mode approximation using a DFT method. Fig. 33–38,† ESI show DFT-based torsional potentials for 1Nac, 2Nac, Aac, 1Nac2, 2Nac2 and Aac2 in the ground and excited states. The torsional potential in the ground state is well approximated by a simple periodic potential of the form:| | 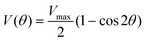 | (1) |
where Vmax is the barrier height of the torsional potential and θ is the ring-to-ring torsional angle. The above equation describes the torsional potential in the ground state well, but describes this in the excited state poorly; the approximation by a Gauss or Lorentz function is much better at an excited state description here (Fig. 38 ESI†). The energy barrier of rotation in the excited state is much higher than in the ground state indicating a hampered rotation of the polyaromatic hydrocarbon units in the excited state, which is attributed to breaking of the cylindrical structure of the electron density along the axis of symmetry61 and the adoption of a partial cummulene-like structure.55 For this reason, the rotation energy barrier for the 1- and 2-naphthalene derivatives in the excited state is high (close to 40 kJ mol−1) contrary to diphenylbutadiyne for which the rotation energy barrier is low (ΔE ∼ 3 kJ mol−1). Therefore replacement of the phenyl group with naphthyl groups eliminates an energy minimum in the orthogonal geometry in the excited state observed for diphenylbuta-1,3-diyne.37 For 1Nac in the ground and excited states, the most stable form is a trans conformation, while the cis conformer is non-planar (the ring-to-ring torsional angle is about 27 degrees) and less stable by about 0.5 kJ mol−1 (Fig. 33 ESI†). Due to the lack of steric hindrance for the acetylene and diacetylene derivatives, the place of substitution in the naphthalene does not differentiate the rotation energy barrier as it does for ethylene derivatives.62 Theoretical calculations at the density functional level of theory indicate that in the ground and excited state, the anthryl derivative of acetylene is not fully planar. An angle between the two anthryl units of about 38 degrees exists in the ground state, with the flat maximum at 90 degrees, in accordance with the data published by Gutierez-Arzaluz et al.51 However, in the excited state this dihedral angle decreases to a value of 20 degrees (Fig. 35 ESI†). For all the studied buta-1,3-diyne derivatives in both the ground and excited states, the most stable are the linear planar conformations (Fig. 36–38 ESI†). The barrier heights of the torsional potential calculated using eqn (1) for all the studied compounds, are gathered in Table 5, ESI.† For the acetylene derivatives the energy barriers of rotation in the ground state are slightly higher than those of the buta-1,3-diyne derivatives and are comparable to the values for diphenylacethylene29,36,37,63 and its symmetrically substituted derivatives.36 The lower values of rotational energy barriers obtained for 1,3-diyne are in conformity with the statement that the insertion of extra –CC– units decreases the barrier by 40–70%.61 The rotational energy barriers calculated for the polycyclic aromatic hydrocarbon derivatives of buta-1,3-diyne are comparable with the data obtained for diphenylbuta-1,3-diyne29,37,46 and its derivatives.37 Moreover, the values of the energy barrier of rotation determined experimentally for covalently-linked multi-porphyrin arrays64 are also lower than those for diphenylacetylene. The DFT method gives overestimated values of the energy barrier of rotation.56 However, the experimentally determined magnitude of the rotational barrier is found to be 40% larger than that in a gas phase,29 thus it may be close to the barrier of rotation in low viscous solvent, especially for the polyaromatic hydrocarbon substituents. The rate constant of the conversion through the energy barrier during the rotation (kc→t) can be calculated on the basis of transition-state theory for unimolecular reactions using the equation:| | 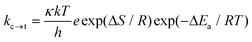 | (2) |
where k is the Boltzmann constant, T is the absolute temperature, h is Planck's constant, R is the gas constant, ΔS is the entropy change, and ΔEa is the energy of activation. It is difficult to assign a value to the transmission coefficient, κ, which incorporates all correction factors and uncertainties. We chose κ = 0.4 as for ring inversion65 and ΔS = 0 and for these values the rate constant and rotamer lifetime were calculated. The rate constants of barrier crossing for the energy barrier of rotation in the ground state at room temperature (300 K), as gathered in Table 4 of the ESI,† are high, and the rotamer lifetimes are as short as tens of femtoseconds, which is tantamount to free rotation in the ground state. The energy barriers of rotation in the excited state (Table 5, ESI†) for acetylene derivatives are in the range of 40 kJ mol−1 to 30 kJ mol−1, and therefore are similar to those for the symmetrically substituted diphnenylacetylene derivatives36 or 1,4-bis(phenylethynyl)benzene.55,56 Moreover, the energy barriers of rotation in the excited state of the studied buta-1,3-diyne derivatives are about 10 kJ mol−1 lower than those of the acetylene derivatives (Table 5 ESI†) and much lower than those for the symmetrically substituted diphenyl derivatives of buta-1,3-diyne.37 The calculated rate constants of rotation for the acetylene derivatives are low, and the rotamer lifetimes are long; a tenth of a nanosecond for the naphthyl derivatives and about 20 ns for the anthryl derivative (Table 5 ESI†). Thus, during the fluorescence lifetime there is no rotation of the aryl substituents in the excited state. However, a lower energy barrier of rotation for buta-1,3-diyne leads to rotamer lifetimes for the naphthyl derivatives that are about 100 ps, while for the anthryl derivative this is about 800 ps, which are comparable to the fluorescence lifetime. Accordingly, rotation in the excited state may lead to the appearance of an additional radiationless channel for excited state deactivation. However, as in the case of the ground state, the energy barrier of rotation in the excited state seems to be overestimated. However, taking into account the size of the substituents and the viscosity of the solvent it can be assumed with a high degree of probability that during the lifetime of the excited state the acetylene derivatives do not change their conformation in contrast to the buta-1,3-diyne derivatives.
A low value of the rotation barriers in the ground state, and at the same time, a high value in the excited state should cause the ground state of the two conformers of 1Nac and 2Nac to be almost equally populated and be distinct isomers in the excited state. Because two conformers belong to different symmetry groups and should have different electronic transitions, consequently they should give different absorption and emission spectra as in the case of trans-1,2-di(2-naphthyl)ethane.66,67 However, theoretically calculated, using a TD DFT method, the vertical transitions in the absorption and emission for a fully optimized structure of both conformers (cis and trans) of 1Nac and 2Nac reveal a great coincidence of the vertical transition in the absorption and emission of the rotamers (Table 3). Moreover, the value of the angle between the long axes of the molecules and transition dipole moments for both rotamers differ slightly (Fig. 39–42 ESI†). Thus, our earlier statement that fluorescence and fluorescence excitation spectra are excitation and monitoring wavelength independent is fully consistent with the calculations.
Table 3 Vertical transition in absorption and emission (in nm), and oscillator strength (f) for the two rotamers of di-naphthylacetylene isomers (1Nac and 2Nac) calculated using a TD DFT method (B3LYP, def2TZVP basis set)
| 1Nc cis |
1Nac trans |
2Nac cis |
2Nac trans |
| Abs |
f
|
Abs |
f
|
Abs |
f
|
Abs |
f
|
| 377 |
0.625 |
378 |
0.655 |
350 |
0.92 |
351 |
1.01 |
| 328 |
0.3 × 10−4 |
326 |
0.2 × 10−5 |
329 |
0.1 × 10−4 |
329 |
0.6 × 10−6 |
| 310 |
0.5 × 10−3 |
309 |
0.5 × 10−6 |
317 |
0.2 × 10−3 |
318 |
0.2 × 10−2 |
| 299 |
0.2 × 10−5 |
300 |
0.3 × 10−4 |
303 |
0.11 |
304 |
1.0 × 10−3 |
| 299 |
0.9 × 10−4 |
299 |
0.6 × 10−6 |
284 |
0.56 |
285 |
0.60 |
| |
| Emission |
| 427 |
0.71 |
427 |
0.74 |
391 |
1.08 |
390 |
1.20 |
According to Zgierski et al.38,39 and Wierzbicka et al.36 a low fluorescence quantum yield of DPA and its derivatives and its strong dependence on temperature is caused by the state switch from initially excited linear fluorescing ππ* to the bent σπ* state. The same phenomenon seems to occur for buta-1,3-diyne and its symmetrically substituted derivatives containing electron-donating substituents.37 Similar calculations to those described in the cited works36–39 were carried out to check the influence of the bending of the CAr–CC angle on the potential energy of the excited state for naphthyl derivatives of acetylene and buta-1,3-diyne. For other studied derivatives (anthryl and pyrenyl substituents) such calculations were not performed due to the cost.
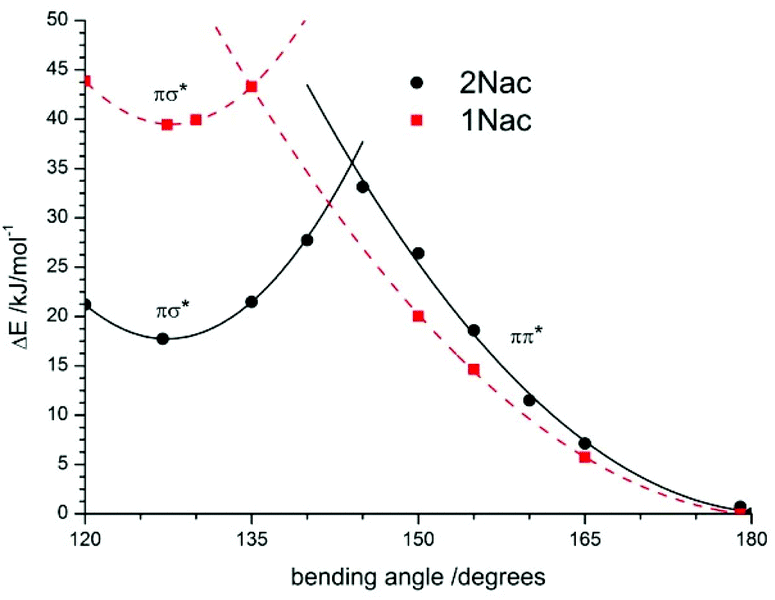
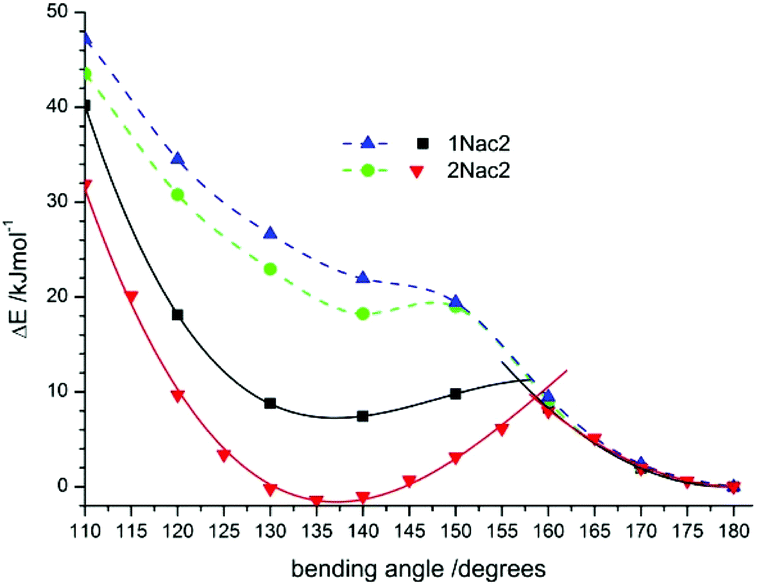
As shown in Fig. 12 for the naphthyl derivatives of acetylene a large excess of excitation energy is needed to achieve a σπ*/ππ* state intersection (about 36 kJ mol−1 at a bending angle equal to 135 degrees, and 42 kJ mol−1 at a bending angle equal to 145 degrees for 2Nac and 1Nac, respectively). Moreover, the σπ* is less stable than the linear ππ* by about 18 kJ mol−1 for 2Nac and 40 kJ mol−1 for 1Nac. Other results were obtained for the buta-1,3-diyne derivatives. In this case, the presence of two acetylene units causes two possible bending modes of the molecule; one comprising atoms CAr–CC, like in the acetylene derivative, and the second one containing CC–C atoms. The results of the calculations are presented in Fig. 13.
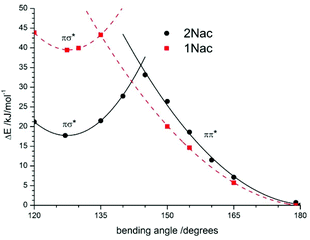 |
| | Fig. 12 The potential-energy profiles of the CPh–CC bending angle in the excited state for 1Nac (red dashed line) and 2Nac (black solid line). | |
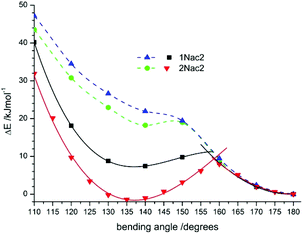 |
| | Fig. 13 The potential-energy profiles of CPh–CC (solid lines) and C–CC (dashed line) bending angles in the excited state for 1Nac2 (black solid line and blue dashed line) and 2Nac2 (red solid line and green dashed line). | |
The bending of the molecule at the site comprising atoms CC–C atoms is less favourable energetically. The potential energy of the bending curve increases without a clear minimum for both 1Nac2 and 2Nac2. However, bending comprising atoms CAr–CC leads to the σπ*/ππ* state intersection with a bending angle of about 160 degrees and an energy barrier of about 11 kJ mol−1 for 1Nac2 and 9 kJ mol−1 for 2Nac2. Moreover, for 1Nac2 the shallow minimum at a bending angle of about 135 degrees is observed and the σπ* state is less stable than the linear ππ* state by about 7 kJ mol−1, while 2Nac2 σπ* is a little more stable (1.7 kJ mol−1). Thus, theoretical calculations explain the differences in the photophysics between acetylene and buta-1,3-diyne derivatives of polycyclic aromatic hydrocarbon derivatives.
Conclusions
Polycyclic aromatic hydrocarbon derivatives of acetylene possess high fluorescence quantum yields and a weak dependence of fluorescence intensity on temperature. This is due to stiffening of the molecule in an excited state as a result of a strong electron coupling between polycyclic aromatic substituents. As a consequence the bending of the molecule leading to a σπ* state is highly energetically unfavourable, as confirmed by theoretical calculations. Thus, they do not show any anomalous dynamic behaviour but act as usual aromatic compounds. In the case of buta-1,3-diyne derivatives, the size of substituent has influence on their photophysical behaviour. For big substituents like pyrenyl and to some extent anhthryl, buta-1,3-diacetylene derivatives behave as the corresponding acetylene derivatives. However, bicyclic aromatic substituents (naphthyl) exhibit all the characteristics of a phenyl derivative of buta-1,3-diyne or acetylene. For 1- and 2-naphthyl isomers and 9-anthracene derivatives of acetylene derivatives, fluorescence and fluorescence excitation spectra are excitation and monitoring wavelength independent because of a coincidence of absorption and emission spectra of the rotamers.
Acknowledgements
This work was financially supported under grant UMO-2011/01/B/ST4/06094 from Polish National Science Centre (NCN).
Notes and references
- A. Okamoto, K. Tainaka, T. Unzai and I. Saito, Synthesis and fluorescence properties of dimethylaminonaphthalene-deoxyuridine conjugates as polarity-sensitive probes, Tetrahedron, 2007, 63, 3465–3470 CrossRef CAS.
- G.-S. Jiao, L. H. Thoresen and K. Burgess, Fluorescence, through-bond energy transfer cassettes for labelling multiple biological molecules in one experiment, J. Am. Chem. Soc., 2003, 125, 14668–14669 CrossRef CAS PubMed.
- R. Bandichhor, A. D. Petrescu, A. Vespa, A. B. Kier, F. Schroeder and K. Burgess, Water-soluble through-bond energy transfer cassettes for intramolecular imaging, J. Am. Chem. Soc., 2006, 128, 10688–10689 CrossRef CAS PubMed.
- N. N. Dioubankova, A. D. Malakov, Z. O. Shenkarev and V. A. Korshum, Oligonucleotides containing new fluorescent 1-phenylethynylpyrene and 9,10-bis(phenylethynyl)anthracene uridine-2′-carbamates: synthesis and properties, Tetrahedron, 2004, 60, 4617–4626 CrossRef CAS.
- K. M.-C. Wong, W.-S. Tang, X.-X. Lu, N. Zhu and V. W.-W. Yam, Functionalized platinium(II) tetrapyridylalkynyl complexes as colorimetric and luminescence pH sensor, Inorg. Chem., 2005, 44, 1492–1498 CrossRef CAS PubMed.
- S. Leroy, T. Soujanaya and F. Fages, Zinc(II)-operated intramolecular charge transfer fluorescence emission in pyrene-2,2′-bipyridine conjugated molecular rods, Tetrahedron Lett., 2001, 42, 1665–1667 CrossRef CAS.
- H. S. Joshi, R. Jamashidi and Y. Tor, Conjugated 1,10-phenantrolines as tunable fluorophore, Angew. Chem., Int. Ed., 1999, 38, 2722–2725 CrossRef CAS.
- C.-B. Huanga, J. Huang and L. Xu, A highly selective fluorescent probe for fast detection of nitric oxide in aqueous solution, RSC Adv., 2015, 5, 13307–13310 RSC.
- E. L. Spitler, S. P. McClintock and M. M. Haley, Dynamic proton-induced emission switching in donor-functionalized dehydrobenzopyrid[15]annulenes, J. Org. Chem., 2007, 72, 6692–6699 CrossRef CAS PubMed.
- T. A. Golovkova, D. V. Kozlov and D. C. Neckers, Synthesis and properties of novel switches, J. Org. Chem., 2005, 70, 5545–5549 CrossRef CAS PubMed.
- A. Elangowan, J.-H. Lin, H.-Y. Hsu and T.-I. Ho, Synthesis and electrogenerated chemiluminescence of donor-substituted phenylethynylcoumarins, J. Org. Chem., 2004, 69, 8086–8092 CrossRef PubMed.
- A. Elangovan, K.-M. Kao, S.-W. Yang, Y.-L. Chen, T.-I. Ho and Y. O. Su, Synthesis, electronic properties and electrochemiluminescence of donor-substituted phenylethynylanthronitriles, J. Org. Chem., 2005, 70, 4460–4469 CrossRef CAS PubMed.
- T. Zhao, Z. Liu, Y. Song, W. Xu, D. Zhang and D. Zhu, Novel diethynylcarbazolemacrocycles: synthesis and optoelectronic properties, J. Org. Chem., 2006, 71, 7422–7432 CrossRef CAS PubMed.
- R. Misra, R. Kumar, T. K. Chandrashekar and A. Nag D, Goswani, Org. Lett., 2006, 8, 629–631 CrossRef CAS PubMed.
- A. Harriman, G. Izzel and R. Ziessel, Rapid energy transfer in cascade-type Bodipy dyes, J. Am. Chem. Soc., 2006, 128, 10868–10875 CrossRef CAS PubMed.
- C.-H. Chen, J. T. Lin and M.-C. Yeh, Nonconjugated red-emitting dendrimers with p-type and/or n-type peripheries, Org. Lett., 2006, 8, 2233–2236 CrossRef CAS PubMed.
- Y. Yamaguchi, T. Ochi, T. Wakamiya, Y. Matsubara and Z.-I. Yoshida, New Fluorophores with Rod-Shaped Polycyano π-Conjugated Structures: Synthesis and Photophysical Properties, Org. Lett., 2006, 8, 717–720 CrossRef CAS PubMed.
- M. Kivala and F. Diederich, Acetylene-derived strong organic acceptors for planar and nonplanar push–pull chromophores, Acc. Chem. Res., 2009, 42, 235–248 CrossRef CAS PubMed.
- D. R. Kanis, M. A. Ratner and T. Marks, Design and construction of molecular assemblies with large second-order optical nonlinearities. Quantum chemical aspects, Chem. Rev., 1994, 94, 195–242 CrossRef CAS.
- I. Bylińska, M. Wierzbicka, C. Czaplewski and W. Wiczk, Solvatochromic studies of pull–push molecules containing dimethylaniline and aromatic hydrocarbon linked by an acetylene unit, RSC Adv., 2014, 4, 48783–48795 RSC.
- U. H. F. Bunz, Poly(aryleneethynylene)s: synthesis, properties, structures and applications, Chem. Rev., 2000, 100, 1605–1644 CrossRef CAS PubMed.
- J. M. Tour, Conjugated macromolecules of precise length and constitution, organic synthesis for construction of nanoarchitectures, Chem. Rev., 1996, 96, 537–554 CrossRef CAS PubMed.
- T.-A. V. Khuong, J. E. Nunez, C. E. Gondinez and M. A. Gracia-Garibay, Crystalline Molecular Machines: A Quest Toward Solid-State Dynamics and Function, Acc. Chem. Res., 2006, 39, 413–422 CrossRef CAS PubMed.
- S. Khatua, J. M. Guerrero, K. Claytor, G. Vives, A. B. Kolomeisky, J. M. Tour and S. Link, Micrometer-scale translation and monitoring of individual nanocars on glass, ACS Nano, 2009, 3, 351–356 CrossRef CAS PubMed.
- G. Vives and J. M. Tour, Synthesis of single-molecule nanocars, Acc. Chem. Res., 2009, 42, 473–487 CrossRef CAS PubMed.
- S. Sankararaman, G. Venkataramana and B. Vrghese, Conformational isomers from ratation of diacetylenic bond in an ethynylpyrene molecular hinge, J. Org. Chem., 2008, 73, 2404–2407 CrossRef CAS PubMed.
- A. Sakar and S. S. Talwar, Heteroaryl functionalised diacetylene: preparation and solid-state reactivity, J. Chem. Soc., Perkin Trans. 1, 1998, 4141–4146 RSC.
- D. Y. Ahn and J. M. Kim, Fluorogenic polydiacetylene supramolecules: immobilization, micropattering and application to label-free chemosensors, Acc. Chem. Res., 2008, 47, 805–816 CrossRef PubMed.
- K. Okuyama, T. Hasegawa, M. Ito and N. Mikami, Electronic spectra of tolan in supersonic free jet; large-amplitude torsional motion, J. Phys. Chem., 1984, 88, 1711–1716 CrossRef CAS.
- Y. Hirata, T. Okada and N. Mataga, Picosecond time-resolved absorption spectrum measurements of the higher excited singlet state of diphenylacetylene in the solution phase, J. Phys. Chem., 1992, 96, 6559–6563 CrossRef CAS.
- C. Ferrante, U. Kensy and B. Dick, Does diphenylacetylene (tolan) fluoresce from its second excited singlet state? Semiempirical MO calculations and fluorescence quantum yield measurements, J. Phys. Chem., 1993, 97, 13457–13463 CrossRef CAS.
- Y. Hirata, T. Okada and T. Nomoto, Photophysical properties of diphenylacetylene derivatives in the solution phase; comparison with the dynamical properties of diphenylbutadiyne, Chem. Phys. Lett., 1998, 293, 371–377 CrossRef CAS.
- Y. Hirata, T. Okada and T. Nomoto, Photophysical properties of diphenylacetylene and diphenylbutadiyne derivatives in solution phase, Acta Phys. Pol., A, 1998, 94, 627–636 CAS.
- Y. Hirata, Photophysical and photochemical primary processes of diphenylacetylene derivatives and related compounds in liquid phase, Bull. Chem. Soc. Jpn., 1999, 72, 1647–1664 CrossRef CAS.
- Y. Nagano, T. Ikoma, K. Akiyama and S. Tero-Kubota, Symmetry switching of the fluorescence excited state in α,ω-diphenylpolyynes, J. Am. Chem. Soc., 2003, 125, 14103–14112 CrossRef CAS PubMed.
- M. Wierzbicka, I. Bylińska, C. Czaplewski and W. Wiczk, Experimental and theoretical studies of spectroscopic properties of simple symmetrically substituted diphenylacetylene derivatives, RSC Adv., 2015, 5, 29294–29303 RSC.
- M. Wierzbicka, I. Bylińska, A. Sikorski, C. Czaplewski and W. Wiczk, Experimental and theoretical studies of spectroscopic properties of simple symmetrically substituted diphenylbuta-1,3-diyne derivatives, Photochem. Photobiol. Sci., 2015, 14, 2251–2260 CAS.
- M. Z. Zgierski and E. C. Lim, Nature of the “dark” state in diphenylacetylene and related molecules: state switch from linear ππ* state to the bent πσ* state, Chem. Phys. Lett., 2004, 387, 353–355 CrossRef.
- M. Z. Zgierski, T. Fujiwara and E. C. Lim, Role of the πσ* state in molecular photophysics, Acc. Chem. Res., 2010, 43, 806–817 CrossRef PubMed.
- J. Saltiel and V. K. R. Kumar, Photophysics of diphenylacetylene: light from the “dark state”, J. Phys. Chem. A, 2012, 116, 10548–10558 CrossRef CAS PubMed.
- R. Gleiter and D. B. Werz, Alkynes between main group elements: from dumbbells via rods to squares and tubes, Chem. Rev., 2010, 110, 4447–4488 CrossRef CAS PubMed.
- U. H. F. Bunz, K. Seehafer, M. Bendera and M. Porza, Poly(aryleneethynylene)s (PAE) as paradigmatic sensor cores, Chem. Soc. Rev., 2015, 44, 4322–4336 RSC.
- K. Sonogashira, Development of Pd–Cu catalyzed cross-coupling of terminal acetylenes with sp2-carbon halides, J. Organomet. Chem., 2002, 653, 46–49 CrossRef CAS.
- K. Sonogashira, Y. Tohda and N. Hagihara, Convinient synthesis of acetylenes – catalytic substitutions of acetylenic hydrogen with bromoalkanes, iodoarenes and bromopyridines, Tetrahedron Lett., 1975, 16, 4467–4470 CrossRef.
- C. A. Glaser, BeiträgezurKenntniss des Acetenylbenzols, Ber. Dtsch. Chem. Ges., 1869, 2, 422–424 CrossRef.
- P. W. Thulstrup, S. V. Hoffmann, B. K. V. Hansen and J. Spanget-Larsen, Unique interplay between electronic states and dihedral angle for the molecular rotor diphenyldiacetylene, Phys. Chem. Chem. Phys., 2011, 13, 16168–16174 RSC.
- A. Beeby, K. Findlay, P. J. Low and T. B. Marder, A re-evaluation of the photophysical properties of 1,4-bis(phenylethynyl)benzene: a model for poly(phenyleneethynylene), J. Am. Chem. Soc., 2002, 124, 8280–8284 CrossRef CAS PubMed.
- P. V. James, P. K. Sudeep, C. H. Suresh and K. G. Thomas, Photophysical and theoretical investigation of oligo(p-phenyleneethynylene)s: effect of aloxy substitution and alkyne-aryl bond rotations, J. Phys. Chem. A, 2006, 110, 4329–4337 CrossRef CAS PubMed.
- A. Beeby, K. S. Findlay, A. E. Goeta, L. Porres, S. R. Rutter and A. L. Thomson, Engineering a twist in 9,10-diethynylanthracenes by steric interactions, Photochem. Photobiol. Sci., 2007, 6, 982–986 CAS.
- G. Duvanel, J. Grilj, A. Schuvey, A. Gossauer and E. Vauthey, Ultrafast excited-state dynamics of phenyleneethylene oligomers in solution, Photochem. Photobiol. Sci., 2007, 6, 952–963 Search PubMed.
- L. Gutierez-Arzaluz, C. A. Guarin, W. Rodrigez-Cordoba and J. Peon, Dynamics of the formation of a charge transfer state in 1,2-bis(9-anthryl)acetylene in polar solvents: symmetry reduction with the participation of an intramolecular torsion coordinate, J. Phys. Chem. B, 2013, 117, 12175–12183 CrossRef PubMed.
- M. I. Sluch, A. Godt, U. H. F. Bunz and M. A. Berg, Excited-state dynamics of oligo(p-phenyleneethynylene): quadratic coupling and torsion motions, J. Am. Chem. Soc., 2001, 123, 6447–6448 CrossRef CAS PubMed.
- L. T. Liu, D. Yaron, M. I. Sluch and M. A. Berg, Modeling the effects of torsional disorder on the spectra of poly- and oligo-(p-phenyleneethynylenes), J. Phys. Chem. B, 2006, 110, 18844–18852 CrossRef CAS PubMed.
- L. T. Liu, D. Yaron and M. A. Berg, Electron–phonon coupling in phenyleneethynylene oligomers: a nonlinear one-dimensional configuration-coordinate model, J. Phys. Chem. C, 2007, 111, 5770–5782 CAS.
- K. Schmieder, M. Levitus, H. Dang and M. A. Gracia-Garibay, Photophysical properties of coplanar and twisted 1,4-Bis(9-ethynylanthracenyl)benzene. Rotational equilibration in the excited states of diaryalkynes, J. Phys. Chem. A, 2002, 106, 1551–1556 CrossRef CAS.
- T. Fujiwara, M. Z. Zgierski and E. C. Lim, Spectroscopy and photophysics of 1,4-bis(phenylethynyl)benzene: effects of ring torsion and dark σπ* state, J. Phys. Chem. A, 2008, 112, 4736–4741 CrossRef CAS PubMed.
- Y. Hirata, T. Okada and T. Nomoto, Photophysical properties of diphenylacetylene derivatives in solution phase III. Thermal repopulation of the S2 state of (aminophenyl)phenylacetylene, J. Phys. Chem., 1993, 97, 9677–9681 CrossRef CAS.
- Y. Hirata, T. Okada and T. Nomoto, Higher excited singlet state of diphenylacetylene in solution phase, Chem. Phys. Lett., 1993, 209, 397–402 CrossRef CAS.
- Y. Hirata, T. Okada and T. Nomoto, Photophysical properties of diphenylacetylene derivatives in solution phase. IV. N,N-dimethylanilino(phenyl)acetylene in n-hexane, Chem. Phys. Lett., 1996, 257, 513–518 CrossRef CAS.
- X. Yin, Y. Li, Y. Zhu, X. Jing, Y. Li and D. Zhu, A highly sensitive viscosity probe based on ferrocene-BODIPY dyes, Dalton Trans., 2010, 39, 9929–9935 RSC.
- S. Toyota, Rotational isomerism involving acetylene carbon, Chem. Rev., 2010, 110, 5398–5424 CrossRef CAS PubMed.
- U. Mazzucato and F. Mmomicchioli, Rotational Isomerism in trans-1,P-Diarylethylenes, Chem. Rev., 1991, 91, 1679–1719 CrossRef CAS.
- Y. Li, J. Zhao, X. Yin, H. Liu and G. Yin, Conformational analysis of diphenylacetylene under the influence of an external electric field, Phys. Chem. Chem. Phys., 2007, 9, 1186–1193 RSC.
- A. A. Bothner-By, J. Dadok, T. E. Johnson and J. S. Lindsey, Molecular dynamics of covalently-linked multi-porphyrinarrays, J. Phys. Chem., 1996, 100, 17551–17557 CrossRef CAS.
- W. Wiczk, K. Stachowiak, P. Skurski, L. Łankiewicz, A. Michniewicz and A. Rój, Photophysics of 7-Hydroxytetrahydroisoquinoline-3-carboxylic acid and its derivatives, J. Am. Chem. Soc., 1996, 118, 8300–8307 CrossRef CAS.
- Y.-P. Sun, D. F. Sears Jr., J. Saltiel, F. B. Mallory, C. W. Mallory and C. A. Buser, Principal component-three component self modelling analysis applied to trans-1,2-di-(2-naphthyl)ethane fluorescence, J. Am. Chem. Soc., 1988, 110, 6974–6984 CrossRef CAS.
- J. Saltiel, D. F. Sears Jr., J.-O. Choi, Y.-P. Sun and D. W. Eaker, The fluorescence, fluorescence-excitation and UV absorption spectra of trans-1-(2-naphthyl)-2-phenylethene conformers, J. Phys. Chem., 1994, 98, 35–446 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Scheme of synthesis and identification data, absorption and emission spectra in 2-MeTHF and acetonitrile, and low temperature luminescence data, fluorescence quantum yield and lifetime, potential of rotation energy in the ground and excited state. See DOI: 10.1039/c5pp00197h |
| ‡ The first two authors contributed equally to the work. |
|
| This journal is © The Royal Society of Chemistry and Owner Societies 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.