
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Phage-displayed macrocyclic glycopeptide libraries†
Simon
Ng
and
Ratmir
Derda
*
Alberta Glycomics Centre and Department of Chemistry, University of Alberta, Edmonton, AB T6G 2G2, Canada. E-mail: ratmir@ualberta.ca
First published on 4th February 2016
Abstract
In this report, we describe an efficient way to generate libraries of macrocyclic glycopeptides in one step by reacting phage-displayed libraries of peptides with dichloro-oxime derivatives. We showed that the reactions do not interfere with the ability of phage to replicate in bacteria. The reactions are site-selective for phage-displayed peptides and they do not modify any other proteins of phage. The technology described in this report will be instrumental for genetic selection of macrocyclic glycopeptides for diverse glycan-binding proteins.
Synthetic macrocycles hold great promise for the development of drugs targeting proteins which are difficult to target by conventional small-molecule drugs.1 The pharmacodynamic and pharmacokinetic properties of macrocyclic peptides are generally more favorable as compared to their linear peptide precursors.2 Flexibility in linear peptides makes them susceptible to enzymatic degradation. In contrast, restricting the conformation of peptides by macrocyclization improves their proteolytic stability.3 Macrocyclization of peptides can also increase their binding affinity by preorganizing the ligands into their biologically active conformations and minimizing the contribution of unproductive conformations.4 This preorganization allows macrocycles to interact with a larger surface area of the target protein while maintaining productive conformation.5 Although linear peptide ligands of similar size can span similar area, maintenance of one productive conformation and elimination of all other conformations result in large entropic penalty and often decrease free energy of ligand–receptor interaction.
Discovery and development of new macrocyclic ligands for proteins usually mandate synthesis of libraries of macrocycles. Amongst many library syntheses available, technologies that permit genetic encoding of macrocyclic libraries are the most attractive because they allow identification of target-binding compounds from a mixture of 108–1015 derivatives present in the same solution. This selection approach is technologically simpler than screening of libraries of compounds in multiwell-plates and higher throughput than one-bead-one-compound6 approaches. Of several synthetic approaches available for constructing genetically encoded libraries, we chose the approach that modifies the existing, biologically produced, and genetically encoded linear libraries using chemical cross-linkers.7 Such approach bypasses the need for complex multi-step synthesis of DNA-conjugated molecules,8,9 proximity-driven ligations,9,10 and bottom-up synthesis of peptide oligomers assisted by genetically encoded reaction routing.11
Since its inception in 1985,12 phage display has been one of the dominant technologies for the discovery of biological drugs.13 Antibodies and peptides derived from this technology have been one of the fastest growing contributors to FDA-approved drugs.14 Recently, we have developed a novel approach, termed “genetically encoded fragment-based discovery” (GE-FBD),15,16 for the identification of potent ligands for glycan-binding proteins from linear glycopeptide libraries displayed on M13 phage.15 The chemistry we used to generate these libraries—N-terminal serine-mediated oxime ligation—is suitable for the production of linear glycopeptide libraries.17 In this report, we aim to generate cyclic glycopeptide library, through only one-step and rapid chemical modification of phage-displayed peptide libraries.
Known synthetic approaches that convert peptides to cyclic glycopeptides employ head-to-tail macrocyclization of glycosylated peptide via thioester formation by thioesterase,18 or lactamization between N- and C-termini.19 While useful for the generation of synthetic cyclic glycopeptides, they are not suitable for the modification of phage library because the peptides displayed on phage lack free C-terminus. Mihara and co-workers reported pseudo-cyclic glycopeptide libraries on phage based on a β-loop stabilized by non-covalent interactions between antiparallel β-strands.20 The authors decorated the library with mannose via a disulfide bond with a constant Cys in the peptide library. Such disulfide linkage, however, is susceptible to reduction and disulfide exchange. Furthermore, expression of peptides with single or odd number of Cys on phage is known to be challenging21 due to (i) trapping of phage in periplasm of E. coli, (ii) mis-folding of pIII proteins by forming disulfides with structural Cys in the pIII and (iii) enforced enrichment of library clones with even number of cysteine.22 The authors bypassed the problem (ii) by expressing the library in phage with Cys-free pIII protein, but the drawback of this variant is >100-fold diminished infectivity of phage.23 Multiple strategies for cyclization of peptides on phage have been reported by our group24 and others,25,26 but introducing a carbohydrate fragment using these approaches is not obvious.
Our design was inspired by cyclization of peptides by 1,3-dichloroacetone (DCA) recently reported by Dawson and co-workers.27 An attractive aspect of this approach is the possibility for functionalization of macrocycles via the ketone handle. For example, Dawson combined DCA-mediated macrocyclization with oxime ligation to yield a labeled cyclic peptide (Fig. 1A). Unfortunately, we found the original approach to be sub-optimal for the modification of phage-displayed libraries because it requires exposure of peptide (i.e., phage) to two consecutive reactions. One of them—a 17 hour long oxime ligation of ketone in anilinium buffer (pH 4.5)—is detrimental to the infectivity of phage particles when reaction is performed in one pot (see Fig. 6).
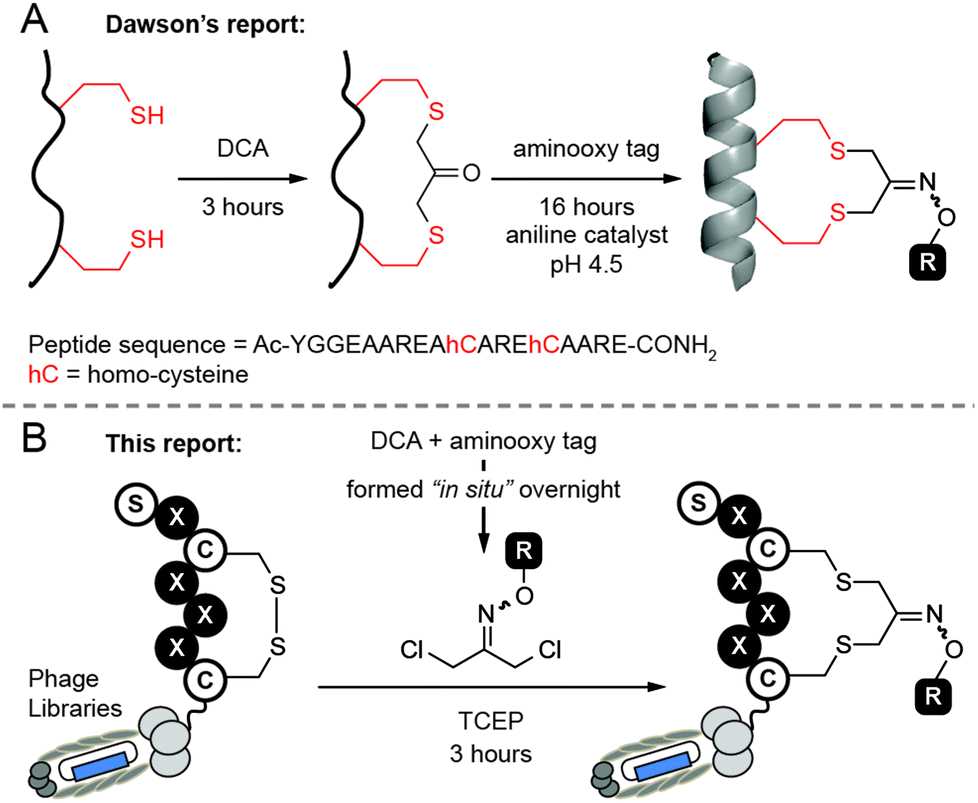 | ||
| Fig. 1 Macrocyclization of peptides and genetically encoded peptide libraries with dichloro-oxime derivatives. (A) Approach reported by Dawson and co-workers. DCA = 1,3-dichloroacetone. (B) We propose to use dichloro-oxime derivatives (DCO) which were formed “in situ” to functionalize phage-displayed peptide libraries in one step. | ||
In this report, we modified this methodology to make functionalized phage libraries in one, rapid, biocompatible step (Fig. 1B). We found that dichloro-oxime (DCO) derivatives, such as DCO-biotin can be pre-formed in >99% conversion by overnight incubation of 1,3-dichloroacetone with 1.1 equivalent of aminooxy-biotin in DMF in the absence of any catalysts (see ESI† for the characterization). We were pleased to find that DCO-biotin formed “in situ” and used without any purification, afforded clean macrocyclization in model synthetic peptide H2N-Ser-Trp-Cys-Ser-Cys-CONH2 (SWCSC). In 20 min, 0.1 mM of SWCSC peptide reacted quantitatively with 1 mM of DCO-biotin in Tris buffer (pH 8.5) in the presence of tris(2-carboxyethyl)phosphine (TCEP) to form the macrocyclic peptide 1 as the sole product (Fig. 2A). Reaction reached >99% conversion in 20 min, according to UPLC-MS analysis, and exhibited second-order rate constant k = 3.8 M−1 s−1 (Fig. 2B). We detected no by-products after the completion of the reaction (20 min). Although both the starting peptide and the macrocyclic product 1 contain nucleophilic N-terminal amine, even after incubation for 72 hours in the presence of 10 equivalents of crude DCO-biotin, macrocyclic peptide 1 remained as the major product (Fig. 2C). We note that the macrocyclic peptide 1 is a mixture of E/Z isomers of oxime, but this mixture appeared as a single peak on UPLC traces because the isomers are inseparable. Accumulated minor by-products constituted less than 50% as determined by the integration of all peaks above the baseline.28 These observations suggest that any side reaction of the peptide, such as the undesired alkylation of N-terminal amine with DCO-biotin, has half-life (t1/2) >2.6 × 105 s ≈ 72 h. It is over 1000 times slower than alkylation of thiol (t1/2 = 182 s) under the same conditions. Unlike the original report,27 macrocyclization of peptide with DCO derivatives was completed in one step and bypassed the need for purification of the peptide–ketone intermediate and subsequent prolonged oxime ligation (16 hours) of the ketone intermediate at pH 4.5.
 | ||
| Fig. 2 Macrocyclization of linear peptide. (A) The reaction of SWCSC with DCO formed the macrocyclic products (1–5). Percentages are the conversion yields determined by UV absorbance measured with UPLC instrument. Reaction time is showed in parentheses. (B) The reaction of SWCSC with DCO-biotin under pseudo-first-order condition gave second-order rate constant k = 3.8 M−1 s−1 (percentage error in bracket). The kinetics of the reaction was monitored by UPLC-MS. Red line represents the best-fit curve and dashed lines encompass the 95% confidence interval of the fit. (C) UPLC traces of starting material (SWCSC, t = 0 min), and UPLC traces of the reaction between SWCSC and DCO-biotin after 20 min, 3 h or 72 h. *: DCO-biotin (weak absorbance at 280 nm). | ||
Encouraged by the success of the model study with DCO-biotin, we extended this approach to four different DCO-carbohydrate derivatives—DCO-ManS, DCO-ManL, DCO-Fuc and DCO-Gal—to form the desired macrocyclic peptide–carbohydrate 2, 3, 4 and 5 respectively (Fig. 2A). Peptide SWCSC was incubated with each DCO-derivative at room temperature for 5, 15, or 30 min, and then quenched by the addition of acetic acid, followed by analysis using UPLC-MS. The reactions with DCO-carbohydrate derivatives were ∼2.5 times slower than that with DCO-biotin (k = 3.8 M−1 s−1, Fig. 2B). Still, these reactions proceeded to near completion (>90%) in 30 min with second-order rate constant k = 1.5–1.8 M−1 s−1 and formed no by-products (see ESI Fig. S1–S4†).
Next, we investigated whether the reaction could be applied to disulfide-containing peptide libraries SXCX3C and SXCX4C displayed on M13 phage (X represents 19 any natural amino acids and no Cys) (Fig. 3A). Three independent on-phage reactions were performed on separate days to assure the reproducibility. All on-phage reactions described in this report were performed with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of phage that displays a library of peptides (“Lib”), and wild-type (WT) phage which lacks the displayed disulfide-peptide, but otherwise it is identical to Lib. Two phage populations can be evaluated together because Lib and WT form blue and white plaques respectively in agar overlay supplemented by X-Gal. We used biotin-capture assay17,29 developed in our lab to quantify the efficiency of the reaction and perform the kinetic studies. Phage libraries were biotinylated and reached saturation at ∼85% of capture within 3 hours (Fig. 3B). Fitting the kinetic curve afforded the rate constant k = 1.1 M−1 s−1 which is ∼3.5-fold slower than the reaction of DCO-biotin with model peptide (k = 3.8 M−1 s−1). Difference in rate is not surprising because phage libraries contain peptides with varying reactivity. This observation highlights the importance of monitoring the reaction progress in the context of phage library even after the reaction has been well optimized on model peptides or proteins.
:1 mixture of phage that displays a library of peptides (“Lib”), and wild-type (WT) phage which lacks the displayed disulfide-peptide, but otherwise it is identical to Lib. Two phage populations can be evaluated together because Lib and WT form blue and white plaques respectively in agar overlay supplemented by X-Gal. We used biotin-capture assay17,29 developed in our lab to quantify the efficiency of the reaction and perform the kinetic studies. Phage libraries were biotinylated and reached saturation at ∼85% of capture within 3 hours (Fig. 3B). Fitting the kinetic curve afforded the rate constant k = 1.1 M−1 s−1 which is ∼3.5-fold slower than the reaction of DCO-biotin with model peptide (k = 3.8 M−1 s−1). Difference in rate is not surprising because phage libraries contain peptides with varying reactivity. This observation highlights the importance of monitoring the reaction progress in the context of phage library even after the reaction has been well optimized on model peptides or proteins.
 | ||
| Fig. 3 Macrocyclization of the phage-displayed peptide libraries. (A) The reaction of 1 mM DCO-biotin with disulfide-containing phage library was monitored by biotin-capture assay in the presence of wild-type (WT) phage as control. (B) The kinetics of reaction between phage and DCO-biotin. Reaction of WT phage is insignificant even after 3 hours of incubation. In the absence of TCEP, reaction of neither library nor WT phage proceeded to any detectable extent (>5%) even after 3 hours of incubation. (C) After 3 hours of modification, more than 80% of phage population retained the ability to infect bacterial host. Data in panel B and C are mean values from three independent experiments; error bars represent one standard deviation. | ||
Importantly, DCO-biotin did not react with WT phage present in the same solution; biotinylation of WT was negligible (<5%). DCO-biotin reacted specifically with peptide sequences displayed on Lib-phage and it did not have any detectable reactivity with other nucleophilic functionalities common to Lib and WT phage. For example, both WT and Lib phage contain four pairs of internal disulfide bonds within pIII coat proteins30 and a vast number of other nucleophilic functionalities on the pVIII coat proteins (e.g., ∼2700 copies of N-terminus amines, similar number of ε-amine of Lys, phenol of Tyr, etc.). Lack of biotinylation of WT proved that none of these functionalities reacted with DCO; it alkylated only externally displayed disulfide. Moreover, in the absence of TCEP, neither Lib nor WT phage were biotinylated. This experiment highlights the excellent regio-selectivity of the reduction of external disulfides by TCEP. To summarize, modification with DCO takes place only when two conditions are satisfied: (1) disulfide displayed on the N-terminus of pIII protein is solvent-accessible;24,30 and (2) free thiol is available through TCEP reduction.
The chemical post-translational modification by DCO-biotin is benign to phage. After 3 hours of treatment with DCO-biotin, more than 80% of phage population still retained its ability to infect bacterial host (Fig. 3C). This result is in contrast to the report of Heinis and Winter,25 in which the modification of phage with 10 μM of 1,3,5-tris(bromomethyl)benzene (TBMB) for 1 hour resulted in ∼80% loss of phage infectivity and much greater losses at higher concentration of TBMB. Heinis and co-workers stated in their report that the loss of infectivity is due to the cross-linking of phage coat proteins.25 In this study, we observed excellent viability of phage after modification with DCO, suggesting that the cross-linking is negligible.
To assess the efficiency of on-phage reaction with DCO derivatives lacking the biotin tag, we employed a “pulse-chase” reaction developed in our group.17,24 We first validated the pulse-chase approach by monitoring the reaction on synthetic peptide by UPLS-MS (Fig. 4A). We pulsed the peptide with DCO-ManL and chased with excess of biotin-PEG2-iodoacetamide (BIA), a thiol-reactive probe with high reactivity (k = 10.8 M−1 s−1, see ESI Fig. S5†). Peptide pulsed with DCO-ManL for 5 min and chased with BIA formed only two products (Fig. 4A): 51% of the desired macrocyclic product 3 and 49% of peptide with two biotin moieties (Fig. 4B, middle spectrum). UPLC-MS did not detect any peptide that contained DCO and biotin simultaneously, suggesting that the intramolecular cyclization was rapid and the open-chain peptide intermediates with mono-reacted Cys did not accumulate. Reaction proceeded to near 99% conversion when pulsing with DCO-ManL for 60 min and BIA did not react with the product 3 after 30 min of exposure (Fig. 4B, bottom spectrum).
 | ||
| Fig. 4 Macrocyclization of linear peptide under pulse-chase condition. (A) SWCSC is pulsed with 10 eq. of DCO-ManL for 5 or 60 min; and then chased immediately with 10 eq. of BIA for 30 min. (B) The pulse-chase reactions were monitored by UPLC-MS. After pulsing with DCO-ManL for 5 min, 51% of peptide formed the desired macrocyclic product 3 (green peak, middle spectrum) and 49% of unreacted peptide was biotinylated twice to give the peptide–biotin conjugate 6 (blue peak, middle spectrum). In contrast, when pulsing with DCO-ManL for 60 min, no peptide–biotin conjugate 6 was observed and nearly all SWCSC formed the desired product 3 (green peak, bottom spectrum). Red peaks: BIA. | ||
We then applied the validated “pulse-chase” method to monitor the modification of phage libraries with different DCO-carbohydrate derivatives (Fig. 5). First, the mixture of phage libraries (SXCX3C and SXCX4C) and WT phage were incubated with four different DCO-carbohydrate derivatives (1 mM) in Tris buffer (pH 8.5) in the presence of 0.5 mM TCEP (Fig. 5A). After pulsing with DCO for 3 hours, the solutions of phage were diluted by 100-fold to halt the modification. To quantify the unreacted phage, we incubated the phage with 1 mM BIA for 30 min. Phage library which was not exposed to any DCO for 3 hours had unreacted thiols, thereby leading to ∼87% of capture after exposure to BIA. This observation was consistent with ∼85% capture observed in alkylation of phage libraries by DCO-biotin (Fig. 3B). In contrast, “pulsing” the phage library with various DCO-carbohydrate derivatives for 3 hours, followed by BIA “chasing”, resulting in decrease of phage capture (blue bars, Fig. 5B). The results suggested that 33%, 50%, 53% and 52% of phage libraries were modified with DCO-ManS, DCO-ManL, DCO-Fuc and DCO-Gal respectively after 3 hours (black bars in Fig. 5B). The incomplete modifications were consistent with our observation that DCO-carbohydrate derivatives (k = 1.5–1.8 M−1 s−1) have slower reactivity than DCO-biotin (k = 3.8 M−1 s−1) in the reactions with model peptide (see Fig. 2B). We note that WT phage presents in the same solution was not captured in any experiments (Fig. 5B).
 | ||
| Fig. 5 Macrocyclization of phage libraries with four different DCO-carbohydrate derivatives. (A) Mixture of phage libraries and WT phage was pulsed with DCO derivatives for 3 hours, diluted by 100-fold, and then chased immediately with BIA for 30 min. Under these conditions, any unreacted free thiol would be biotinylated, resulting in the capture of phage. (B) Bar graph represents the efficiency of modification. About 87% of phage population was captured when no DCO was added. The black bars quantified the population of phage libraries (white labels) that were fully modified with DCO-carbohydrate derivatives. | ||
As a secondary validation of “pulse-chase” method, we pulsed the mixture of Lib and WT phage with DCA for 3 h and then chased with BIA. We observed no biotinylation of phage during the process indicating that in 3 hours all thiols were converted to cyclic peptide–ketone derivatives. We further validated it by treating the DCA-reacted phage with aminooxy-biotin (AOB) in anilinium acetate buffer (pH 4.7) for 17 h (Fig. 6A). After this treatment, ∼72% of the phage libraries contained biotin and could be captured, whereas, WT phage exhibited insignificant capture (Fig. 6B). As noted in the introduction, this one-pot two-step manipulation was detrimental to phage viability. Only ∼12% of the Lib and WT population survived the 17 hour-long reaction (Fig. 6B). The reason for the decrease in viability is not clear but we hypothesize that the imine formed by DCA and aniline could be susceptible to attack by side-chain nucleophiles,31 leading to cross-linking of phage coat proteins.
 | ||
| Fig. 6 Two-step macrocyclization and functionalization of phage libraries with biotin. (A) Scheme showing the one-pot two-step reaction on phage. (B) We observed ∼72% of phage libraries, but not WT phage, were biotinylated under such conditions. However, this two-step approach might generate side products which are detrimental to phage infectivity as demonstrated by the significant drop of viability of both library and WT phage down to ∼12%. Data are mean values of two independent experiments; error bars represent one standard deviation. | ||
In conclusion, we have demonstrated the use of DCO, for one-step macrocyclization and labeling of a model peptide and phage-displayed peptide libraries with biotin and carbohydrates. Our strategy bypasses the need for the isolation of intermediate and minimizes the losses of valuable starting materials, e.g., peptides, proteins, or phage libraries, in multi-steps reactions. The reaction is highly chemo- and regio-selective and it does not interfere with the infectivity of phage libraries. The macrocyclic peptide libraries contain an additional fragment, in this work, carbohydrate which could be useful for selection with glycan-binding proteins.15 The fragment is not limited to carbohydrates; any known inhibitors, enzyme substrates or small-molecule leads could be incorporated into the libraries and serve as an anchor fragment to facilitate the search of more potent compounds using genetically encoded fragment-based design.15,16,20,32 We anticipate that this strategy will find wide application for fragment-based selection of conformationally constrained, proteolytically stable, and potent macrocyclic peptide-fragment conjugates for many protein targets.
Acknowledgements
This work was supported by research grants from the National Science and Engineering Research Council of Canada (NSERC, #402511) and Alberta Glycomics Centre. Infrastructure support was provided by Canadian Foundation for Innovation (CFI) New Leaders Opportunity. S. Ng thanks Alberta Innovates Technology Future for the scholarship. We thank Pavel Kitov for the synthesis of carbohydrate-hydroxylamine derivatives and Katrina Tjhung for the expression of phage libraries. We are grateful to Ferring Pharmaceuticals for funding the purchase of DNA for these libraries.Notes and references
- E. M. Driggers, S. P. Hale, J. Lee and N. K. Terrett, Nat. Rev. Drug Discovery, 2008, 7, 608–624 CrossRef CAS PubMed; E. A. Villar, D. Beglov, S. Chennamadhavuni, J. A. Porco Jr., D. Kozakov, S. Vajda and A. Whitty, Nat. Chem. Biol., 2014, 10, 723–731 CrossRef PubMed.
- E. Marsault and M. L. Peterson, J. Med. Chem., 2011, 54, 1961–2004 CrossRef CAS PubMed; F. Giordanetto and J. Kihlberg, J. Med. Chem., 2014, 57, 278–295 CrossRef PubMed.
- Z. Szewczuk, B. F. Gibbs, S. Y. Yue, E. O. Purisima and Y. Konishi, Biochemistry, 1992, 31, 9132–9140 CrossRef CAS PubMed; R. J. Clark, H. Fischer, L. Dempster, N. L. Daly, K. J. Rosengren, S. T. Nevin, F. A. Meunier, D. J. Adams and D. J. Craik, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 13767–13772 CrossRef PubMed; Y. H. Lau, P. de Andrade, S.-T. Quah, M. Rossmann, L. Laraia, N. Skold, T. J. Sum, P. J. E. Rowling, T. L. Joseph, C. Verma, M. Hyvonen, L. S. Itzhaki, A. R. Venkitaraman, C. J. Brown, D. P. Lane and D. R. Spring, Chem. Sci., 2014, 5, 1804–1809 RSC.
- H. Kessler, Angew. Chem., Int. Ed. Engl., 1982, 21, 512–523 CrossRef CAS; L. D. Walensky, A. L. Kung, I. Escher, T. J. Malia, S. Barbuto, R. D. Wright, G. Wagner, G. L. Verdine and S. J. Korsmeyer, Science, 2004, 305, 1466–1470 CrossRef PubMed; J. Gavenonis, B. A. Sheneman, T. R. Siegert, M. R. Eshelman and J. A. Kritzer, Nat. Chem. Biol., 2014, 10, 716–722 CrossRef PubMed; T. A. Hill, N. E. Shepherd, F. Diness and D. P. Fairlie, Angew. Chem., Int. Ed., 2014, 53, 13020–13041 CrossRef PubMed.
- C. J. White and A. K. Yudin, Nat. Chem., 2011, 3, 509–524 CrossRef CAS PubMed; A. K. Yudin, Chem. Sci., 2015, 6, 30–49 RSC.
- K. S. Lam, R. Liu, S. Miyamoto, A. L. Lehman and J. M. Tuscano, Acc. Chem. Res., 2003, 36, 370–377 CrossRef CAS PubMed.
- C. Heinis and G. Winter, Curr. Opin. Chem. Biol., 2015, 26, 89–98 CrossRef CAS PubMed.
- S. Melkko, J. Scheuermann, C. E. Dumelin and D. Neri, Nat. Biotechnol., 2004, 22, 568–574 CrossRef CAS PubMed.
- Z. J. Gartner, B. N. Tse, R. Grubina, J. B. Doyon, T. M. Snyder and D. R. Liu, Science, 2004, 305, 1601–1605 CrossRef CAS PubMed.
- Z. J. Gartner and D. R. Liu, J. Am. Chem. Soc., 2001, 123, 6961–6963 CrossRef CAS PubMed.
- D. R. Halpin and P. B. Harbury, PLoS Biol., 2004, 2, 1022–1030 CAS.
- G. P. Smith, Science, 1985, 228, 1315–1317 CAS.
- C. M. Y. Lee, N. Iorno, F. Sierro and D. Christ, Nat. Protocols, 2007, 2, 3001–3008 CAS; S. S. Sidhu and C. R. Geyer, Phage display in biotechnology and drug discovery, CRC Press, Boca Raton, 2015 Search PubMed.
- A. E. Nixon, D. J. Sexton and R. C. Ladner, mAbs, 2014, 6, 73–85 CrossRef CAS PubMed; K. Omidfar and M. Daneshpour, Expert Opin. Drug Discovery, 2015, 10, 651–669 CrossRef PubMed; M. Hamzeh-Mivehroud, A. A. Alizadeh, M. B. Morris, W. Bret Church and S. Dastmalchi, Drug Discovery Today, 2013, 18, 1144–1157 CrossRef PubMed.
- S. Ng, E. Lin, P. I. Kitov, K. F. Tjhung, O. O. Gerlits, L. Deng, B. Kasper, A. Sood, B. M. Paschal, P. Zhang, C.-C. Ling, J. S. Klassen, C. J. Noren, L. K. Mahal, R. J. Woods, L. Coates and R. Derda, J. Am. Chem. Soc., 2015, 137, 5248–5251 CrossRef CAS PubMed.
- K. F. Tjhung, P. I. Kitov, S. Ng, E. N. Kitova, L. Deng, J. S. Klassen and R. Derda, J. Am. Chem. Soc., 2016, 138, 32–35 CrossRef CAS PubMed.
- S. Ng, M. R. Jafari, W. L. Matochko and R. Derda, ACS Chem. Biol., 2012, 7, 1482–1487 CrossRef CAS PubMed.
- H. Lin, D. A. Thayer, C.-H. Wong and C. T. Walsh, Chem. Biol., 2004, 11, 1635–1642 CrossRef CAS PubMed.
- H. Hu, J. Xue, B. M. Swarts, Q. Wang, Q. Wu and Z. Guo, J. Med. Chem., 2009, 52, 2052–2059 CrossRef CAS PubMed.
- K. Arai, H. Tsutsumi and H. Mihara, Bioorg. Med. Chem. Lett., 2013, 23, 4940–4943 CrossRef CAS PubMed.
- B. K. Kay, N. B. Adey, H. Yun-Sheng, J. P. Manfredi, A. H. Mataragnon and D. M. Fowlkes, Gene, 1993, 128, 59–65 CrossRef CAS PubMed; S. McConnell, A. Uveges, D. Fowlkes and D. Spinella, Mol. Diversity, 1996, 1, 165–176 CrossRef PubMed; T. Wind, S. Kjær and B. F. C. Clark, Biochimie, 1999, 81, 1079–1087 CrossRef PubMed.
- S. Chen, I. Rentero Rebollo, S. A. Buth, J. Morales-Sanfrutos, J. Touati, P. G. Leiman and C. Heinis, J. Am. Chem. Soc., 2013, 135, 6562–6569 CrossRef CAS PubMed.
- S. Chen and C. Heinis, in Peptide Libraries, ed. R. Derda, Springer, New York, 2015, vol. 1248, ch. 9, pp. 119–137 Search PubMed.
- M. R. Jafari, L. Deng, P. I. Kitov, S. Ng, W. L. Matochko, K. F. Tjhung, A. Zeberoff, A. Elias, J. S. Klassen and R. Derda, ACS Chem. Biol., 2014, 9, 443–450 CrossRef CAS PubMed.
- C. Heinis, T. Rutherford, S. Freund and G. Winter, Nat. Chem. Biol., 2009, 5, 502–507 CrossRef CAS PubMed.
- S. Chen, D. Bertoldo, A. Angelini, F. Pojer and C. Heinis, Angew. Chem., Int. Ed., 2014, 53, 1602–1606 CrossRef CAS PubMed.
- N. Assem, D. J. Ferreira, D. W. Wolan and P. E. Dawson, Angew. Chem., Int. Ed., 2015, 54, 8665–8668 CrossRef CAS PubMed.
- The by-products could not be identified unambiguously based on the m/z signals.
- S. Ng, K. Tjhung, B. Paschal, C. Noren and R. Derda, in Peptide Libraries, ed. R. Derda, Springer, New York, 2015, vol. 1248, ch. 11, pp. 155–172 Search PubMed.
- S. Ng, M. R. Jafari and R. Derda, ACS Chem. Biol., 2012, 7, 123–138 CrossRef CAS PubMed.
- P. I. Kitov, D. F. Vinals, S. Ng, K. F. Tjhung and R. Derda, J. Am. Chem. Soc., 2014, 136, 8149–8152 CrossRef CAS PubMed; F. Saito, H. Noda and J. W. Bode, ACS Chem. Biol., 2015, 10, 1026–1033 CrossRef PubMed.
- S. Li and R. W. Roberts, Chem. Biol., 2003, 10, 233–239 CrossRef CAS PubMed; S. C. Meyer, C. D. Shomin, T. Gaj and I. Ghosh, J. Am. Chem. Soc., 2007, 129, 13812–13813 CrossRef PubMed; D. A. Erlanson, ACS Chem. Biol., 2007, 2, 779–782 CrossRef PubMed; B. Santoso, S. Lam, B. W. Murray and G. Chen, Bioorg. Med. Chem. Lett., 2013, 23, 5680–5683 CrossRef PubMed; W. Wang, Y. Hirano, T. Uzawa, M. Liu, M. Taiji and Y. Ito, MedChemComm, 2014, 5, 1400–1403 RSC; Y. Tokunaga, Y. Azetsu, K. Fukunaga, T. Hatanaka, Y. Ito and M. Taki, Molecules, 2014, 19, 2481 CrossRef PubMed; M. Wichert, N. Krall, W. Decurtins, R. M. Franzini, F. Pretto, P. Schneider, D. Neri and J. Scheuermann, Nat. Chem., 2015, 7, 241–249 CrossRef PubMed; J. Beech, L. Saleh, J. Frentzel, H. Figler, I. R. Corrêa, B. Baker, C. Ramspacher, M. Marshall, S. Dasa, J. Linden, C. J. Noren and K. A. Kelly, Bioconjugate Chem., 2015, 26, 529–536 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S5, synthesis and characterization of DCO derivatives, experimental procedures for reaction on model peptide and phage libraries. See DOI: 10.1039/c5ob02646f |
| This journal is © The Royal Society of Chemistry 2016 |