
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Diazaoxatriangulenium: synthesis of reactive derivatives and conjugation to bovine serum albumin†
Ilkay
Bora
,
Sidsel A.
Bogh
,
Martin
Rosenberg
,
Marco
Santella
,
Thomas Just
Sørensen
* and
Bo W.
Laursen
*
Nano-Science Center & Department of Chemistry, University of Copenhagen, Universitetsparken 5, 2100 København Ø, Denmark. E-mail: TJS@chem.ku.dk; bwl@nano.ku.dk
First published on 27th November 2015
Abstract
The azaoxa-triangulenium dyes are characterised by emission in the red and a long fluorescence lifetime (up to 25 ns). These properties have been widely explored for the azadioxatrianguelnium (ADOTA) dye. Here, the syntheses of reactive maleimide and NHS-ester forms of the diazaoxatriangulenium (DAOTA) system are reported. The DAOTA fluorophore was conjugated to bovine serum albumin (BSA) and investigated in comparison to the corresponding ADOTA-BSA conjugate. It was found that the fluorescence of DAOTA experienced a significantly higher degree of solvent quenching if compared to ADOTA as non-conjugated dyes in aqueous solution, while the fluorescence quenching observed upon conjugation to BSA was significantly reduced for DAOTA when compared to ADOTA. The differences in observed quenching for the conjugates can be explained by the different electronic structures of the dyes, which renders DAOTA significantly less prone to reductive photoinduced electron transfer (PET) quenching from e.g. tryptophan. We conclude that DAOTA, with emission in the red and inherent resistance to PET quenching, is an ideal platform for the development of long fluorescence lifetime probes for time-resolved imaging and fluorescence polarisation assay.
Introduction
Fluorescence technology is prevalent in high-tech applications from diagnostics,1,2 point-of-care devices,3,4 and display technology to DNA sequencing,5 drug-discovery,6 and imaging.7,8 Each application relies on dye development, be it bioengineered fluorescent proteins,9 emissive nanoparticles,10 inorganic luminophores,11,12 or organic dyes.13–16 Organic dye development is limited by fluorophore design, where many of the existing molecular frameworks have been known for more than a century.17 Many dye systems have been synthesised using these well-known scaffolds,18–21 and have been optimised to show significantly enhanced performance.22–31 In particular, photostability, absorption cross-section, fluorescence quantum yield, and emission wavelength have been enhanced to the extent possible for the available fluorophores.In the group of organic dyes, the triangulenium dyes are different.32 In these molecules, a small absorption cross-section, and the resulting low fluorescence rate constant, does not infer low photostability and low quantum yield. While donor-substituted triangulenium dyes are bright emitters similar to rhodamine and fluorescein dyes,33–36 the azaoxa-triangulenium dyes are highly photostable, highly emissive, long fluorescence lifetime dyes.37–41 This group of triangulenium dyes includes azadioxatriangulenium (ADOTA) and diazaoxatriangulenium (DAOTA) shown in Chart 1 (for details on the triangulenium nomenclature see ESI†). The aza-bridges are readily functionalised with groups compatible, with the reaction conditions used to form the aromatic core.
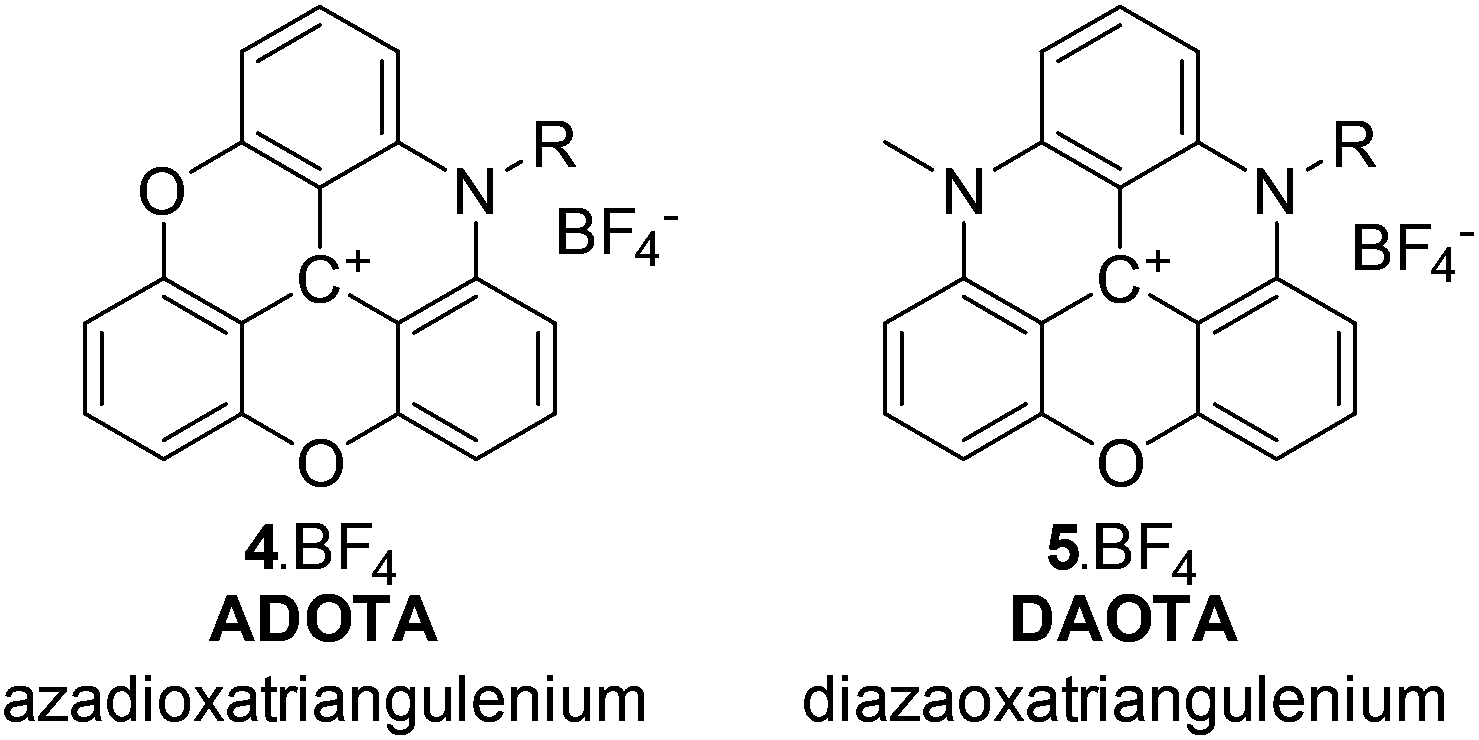 | ||
| Chart 1 The azaoxa-triangulenium dyes: ADOTA and DAOTA. | ||
The triangulenium dyes are synthesised from common precursors using sequences of highly selective nucleophilic aromatic substitution reactions (SNAr).33–35,38,42 For the azaoxa-triangulenium dyes, each substitution step occurs in a cascade of two SNAr-reactions forming a heteroatom bridge.37,38 A similar approach has recently been used to form the aza-bridge in carbazoles.43 Alternatively, ether-cleaving reaction conditions can be used to initiate an intramolecular SNAr-reaction with the formation of an oxygen bridge.44 Here, we elaborate on the synthesis of diazaoxatriangulenium (DAOTA) in an effort to make DAOTA derivatives, with reactive linkers for conjugation of the DAOTA fluorophore to biomolecules.
We chose to use bovine serum albumin (BSA) as a demonstrator for bioconjugation, although native BSA is not an ideal model system,26,29,45 the results allow for a direct comparison between DAOTA and ADOTA BSA-conjugates.46 We have previously investigated bioconjugates of ADOTA to BSA and IgG, and used both of the azaoxa-triangulenium dyes in bioimaging.46–49 When conjugated to a biomolecule, the ADOTA fluorescence is significantly quenched. This has also been observed for other fluorophores and rationalised as photoinduced electron transfer (PET) quenching by tryptophan.50–52 As DAOTA is less electron deficient than ADOTA,38,53 it was expected that DAOTA would be much less prone to reductive PET quenching by tryptophan. This difference in PET activity between ADOTA and DAOTA was recently highlighted in a study of a DAOTA based DNA G-quadruplex fluorescence lifetime probe.41 Here, we report a significant reduction in fluorescence quenching of DAOTA upon bio-conjugation, but also that the DAOTA dyes undergo significant solvent quenching, which leaves room for further improvements of this long fluorescent lifetime fluorophore. In particular, the solubility of DAOTA in water needs to be improved for several applications, in close analogy to what was achieved with the Alexa dyes®.54–57
Experimental
Materials and methods
Absorption spectroscopy was recorded with a double-beam spectrophotometer using the pure solvent as baseline. Steady state fluorescence spectra were recorded with a standard L-configuration fluorimeter equipped with single grating monochromators. All solvents used for spectroscopic experiments were of HPLC grade and used as received. Phosphate buffered saline (PBS) was prepared from slats according to common protocols. The pH value of the buffer was determined and subsequently adjusted to 7.4. Molar absorption coefficients were determined for each of the dyes using Lambert–Beers law by measuring the absorption spectrum of three stock solutions with different concentrations of the dye. Quantum yields were determined using the relative method,58 using rhodamine 6G as standard (ϕfl = 0.95).59 Details on the quantum yield measurements are given in the ESI.† Fluorescence lifetimes were measured using a FluoTime 300 (PicoQuant, Berlin, Germany) system. The emission signal was measured with a Hybrid-PMT detector with a spectral range of 220–650 nm. The dyes were excited at 510 nm using a solid-state laser excitation source. The instrument response function was recorded at the excitation wavelength using a dilute solution of Ludox®. The fluorescence decays were analyzed using the FluoFit software package. The decay data were all found to be monoexponential and was fitted by iterative reconvolution with a single exponential| If(t) = α\;exp(−t/τ) | (1) |
In eqn (1)α is the amplitude and τ is the fluorescence lifetime. All time-resolved emission decay profiles and fits are shown in the ESI.†
Synthetic procedures
Unless otherwise stated, all starting materials were obtained from commercial suppliers and used as received. Solvents were of HPLC grade for reactions and recrystallisations and technical grade for column chromatography and were used as received. 1H NMR and 13C NMR spectra were recorded on a 500 MHz or a 300 MHz instrument (500/300 MHz for 1H NMR and 126 MHz for 13C NMR). Proton chemical shifts are reported in ppm downfield from tetramethylsilane (TMS) and carbon chemical shifts in ppm downfield of TMS, using the resonance of the residual solvent peak as internal standard. High-resolution mass spectra (HRMS) were recorded with an ESP-MALDI-FT-ICR spectrometer equipped with a 7 T magnet (prior to experiments, the instrument was calibrated using NaTFA cluster ions) using dithranol as matrix. Column chromatographic purifications were performed on Kieselgel 60 (0.040–0.063 mm particle size). Dry column vacuum chromatography was performed on Kieselgel 60 (0.015–0.040 mm particle size). Thin layer chromatography was carried out using aluminum sheets pre-coated with silica gel 60F.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to yield the compound as a red powder, which is washed with dichloromethane and dried in vacuum (0.11 g, 45%). 1H NMR (500 MHz, Acetonitrile-d3) δ 8.20 (t, J = 8.6 Hz, 1H), 8.04–8.01 (m, 1H), 8.01–7.97 (m, 1H), 7.57 (d, J = 8.7 Hz, 1H), 7.50 (d, J = 8.6 Hz, 1H), 7.44 (d, J = 8.8 Hz, 1H), 7.36 (d, J = 8.6 Hz, 1H), 7.19 (d, J = 7.7 Hz, 1H), 7.15 (d, J = 7.8 Hz, 1H), 4.43–4.35 (m, 2H), 3.84 (s, 4H), 3.73 (s, 3H), 2.66 (t, J = 6.6 Hz, 2H), 2.12–2.09 (m, 2H). 13C NMR (126 MHz, Acetonitrile-d3) δ 174.4, 153.3, 153.2, 142.4, 141.6, 141.2, 140.7, 140.6, 140.3, 139.6, 139.5, 118.0, 112.1, 110.3, 110.1, 109.5, 109.4, 108.3, 108.1, 107.0, 106.8, 52.5, 48.1, 36.4, 30.8, 21.2. HRMS (MALDI-TOF):m/z calcd for C28H22N3O5+, 480.1554; found, 480.1550.
:1) to yield the compound as a red powder, which is washed with dichloromethane and dried in vacuum (0.11 g, 45%). 1H NMR (500 MHz, Acetonitrile-d3) δ 8.20 (t, J = 8.6 Hz, 1H), 8.04–8.01 (m, 1H), 8.01–7.97 (m, 1H), 7.57 (d, J = 8.7 Hz, 1H), 7.50 (d, J = 8.6 Hz, 1H), 7.44 (d, J = 8.8 Hz, 1H), 7.36 (d, J = 8.6 Hz, 1H), 7.19 (d, J = 7.7 Hz, 1H), 7.15 (d, J = 7.8 Hz, 1H), 4.43–4.35 (m, 2H), 3.84 (s, 4H), 3.73 (s, 3H), 2.66 (t, J = 6.6 Hz, 2H), 2.12–2.09 (m, 2H). 13C NMR (126 MHz, Acetonitrile-d3) δ 174.4, 153.3, 153.2, 142.4, 141.6, 141.2, 140.7, 140.6, 140.3, 139.6, 139.5, 118.0, 112.1, 110.3, 110.1, 109.5, 109.4, 108.3, 108.1, 107.0, 106.8, 52.5, 48.1, 36.4, 30.8, 21.2. HRMS (MALDI-TOF):m/z calcd for C28H22N3O5+, 480.1554; found, 480.1550.
Results and discussion
Synthesis
The synthesis of azaoxa-triangulenium dyes was developed in our lab,37,38 based on the work of Martin and Smith,44 who first synthesised the trioxatriangulenium (TOTA) system. In our early work, azadioxa-, diazaoxa-, and triaza-triangulenium (ADOTA, DAOTA, and TATA) was reported.37,38 Lacour and co-workers have later expanded the series of triangulenium salts to also include derivatives with a single sulfur bridge.60 The incorporation of more than one sulfur atom in the triangulenium core seems not to be possible due to the distortion enforced by the significantly different C–S bond length as compared to C–N and C–O bonds.61 The azaoxa-triangulenium dyes are all made from a common precursor; tris(2,6-dimethoxyphenyl)-carbenium tetrafluoroborate (1) which, can be reacted stepwise with primary amines to form between one and three aza-bridges. 1 or each intermediate may be reacted under ether cleaving conditions at elevated temperatures to form the fully ring closed triangulenium core with one, two, or three oxa-bridges. The oxa-bridges are themselves reactive towards primary amines. However, this substitution reaction is slow compared to attack on the methoxy groups of the open precursors, but it is still highly selective.38Post-functionalisation of the substituents on the aza-bridges have previously been reported,62,63 and we have synthesised and explored reactive derivatives of ADOTA for bioconjugation.46,48,49
The syntheses of ADOTA with reactive NHS esters and maleimide groups (Scheme 1) are straightforward, and proceed by a SNAr reaction between 1 and a suitable amino acid or diamine. The primary amine attack one of the methoxy substituted carbon atoms in 1 followed by elimination of methanol upon formation of a transient intermediate set up for an intramolecular SNAr reaction, eliminating methanol and forming an aza-bridge. The product is an N-substituted tetramethoxy-acridinium salt (2). Reacting 2 in molten pyridinium chloride yields the fully ring closed azadioxa-triangulenium (4, ADOTA).37,38 Alternatively, a second aza-bridge can be introduced by reacting 2 with a second primary amine forming a [4]helicenium ion. The product is an N,N′-substituted 1,13-dimethoxyquinacridinium (DMQA) salt (3, for details on this nomenclature see ESI†), which in molten pyridinium chloride yields the fully ring closed diazaoxa-triangulenium core (5, DAOTA).
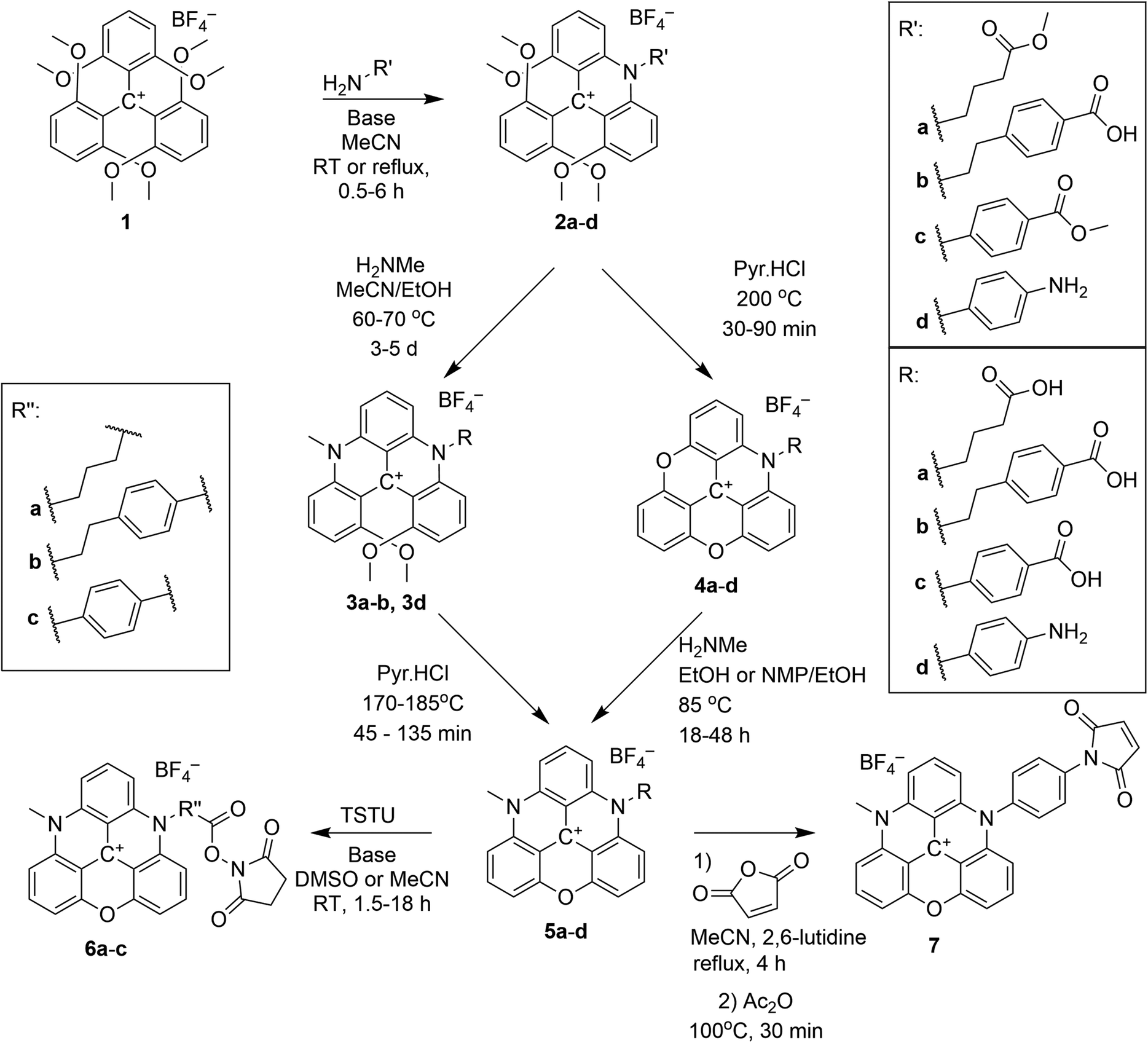 | ||
| Scheme 1 Synthetic route to DAOTA compounds: 5a–d, 6a–c, and 7. | ||
The steps to ADOTA-derivatives suitable for bioconjugation as active esters and maleimides follow the direct path from 1 over 2 to 4.47 Similarly, the 3-carboxypropyl derivative of DAOTA (2a) could be synthesised by introducing first one (2a), and then a second aza-bridge to give the DMQA derivative (3c). Subsequent reaction of the DMQA compounds in molten pyridinium chloride followed by basic hydrolysis of the intermediate amide yielded the desired N-(3-carboxypropyl)-N′-methyl-DAOTA tetrafluoroborate (5a, Scheme 1) as a red powder in a good yield.49 The synthesis of the 4-aminophenyl DAOTA derivative (5d) was also performed via the route through the DMQA derivative (3d, Scheme 1). This yielded 3d in a high yield, while the ring closure reaction for conversion of 3d into 5d yielded 5d in a low yield. In attempts to prepare the 2-(4-carboxyphenyl)-ethyl and 4-carboxyphenyl DAOTA derivatives (5b and 5c, respectively) via the same synthetic route as described for 5a and 5d, we found that this was associated with difficulties. The compound 3b was isolated in a high yield. However, the following ring-closure reaction in molten pyridinium chloride yielded the desired product (5b) in combination with impurities, which were inseparable from 5b. Similarly, the DMQA derivative 3c could not be prepared as the basic hydrolysis of the intermediate amide derivative (3c′, Scheme 2) was not successful.
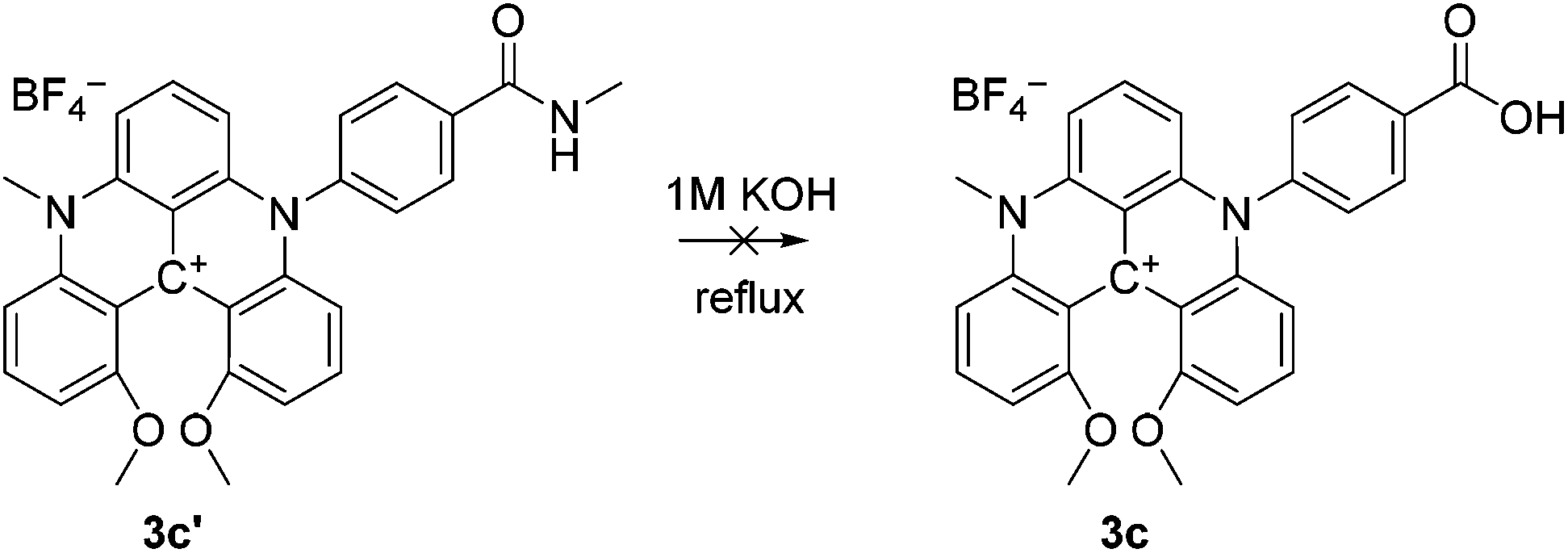 | ||
| Scheme 2 Hydrolysis of DMQA amide 3c’. | ||
Thus, 5b and 5c were synthesised via an alternative route, which to our knowledge has only been reported for the N,N′-dipropyl-DAOTA salt by Laursen and Krebs.38 The compounds 5b and 5c were obtained in good yields via reaction of the ADOTA derivatives (4b and 4c) with methylamine in acetonitrile/ethanol or NMP/ethanol mixtures. The acid derivatives 5a–c were reacted with N,N,N′,N′-Tetramethyl-O-(N-succinimidyl)uronium tetrafluoroborate (TSTU) in acetonitrile or DMSO solution to form the reactive NHS esters (6a–c) in good yields. The NHS ester functional group is used to conjugate fluorescent dyes to biomacromolecules via coupling to a primary amine residue of the biomacromolecule.64
Compound 5d was reacted with maleic acid anhydride to form the corresponding maleimide 7 in a two-step/one pot reaction (Scheme 1).47 The maleimide group is used to selectively label thiol groups in biomolecules.64
Optical spectroscopy
In the following N-(4-carboxyphenyl)-N′-methyl-DAOTA tetrafluoroborate 5c is used to demonstrate the spectroscopic properties of the reactive DAOTA derivatives. The properties of 5c are compared to those of the corresponding N-(4-carboxyphenyl)-ADOTA tetrafluoroborate 4c.46,47 The spectra displayed in Fig. 1 show that DAOTA (5c) has more desirable absorption and emission properties. These are in the red region of spectrum, which is better for measurements on biological systems as compared to ADOTA (4c).49,53,65Table 1 summarises the photophysical properties of 4c and 5c in acetonitrile, dimethyl sulfoxide (DMSO), and phosphate buffered saline at pH = 7.4 (PBS). The differences between 4c and 5c in organic solvents are closely related to the oscillator strengths of the lowest energy transition of the two parent chromophores. The higher molar absorption coefficient at the lowest energy transition of DAOTA as compared that of ADOTA (ε = 16000 vs. 10000 M−1 cm−1 in acetonitrile solution),65 results in a shorter fluorescence lifetime (τfl = 19 vs. 21 ns in MeCN).65,66 In PBS solution, DAOTA 5c exhibits a significant change in fluorescence lifetime (τfl) and fluorescence quantum yield (ϕfl), where both are reduced; ϕfl from 55% to 35% and τfl from 19 ns to 14 ns. We have found that chloride is not a specific quencher of DAOTA, and suggest that the observed 30% reduction in ϕfl must be due to unspecific solvent quenching, likely due to the high hydrophobicity of the DAOTA core of 5c. ADOTA 4c is less hydrophobic and does not show more than 18% reduction in ϕfl in PBS solution as compared to the organic solvents.
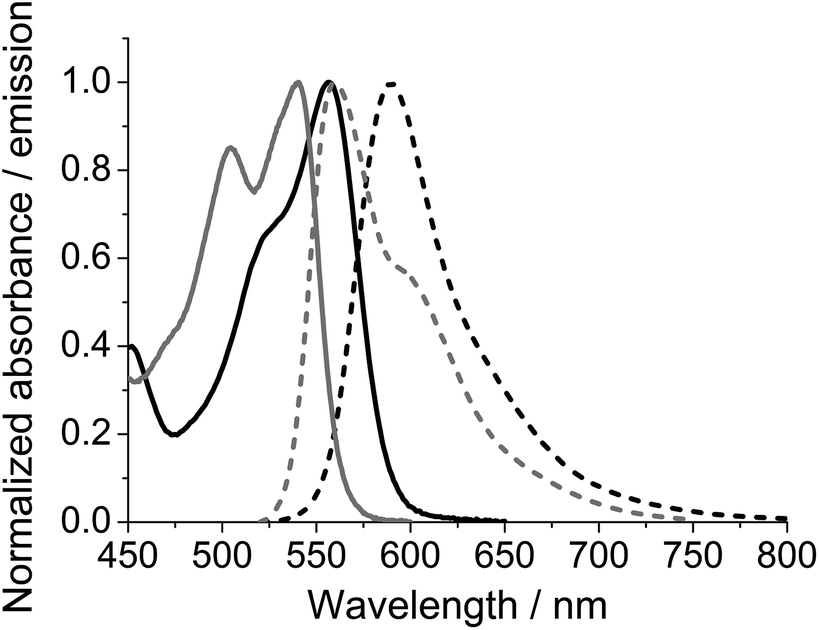 | ||
| Fig. 1 Absorption (full) and emission (dashed) spectra of ADOTA 4c (grey) and DAOTA 5c (black) measured in acetonitrile solution. | ||
| Solvent | ADOTA 4c | DAOTA 5c | ||||
|---|---|---|---|---|---|---|
| MeCN | DMSO | PBS | MeCN | DMSO | PBS | |
| λ abs is the wavelength of the lowest energy absorption maximum given in nm. λem is the wavelength at the emission maximum given in nm, ΔStokes’ is the Stokes’ shift given in nm, τfl is the fluorescence lifetime given in ns, and ϕfl is the measured fluorescence quantum yield using rhodamine 6G as a reference, τ0 is the radiative lifetime: τfl/ϕfl given in ns. | ||||||
| λ abs/nm | 536 | 539 | 539 | 557 | 561 | 556 |
| λ em/nm | 562 | 570 | 560 | 590 | 600 | 592 |
| ΔStokes′/nm | 26 | 31 | 21 | 33 | 39 | 36 |
| τ fl/ns | 21.0 | 17.1 | 17.9 | 18.7 | 15.1 | 14.0 |
| ϕ fl | 0.64 | 0.64 | 0.57 | 0.55 | 0.45 | 0.35 |
| τ 0/ns | 32.8 | 26.7 | 31.4 | 34.0 | 33.6 | 40.0 |
The fluorescence lifetime (τfl) for DAOTA 5c is exceptionally long for a red emitting organic dye, even when reduced to 14 ns by unspecific solvent quenching in PBS solution. The combination of a long fluorescence lifetime (>10 ns) and emission in the red (>600 nm) makes DAOTA ideally suited to monitor rotational correlation times of biomolecules and as a fluorescent probe for fluorescence polarisation based assays.8,47–49
DAOTA-BSA conjugates
To test the ability of DAOTA as a probe for measuring the rotational motion of proteins the reactive ester of DAOTA 5c, N-(4-carboxyphenyl)-N′-methyl-diazaoxatriangulenium NHS ester tetrafluoroborate 6c, was conjugated to BSA. This reaction is expected to predominately result in conjugation of the triangulenium based probe to the N-terminus of the protein.46 The labelling protocol was optimised to give a low degree of labelling (DOL) to ensure that complications arising from multiple labels, such as energy transfer between labels, was minimal. When developing a fluorescence polarisation assay, these effects can be probed by looking for Weber's red-edge effect.67,68 A DOL of 0.9 DAOTA dyes per BSA was used to obtain the results presented below.The Perrin equation (eqn (2)) describes the ideal relationship between: the observed fluorescence anisotropy (r), the fundamental anisotropy of the dye (r0), the rotational correlation time of the rotating volume (θ), and the fluorescence lifetime (τfl). The rotational correlation time (θ) is directly related (eqn (3)) to the rotational volume (V) and the viscosity of the surrounding medium (η). For proteins, the rotational correlation time can be related (eqn (3)) to the molecular mass (M), the specific density (ν), and the average hydration (h) of the protein.66
| r0/r = 1 + τfl/θ | (2) |
| θ = ηV/RT = ηM/RT·(ν + h) | (3) |
Two factors related to the fluorescent probe used are found in the equations: the fluorescence lifetime (τfl) and the fundamental fluorescence anisotropy (r0). The fluorescence lifetime determines the range of rotational correlation times that may be probed, in other words the range of molecular weights that can be investigated. The fundamental anisotropy, ranging from 0.4 to −0.2, determines the dynamic range of the experiments. Dyes with a low r0 value are poor probes for fluorescence polarisation-based methods. DAOTA has r0 = 0.38,65 which is very close to the maximal value (0.4), while the fluorescence lifetime allows for probing biomolecules with a molecular weight up towards 1000 kDa.46
Fig. 2 shows a Perrin plot of 5c conjugated to BSA (5c-BSA), where the steady-state fluorescence anisotropy (r) measured at four different temperatures (T) are plotted as 1/r against T/η and used to determine the θ/V, and the apparent anisotropy (rapp0) by extrapolation of T/η to zero. The latter is a measure for the flexibility of the dye label, when conjugated to the biomolecule. Ideally, if the label upon conjugation loses all degrees of freedoms, except co-rotation with the biomolecule, the apparent and fundamental anisotropy will be identical. Any local flexibility of the dye label will induce a pathway for fast scrambling of the photoselection not related to the motion of the biomolecule. The result is a lowering of the apparent anisotropy rapp0 in a Perrin plot, and a loss of dynamic range in any fluorescence polarisation based experiment. For 5c-BSA the apparent anisotropy is at rapp0 = 0.36 surprisingly high, clearly indicating that the DAOTA label is immobilised on the surface of BSA.
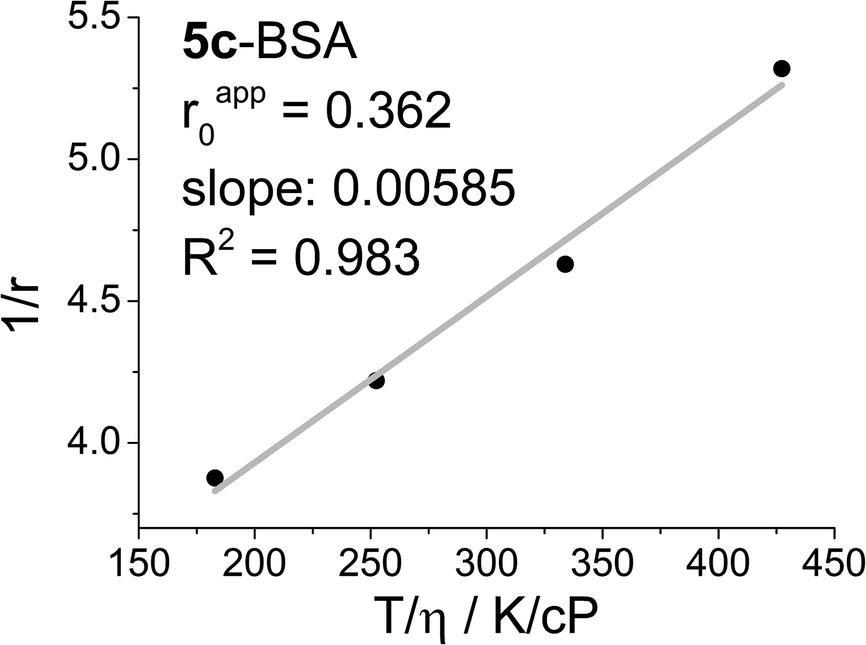 | ||
| Fig. 2 Perrin plot of 5c-BSA in phosphate buffered saline at pH = 7.4 solution. The experiment was performed on a sample with a DOL = 0.9, where the steady state fluorescence anisotropy (r) was determined at four different temperatures (T). | ||
While the effect of the long fluorescence lifetime of 5c may be hard to identify in the steady state spectra, it is directly visible in time-resolved experiments. Fig. 3 (top) shows a time-resolved emission decay profile for 5c and 5c-BSA measured in PBS solution, obtained using time-correlated single photon counting (TCSPC). Cursory inspection of the fluorescence decays profiles in Fig. 3 (top) shows that the 5c-BSA has a longer fluorescence lifetime than 5c in PBS solution (τfl,5c = 14.0 ns vs. τfl,5c-BSA 21.2 ns, see ESI†), and that photons can be detected well beyond 150 ns when using the standard settings of TCSPC with a maximum count of 10.000. By using longer acquisition times (higher maximum number of counts), photons arising from emission of the 5c-BSA conjugates may be detected up towards 250 ns after excitation, this is without equal when considering organic dyes with emission in the red.
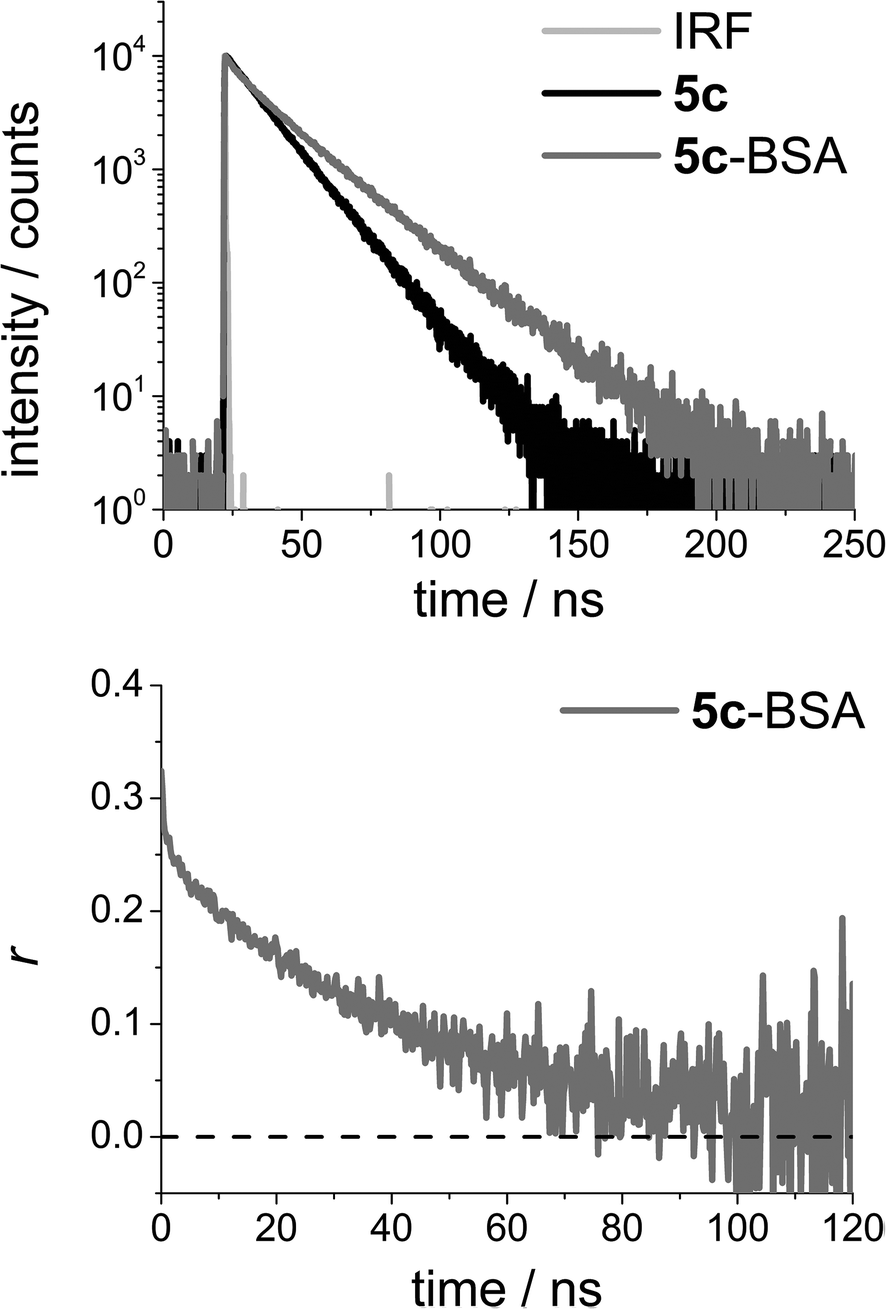 | ||
| Fig. 3 Top: Time-resolved emission decay profile for N-(4-carboxyphenyl)-N′-methyl-diazaoxatriangulenium tetrafluoroborate (DAOTA, 5c) and the corresponding BSA conjugate (5c-BSA) measured in PBS solution. Bottom: Time-resolved anisotropy decay profile for 5c-BSA measured in PBS solution. For details on fits and the resulting parameters see the ESI.† | ||
Fig. 3 (bottom) shows the time-resolved anisotropy decay for 5c-BSA. The data allow for direct determination of the rotational correlation time of the rotating volume (θ). The long fluorescence lifetime is important in obtaining these data, as photons must be emitted in a time interval long enough to describe the rotational motion of the biomolecule. Fig. 3b shows that in the case of 5c-BSA the photoselection is fully scrambled by rotational motion in ∼100 ns. That is, the anisotropy has decayed to zero. The long rotational correlation time determined for BSA from these data is θBSA = 40 ns (see ESI for details†), a number identical to the average literature value of θBSA = 40 ns.69 Note that BSA is not a perfect spherical rotor, and the value determined will be influenced by the position and relative orientation of the dye in the bioconjugate.
While emission from either of the azaoxa-triangulenium dyes can be used to follow the rotational motion of large biomolecules for more than 100 ns,27 there is a clear difference in the behaviour of 4a-BSA and 5c-BSA. Where ADOTA fluorescence is quenched in the conjugates (4a-BSA) when compared to the non-bounded dyes (4a), the DAOTA fluorescence appears to be enhanced upon conjugation, as seen by the significantly increased fluorescence lifetime (Fig. 3). As the fluorescence lifetime only report on the emitting population of dyes, the actual emission intensity of each dye was determined. Fig. 4 shows the results for 4a-BSA and 5c-BSA. The total emission intensity of either dye is decreased upon conjugation to BSA, but the effect is much less pronounced for the DAOTA derivative. We rationalised the quenching of the ADOTA fluorescence as a result of reductive PET quenching by tryptophan,50 a process the increased cation stability of DAOTA makes less favoured (see ESI for details†).53 Thus, we see less quenching of the DAOTA fluorescence in the 5c-BSA conjugates. The significant increase in lifetime upon conjugation of 5c to BSA must be due to a reduction in the non-specific solvent induced quenching of a population of DAOTA dyes that is partially shielded by the protein surface, while the reduction in overall intensity must be due to an almost fully quenched population of 5c-BSA. The quenched population does not contribute to the time-resolved emission decay profile, and will not influence the fluorescence anisotropy. The net result is that the fluorescence quantum yield of the 5c-BSA conjugate at ϕfl = 0.34 is very close to that of the free dye 5c in PBS at ϕfl = 0.35, although with a more complicated time-resolved fluorescence decay profile (see ESI†).
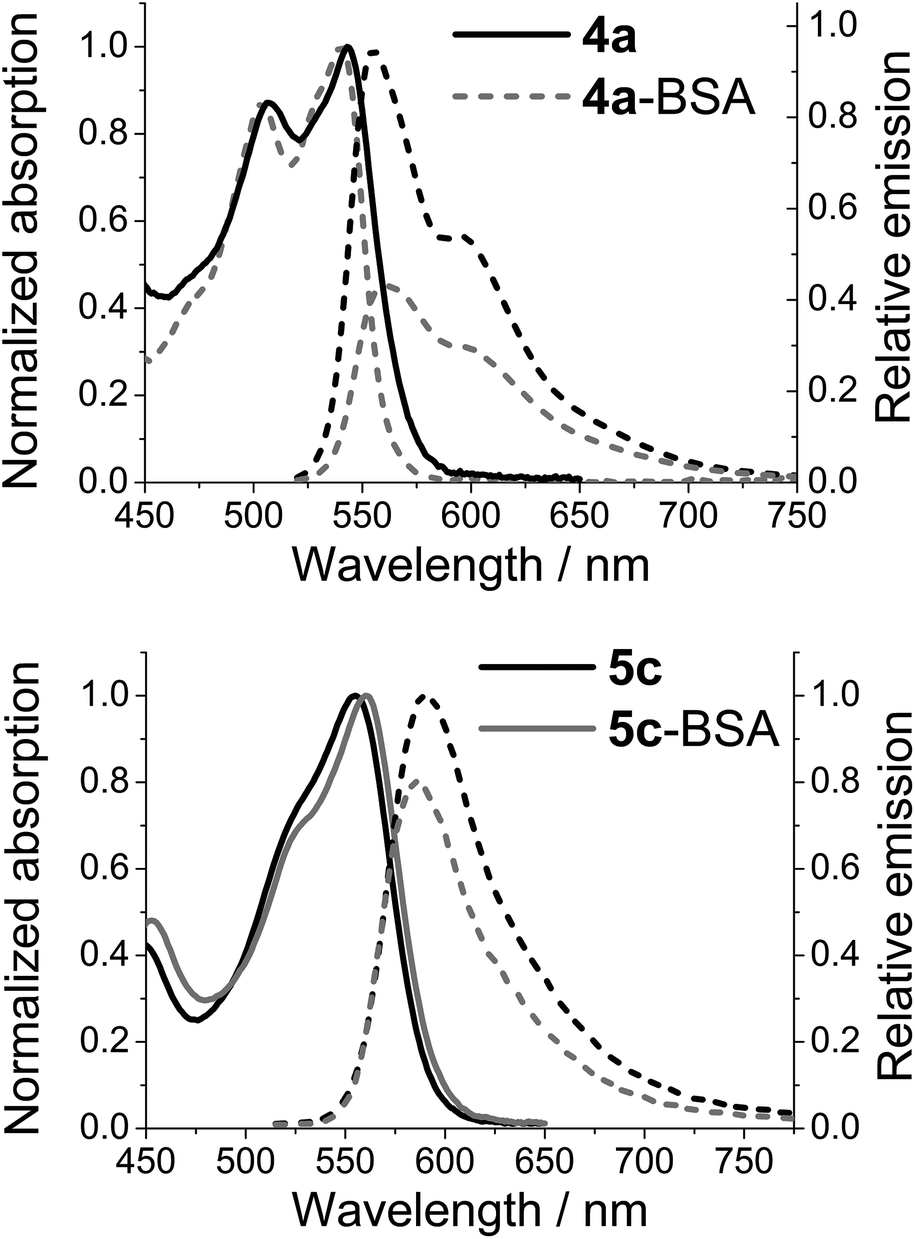 | ||
| Fig. 4 Normalised absorption spectra (full) and relative emission spectra (dashed) of 4a and 4a-BSA (top) and 5c and 5c-BSA (bottom) measured in PBS solution. The absorption spectra are normalised at λmax, while the emission is normalised by the absorption at the point of excitation. | ||
Conclusions
The syntheses of six new derivatives of diazaoxatriangulenium (DAOTA) salts were reported, and it was shown that these dyes can be accessed via two synthetic routes. One set of substituents may favour one route over the other.The photophysical properties of the DAOTA fluorophore were investigated in view of using the red emitting, long fluorescence lifetime dye in fluorescence polarisation assays. We showed that the DAOTA fluorophore undergoes unspecific solvent fluorescence quenching in aqueous buffer, reducing the fluorescence lifetime from 19 ns in acetonitrile solution to 14 ns in PBS solution. 5c was conjugated to BSA, and we found that 5c-BSA had a significantly increased longest fluorescence lifetime component at 21.2 ns (as well as intensity weighted average fluorescence lifetime of 19.2 ns). Furthermore, the overall emission intensity of the conjugates (5c-BSA) was found to be equal to that of the free dye (5c) measured in PBS solution, as 5c is less quenched by tryptophan. Thus the DAOTA fluorophore, with emission further in the red and a longer fluorescence lifetime in biomolecule conjugates, was found to be superior to the ADOTA fluorophore as a probe for developing fluorescence polarisation assays for large biomolecules.
Acknowledgements
The authors thank the University of Copenhagen, the National Institute of Health (USA) NIH Grant: R01EB12003, the Carlsberg Foundation, the Villum Foundation, DFF|FTP, and the Danish National Research Foundation under the Danish-Chinese Centre for Self-Assembled Molecular Electronic Nanosystems for financial support. M.R. is grateful to the Danish Council of Independent Research for financial support.Notes and references
- B. R. Hurley and C. D. Regillo, Fluorescein Angiography: General Principles and Interpretation, Springer, New York, 2009 Search PubMed.
- D. M. Jameson and J. A. Ross, Chem. Rev., 2010, 110, 2685–2708 CrossRef CAS PubMed.
- S. M. Borisov and O. S. Wolfbeis, Chem. Rev., 2008, 108, 423–461 CrossRef CAS PubMed.
- O. S. Wolfbeis, Anal. Chem., 2008, 80, 4269–4283 CrossRef CAS PubMed.
- L. M. Smith, J. Z. Sanders, R. J. Kaiser, P. Hughes, C. Dodd, C. R. Connell, C. Heiner, S. B. H. Kent and L. E. Hood, Nature, 1986, 321, 674–679 CrossRef CAS PubMed.
- J. Inglese, R. L. Johnson, A. Simeonov, M. Xia, W. Zheng, C. P. Austin and D. S. Auld, Nat. Chem. Biol., 2007, 3, 466–479 CrossRef CAS PubMed.
- S. Weiss, Nat. Struct. Biol., 2000, 7, 724–729 CrossRef CAS PubMed.
- J. C. Owicki, J. Biomol. Screening, 2000, 5, 297–306 CrossRef CAS PubMed.
- S. Viswanathan, M. E. Williams, E. B. Bloss, T. J. Stasevich, C. M. Speer, A. Nern, B. D. Pfeiffer, B. M. Hooks, W. P. Li, B. P. English, T. Tian, G. L. Henry, J. J. Macklin, R. Patel, C. R. Gerfen, X. Zhuang, Y. Wang, G. M. Rubin and L. L. Looger, Nat. Methods, 2015, 12, 568–576 CrossRef CAS PubMed.
- K. D. Wegner and N. Hildebrandt, Chem. Soc. Rev., 2015, 44, 4792–4834 RSC.
- E. G. Moore, A. P. Samuel and K. N. Raymond, Acc. Chem. Res., 2009, 42, 542–552 CrossRef CAS PubMed.
- T. Yoshihara, Y. Yamaguchi, M. Hosaka, T. Takeuchi and S. Tobita, Angew. Chem., Int. Ed., 2012, 51, 4148–4151 CrossRef CAS PubMed.
- J. B. Grimm, B. P. English, J. Chen, J. P. Slaughter, Z. Zhang, A. Revyakin, R. Patel, J. J. Macklin, D. Normanno, R. H. Singer, T. Lionnet and L. D. Lavis, Nat. Methods, 2015, 12, 244–250 CrossRef CAS PubMed.
- B. N. G. Giepmans, S. R. Adams, M. H. Ellisman and R. Y. Tsien, Science, 2006, 312, 217–224 CrossRef CAS PubMed.
- I. Johnson, Histochem. J., 1998, 30, 123–140 CrossRef CAS PubMed.
- A. P. de Silva, H. Q. N. Gunaratne, T. Gunnlaugsson, A. J. M. Huxley, C. P. McCoy, J. T. Rademacher and T. E. Rice, Chem. Rev., 1997, 97, 1515–1566 CrossRef CAS PubMed.
- R. Meyer, Z. Phys. Chem., 1897, 24, 468–508 Search PubMed.
- L. M. Wysocki and L. D. Lavis, Curr. Opin. Chem. Biol., 2011, 15, 752–759 CrossRef CAS PubMed.
- S. W. Yun, N. Y. Kang, S. J. Park, H. H. Ha, Y. K. Kim, J. S. Lee and Y. T. Chang, Acc. Chem. Res., 2014, 47, 1277–1286 CrossRef CAS PubMed.
- H. Kobayashi, M. Ogawa, R. Alford, P. L. Choyke and Y. Urano, Chem. Rev., 2009, 110, 2620–2640 CrossRef PubMed.
- A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef CAS PubMed.
- Advanced Fluorescence Reporters in Chemistry and Biology II, ed. A. P. Demchenko, Series ed. O. S. Wolfbeis, Springer, Berlin, Heidelberg, 2010 Search PubMed.
- Advanced Fluorescence Reporters in Chemistry and Biology I, ed. A. P. Demchenko, Series ed. O. S. Wolfbeis, Springer, Berlin, Heidelberg, 2010 Search PubMed.
- T. Myochin, K. Hanaoka, S. Iwaki, T. Ueno, T. Komatsu, T. Terai, T. Nagano and Y. Urano, J. Am. Chem. Soc., 2015, 137, 4759–4765 CrossRef CAS PubMed.
- E. Kim, Y. Lee, S. Lee and S. B. Park, Acc. Chem. Res., 2015, 48, 538–547 CrossRef CAS PubMed.
- E. Heyer, P. Lory, J. Leprince, M. Moreau, A. Romieu, M. Guardigli, A. Roda and R. Ziessel, Angew. Chem., Int. Ed., 2015, 54, 2995–2999 CrossRef CAS PubMed.
- M. Tasior, Y. M. Poronik, O. Vakuliuk, B. Sadowski, M. Karczewski and D. T. Gryko, J. Org. Chem., 2014, 79, 8723–8732 CrossRef CAS PubMed.
- P. Shieh, M. S. Siegrist, A. J. Cullen and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 5456–5461 CrossRef CAS PubMed.
- A. Poirel, P. Retailleau, A. De Nicola and R. Ziessel, Chem. – Eur. J., 2014, 20, 1252–1257 CrossRef CAS PubMed.
- G. Lukinavicius, L. Reymond, E. D'Este, A. Masharina, F. Gottfert, H. Ta, A. Guther, M. Fournier, S. Rizzo, H. Waldmann, C. Blaukopf, C. Sommer, D. W. Gerlich, H. D. Arndt, S. W. Hell and K. Johnsson, Nat. Methods, 2014, 11, 731–733 CrossRef CAS PubMed.
- Y.-L. Huang, A. S. Walker and E. W. Miller, J. Am. Chem. Soc., 2015, 137, 10767–10776 CAS.
- J. Bosson, J. Gouin and J. Lacour, Chem. Soc. Rev., 2014, 43, 2824–2840 RSC.
- B. W. Laursen, F. C. Krebs, M. F. Nielsen, K. Bechgaard, J. B. Christensen and N. Harrit, J. Am. Chem. Soc., 1998, 120, 12255–12263 CrossRef CAS.
- B. W. Laursen and T. J. Sørensen, J. Org. Chem., 2009, 74, 3183–3185 CrossRef CAS PubMed.
- T. J. Sørensen and B. W. Laursen, J. Org. Chem., 2010, 75, 6182–6190 CrossRef PubMed.
- F. Westerlund, C. B. Hildebrandt, T. J. Sørensen and B. W. Laursen, Chem. – Eur. J., 2010, 16, 2992–2996 CrossRef CAS PubMed.
- B. W. Laursen and F. C. Krebs, Angew. Chem., Int. Ed., 2000, 39, 3432–3434 CrossRef CAS.
- B. W. Laursen and F. C. Krebs, Chem. – Eur. J., 2001, 7, 1773–1783 CrossRef CAS.
- T. J. Sørensen, B. W. Laursen, R. Luchowski, T. Shtoyko, I. Akopova, Z. Gryczynski and I. Gryczynski, Chem. Phys. Lett., 2009, 476, 46–50 CrossRef PubMed.
- P. Hammershøj, T. J. Sørensen, B.-H. Han and B. W. Laursen, J. Org. Chem., 2012, 77, 5606–5612 CrossRef PubMed.
- A. Shivalingam, M. A. Izquierdo, A. L. Marois, A. Vysniauskas, K. Suhling, M. K. Kuimova and R. Vilar, Nat. Commun., 2015, 6, 8178 CrossRef PubMed.
- M. B. Smith and J. March, in March's Advanced Organic Chemistry, Wiley, New York, 5th edn, 2001, ch. 13, pp. 850–893 Search PubMed.
- M. Bhanuchandra, K. Murakami, D. Vasu, H. Yorimitsu and A. Osuka, Angew. Chem., Int. Ed., 2015, 54, 10234–10238 CrossRef CAS PubMed.
- J. C. Martin and R. G. Smith, J. Am. Chem. Soc., 1964, 86, 2252–2256 CrossRef CAS.
- M. E. Smith, M. B. Caspersen, E. Robinson, M. Morais, A. Maruani, J. P. Nunes, K. Nicholls, M. J. Saxton, S. Caddick, J. R. Baker and V. Chudasama, Org. Biomol. Chem., 2015, 13, 7946–7949 CAS.
- S. A. Bogh, I. Bora, M. Rosenberg, E. Thyrhaug, B. W. Laursen and T. J. Sørensen, Methods Appl. Fluoresc., 2015, 3, 045001 CrossRef.
- I. Bora, S. A. Bogh, M. Santella, M. Rosenberg, T. J. Sørensen and B. W. Laursen, Eur. J. Org. Chem., 2015, 6351–6358, DOI:10.1002/ejoc.201500888.
- T. J. Sørensen, E. Thyrhaug, M. Szabelski, R. Luchowski, I. Gryczynski, Z. Gryczynski and B. W. Laursen, Methods Appl. Fluoresc., 2013, 1, 025001 CrossRef PubMed.
- B. P. Maliwal, R. Fudala, S. Raut, R. Kokate, T. J. Sørensen, B. W. Laursen, Z. Gryczynski and I. Gryczynski, PLoS One, 2013, 8, e63043 CAS.
- S. Doose, H. Neuweiler and M. Sauer, ChemPhysChem, 2005, 6, 2277–2285 CrossRef CAS PubMed.
- A. C. Vaiana, H. Neuweiler, A. Schulz, J. Wolfrum, M. Sauer and J. C. Smith, J. Am. Chem. Soc., 2003, 125, 14564–14572 CrossRef CAS PubMed.
- N. Marme, J. P. Knemeyer, M. Sauer and J. Wolfrum, Bioconjugate Chem., 2003, 14, 1133–1139 CrossRef CAS PubMed.
- S. Dileesh and K. R. Gopidas, J. Photochem. Photobiol., A, 2004, 162, 115–120 CrossRef CAS.
- M. Sauer, K. T. Han, R. Müller, S. Nord, A. Schulz, S. Seeger, J. Wolfrum, J. Arden-Jacob, G. Deltau, N. J. Marx, C. Zander and K. H. Drexhage, J. Fluoresc., 1995, 5, 247–261 CrossRef CAS PubMed.
- M. Sauer, K. T. Han, R. Müller, A. Schulz, R. Tadday, S. Seeger, J. Wolfrum, J. Arden-Jacob, G. Deltau, N. J. Marx and K. H. Drexhage, J. Fluoresc., 1993, 3, 131–139 CrossRef CAS PubMed.
- R. P. Haugland, Handbook of Fluorescent Probes and Research Chemicals, Molecular Probes, Eugene, Oregon, 11th edn, 2010 Search PubMed.
- N. Panchuk-Voloshina, R. P. Haugland, J. Bishop-Stewart, M. K. Bhalgat, P. J. Millard, F. Mao and W. Y. Leung, J. Histochem. Cytochem., 1999, 47, 1179–1188 CrossRef CAS PubMed.
- C. Würth, M. Grabolle, J. Pauli, M. Spieles and U. Resch-Genger, Nat. Protocols, 2013, 8, 1535–1550 Search PubMed.
- A. M. Brouwer, Pure Appl. Chem., 2011, 83, 2213–2228 CrossRef CAS.
- C. Nicolas, G. Bernardinelli and J. Lacour, J. Phys. Org. Chem., 2010, 23, 1049–1056 CrossRef CAS.
- G. D. Figuly, C. K. Loop and J. C. Martin, J. Am. Chem. Soc., 1989, 111, 654–658 CrossRef CAS.
- F. C. Krebs, H. Spanggaard, N. Rozlosnik, N. B. Larsen and M. Jørgensen, Langmuir, 2003, 19, 7873–7880 CrossRef CAS.
- T. J. Sørensen, C. B. Hildebrandt, M. Glyvradal and B. W. Laursen, Dyes Pigm., 2013, 98, 297–303 CrossRef.
- G. T. Hermanson, Bioconjugate Techniques, Academic Press, Oxford, 2013 Search PubMed.
- E. Thyrhaug, T. J. Sørensen, I. Gryczynski, Z. Gryczynski and B. W. Laursen, J. Phys. Chem. A, 2013, 117, 2160–2168 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer-Verlag, New York Inc., New York, 3. Revised edn, 2006 Search PubMed.
- G. Weber and M. Shinitzky, Proc. Natl. Acad. Sci. U. S. A., 1970, 65, 823–830 CrossRef CAS.
- B. D. Hamman, A. V. Oleinikov, G. G. Jokhadze, R. R. Traut and D. M. Jameson, Biochemistry, 1996, 35, 16680–16686 CrossRef CAS PubMed.
- M. L. Ferrer, R. Duchowicz, B. Carrasco, J. G. D. L. Torre and A. U. Acuna, Biophys. J., 2001, 2422–2430 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Nomenclature of triangulenium ions, data for quantum yield determinations, labelling procedure, parameters describing the time-dependent photophysical properties, tryptophan quenching experiments, and NMR spectra. See DOI: 10.1039/c5ob02293b |
| This journal is © The Royal Society of Chemistry 2016 |