
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Palladium-catalyzed enolate arylation as a key C–C bond-forming reaction for the synthesis of isoquinolines†
Ben S.
Pilgrim‡
a,
Alice E.
Gatland
a,
Carlos H. A.
Esteves
a,
Charlie T.
McTernan
a,
Geraint R.
Jones
a,
Matthew R.
Tatton
a,
Panayiotis A.
Procopiou
b and
Timothy J.
Donohoe
*a
aDepartment of Chemistry, University of Oxford, Chemistry Research Laboratory, Mansfield Road, Oxford, OX1 3TA, UK. E-mail: timothy.donohoe@chem.ox.ac.uk
bGlaxoSmithKline, Medicines Research Centre, Gunnels Wood Road, Stevenage, SG1 2NY, UK
First published on 3rd December 2015
Abstract
The palladium-catalyzed coupling of an enolate with an ortho-functionalized aryl halide (an α-arylation) furnishes a protected 1,5-dicarbonyl moiety that can be cyclized to an isoquinoline with a source of ammonia. This fully regioselective synthetic route tolerates a wide range of substituents, including those that give rise to the traditionally difficult to access electron-deficient isoquinoline skeletons. These two synthetic operations can be combined to give a three-component, one-pot isoquinoline synthesis. Alternatively, cyclization of the intermediates with hydroxylamine hydrochloride engenders direct access to isoquinoline N-oxides; and cyclization with methylamine, gives isoquinolinium salts. Significant diversity is available in the substituents at the C4 position in four-component, one-pot couplings, by either trapping the in situ intermediate after α-arylation with carbon or heteroatom-based electrophiles, or by performing an α,α-heterodiarylation to install aryl groups at this position. The α-arylation of nitrile and ester enolates gives access to 3-amino and 3-hydroxyisoquinolines and the α-arylation of tert-butyl cyanoacetate followed by electrophile trapping, decarboxylation and cyclization, C4-functionalized 3-aminoisoquinolines. An oxime directing group can be used to direct a C–H functionalization/bromination, which allows monofunctionalized rather than difunctionalized aryl precursors to be brought through this synthetic route.
Introduction
It has been over a century since the first synthetic routes to isoquinolines were published. The Bischler–Napieralski,1 Pictet–Spengler2 and Pomeranz–Fritch reactions,3 all based around the lynchpin of electrophilic aromatic substitution, were the mainstay of the synthetic chemist's toolkit for much of the intervening period. However, the ubiquity of the isoquinoline motif in biologically-active natural products,4 alongside its applications in pharmaceuticals,5 functional organic materials6 and ligands for catalysis7 has recently inspired a resurgence in synthetic efforts towards this template.8 A number of notable recent contributions have exploited the versatility provided by modern synthetic methodology to access this motif, in particular the scope afforded by transition metal catalysis,9 and in so doing have vastly expanded the synthetically-accessible isoquinoline motifs, particularly those containing electron-deficient core structures which were unobtainable via the traditional methods.Our contribution to this area began when we embarked on a research program employing the palladium-catalyzed cross-coupling of a ketone enolate with an aryl halide to construct the C4–C4′ bond en route to the isoquinoline nucleus.10 This α-arylation reaction has become a powerful addition to the arsenal of the synthetic chemist11 since its discovery in 199712 and is now a well-established, albeit underutilized, palladium-catalyzed coupling procedure. It had previously been utilized in the synthesis of various five-membered heterocyclic frameworks, including indoles,13 benzofurans14 and benzothiophenes,14b where together the aryl halide and enolate provided all required skeletal carbon atoms. In our work we discovered that aryl halides that were ortho-functionalized with a protected aldehyde or ketone moiety, could be efficiently coupled furnishing a protected 1,5-dicarbonyl, which possessed all necessary carbon atoms to cyclize to an isoquinoline upon subjection to a mildly acidic source of ammonia, forming the C1–N and N–C3 bonds in the process. Later we disclosed how the greater acidity imparted on the α-carbon by the addition of the aryl group meant that the α-arylated intermediate (which sat deprotonated as the anion in situ) could be treated with a reactive electrophile to install additional functionality at the C4 position.15 This resulted in an efficient multi-component coupling procedure which could be applied to the synthesis of a series of highly-substituted isoquinolines. The α-arylation and electrophile trapping of the enolate of tert-butyl cyanoacetate gave an intermediate which could be decarboxylated and cyclized to furnish 3-aminoisoquinolines. We have recently applied this methodology to the synthesis of a number of natural products.16
Herein we present significant extensions of this earlier work, revealing for the first time how this protocol can be applied to the α-arylation of enolates derived from the less acidic nitriles and esters, allowing swift access to 3-amino and 3-hydroxyisoquinolines. We also report the first direct access to an isoquinolinium salt. Furthermore, we disclose here how exchanging the acetal protecting group for an oxime directing group, allows the procedure to be extended to unfunctionalized benzaldehydes and phenyl ketones via the application of C–H functionalization/bromination chemistry. In addition to these previously unexplored areas, we include a number of additional novel examples and single crystal X-ray structures of some of our earlier methods. We have presented these together with our previous results to provide proper context and allow the full scope (and limitations) of this research program to be discussed in detail.
Results and discussion
A classic retrosynthetic disconnection of a pyridine ring involves the condensation of a 1,5-dicarbonyl with a source of ammonia to form a dihydropyridine followed by an oxidation to a pyridine (Scheme 1, part (a)). The addition of a fused benzene ring to the backbone of the 1,5-dicarbonyl engenders the intermediate suitable for isoquinoline synthesis and formally increases the oxidation level, hence removing the need for a separate oxidation step in the synthesis (part (b)).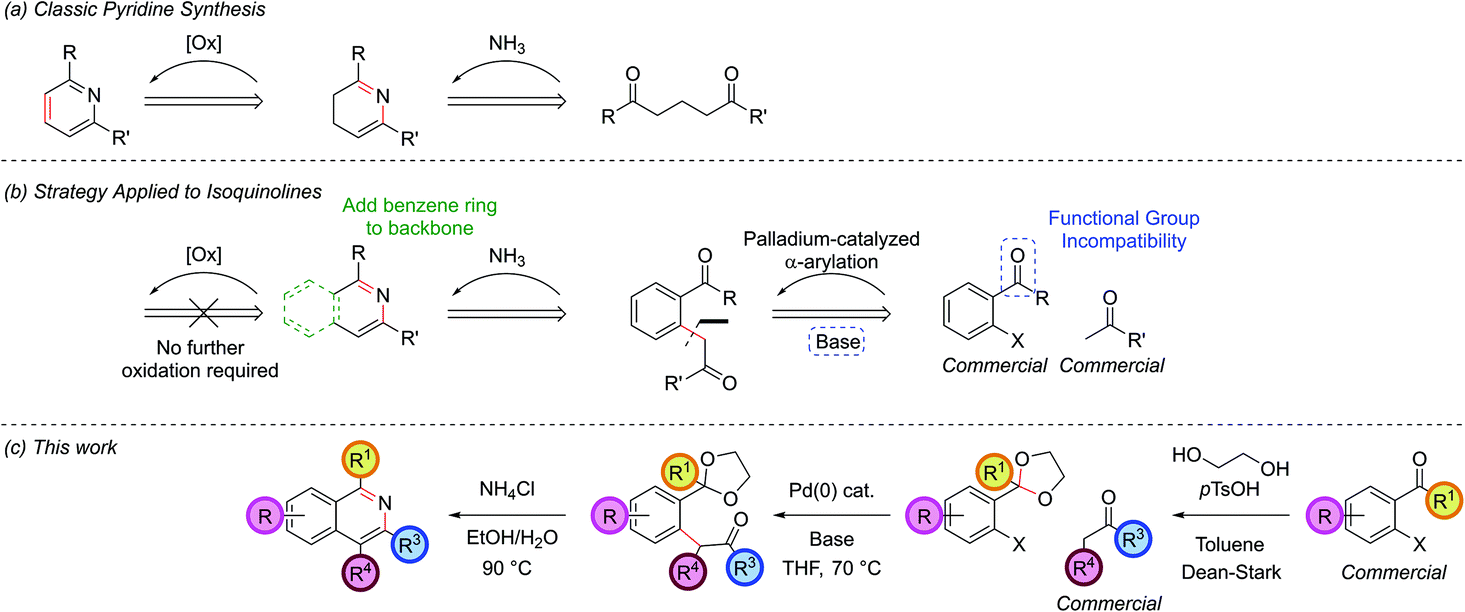 | ||
| Scheme 1 Retrosynthetic strategy for isoquinolines. | ||
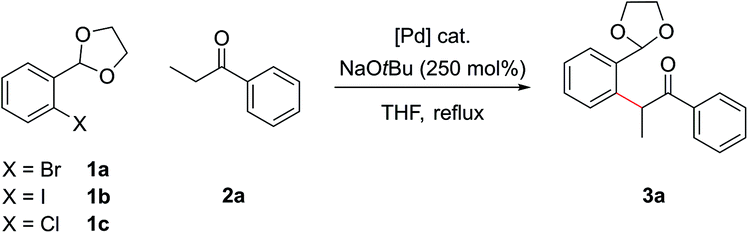
Disconnection of the 1,5-dicarbonyl at the carbon–aryl bond indicated leads to two simple commerically available building blocks, an enolizable ketone and an aryl halide possessing a formyl group or similar at the ortho-position. Benzaldehydes are vulnerable to aldol condensations in the presence of enolizable ketones under the basic reaction conditions of the α-arylation reaction and so a cyclic acetal protecting group was chosen for this moiety (part (c)). While protecting groups in chemical synthesis are always undesirable, the use of an acetal is notable in requiring only inexpensive reagents to both install and remove, the latter of which is able to occur in situ in the final step of our procedure. The prototype acetal 1a is commercially available, however, when the requisite acetals were not they could be synthesized in near quantitative yield by refluxing the benzaldehyde with ethylene glycol and catalytic para-toluenesulfonic acid in toluene in a Dean–Stark apparatus.10,15,16 Dioxolane acetals had been previously shown to be compatible with α-arylation in the meta position,12a but their tolerance in the sterically more encumbering ortho-position, where there was also a possibility of chelation, was unknown at the outset of this project.
A model reaction was chosen, using commercially available aryl bromide 1a and ketone 2a, and a range of palladium catalyst/ligand combinations were screened in addition to various bases and solvents. NaOtBu proved to be the best base and THF the best solvent of a variety screened. It was found that an excess of base (250 mol%) increased product yield, as had been noted in previous studies.17 The most successful ligand/catalyst systems were the air stable Pd(II) precursors (DtBPF)PdCl2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 18 and PdCl2(Amphos)2.19 Employing (DtBPF)PdCl2 (2.0 mol%) in this system gave a yield of 3a of 82%; PdCl2(Amphos)2 (2.0 mol%) was equally as proficient, also giving a yield of 82% (Scheme 2). These two preformed catalysts both contain electron-rich bulky phosphines bearing two tert-butyl groups and one electron-rich arene, giving an indication of the optimal ligand for the reaction. The air stable nature of these catalysts greatly increased the practicality of the synthetic procedure.
18 and PdCl2(Amphos)2.19 Employing (DtBPF)PdCl2 (2.0 mol%) in this system gave a yield of 3a of 82%; PdCl2(Amphos)2 (2.0 mol%) was equally as proficient, also giving a yield of 82% (Scheme 2). These two preformed catalysts both contain electron-rich bulky phosphines bearing two tert-butyl groups and one electron-rich arene, giving an indication of the optimal ligand for the reaction. The air stable nature of these catalysts greatly increased the practicality of the synthetic procedure.
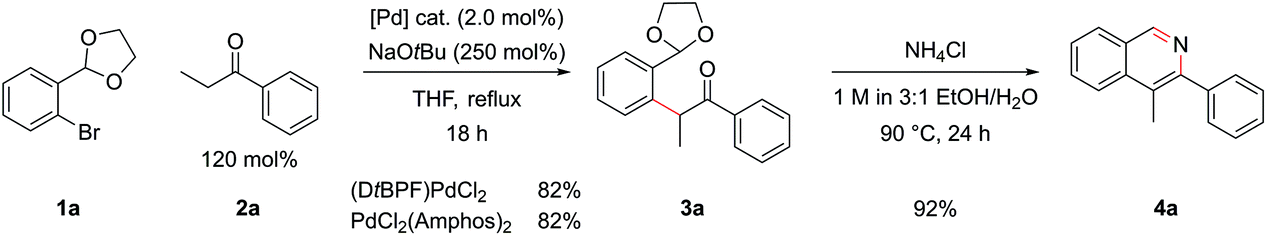 | ||
| Scheme 2 Optimized α-arylation and cyclization conditions for R1 = H. | ||
In order to concurrently promote acetal hydrolysis and the cyclization to form the isoquinoline in one synthetic procedure, both a source of acid and a source of ammonia were needed. Both were provided by a solution of NH4Cl in EtOH/H2O (pH ≈ 5), which enabled the smooth conversion of intermediate 3a to isoquinoline 4a. Further acidifying the NH4Cl solution did not result in increased yields. This procedure could also be extended to employ aryl chlorides and iodides in place of bromides, which greatly increased the scope of commercially available substrates that could be channelled into the reaction (Table 1). Under the optimized α-arylation reaction conditions, aryl iodide 1b gave comparable yields to that of the aryl bromide (entry 10). The chloride 1c was understandably less reactive, but with higher catalyst loadings and longer reaction times improved yields could be obtained with PdCl2(Amphos)2 as the catalyst (entry 3).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | X | Catalyst | Loading | Ketone | Time | Yield |
| 1 | Cl | (DtBPF)PdCl2 | 5.0 mol% | 200 mol% | 18 h | 30% |
| 2 | Cl | PdCl2(Amphos)2 | 5.0 mol% | 200 mol% | 18 h | 45% |
| 3 | Cl | PdCl2(Amphos)2 | 5.0 mol% × 2 | 200 mol% | 96 h | 74% |
| 4 | Br | (DtBPF)PdCl2 | 0.5 mol% | 120 mol% | 18 h | 71% |
| 5 | Br | (DtBPF)PdCl2 | 0.5 mol% | 200 mol% | 18 h | 74% |
| 6 | Br | (DtBPF)PdCl2 | 2.0 mol% | 120 mol% | 18 h | 82% |
| 7 | Br | (DtBPF)PdCl2 | 2.0 mol% | 200 mol% | 18 h | 83% |
| 8 | Br | (DtBPF)PdCl2 | 5.0 mol% | 200 mol% | 18 h | 89% |
| 9 | Br | PdCl2(Amphos)2 | 2.0 mol% | 120 mol% | 18 h | 82% |
| 10 | I | (DtBPF)PdCl2 | 2.0 mol% | 120 mol% | 18 h | 79% |
Employing protected 2′-bromophenyl ketones as the aryl bromide partner allowed access to isoquinolines bearing substitution at the C1 position, where the extra steric hindrance induced by the additional methyl group in 1d did not detrimentally affect the α-arylation reaction of 1d with propiophenone (Scheme 3). In contrast to the benzaldehyde case, where cyclization ensued rapidly after deprotection of the intermediate acetal at pH 5, here cyclization and aromatization at pH 5 was sluggish. The optimum pH for cyclization of these substrates was approximately pH 9. To effect this sequence efficiently, aqueous 2 M NH4HCO3 was added to increase the pH after acetal hydrolysis was complete and then the reaction was heated for a further 24 h at 90 °C.
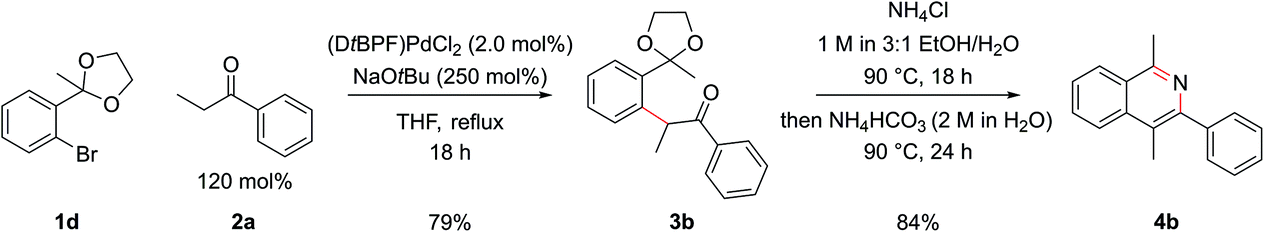 | ||
| Scheme 3 Optimized α-arylation and cyclization conditions for R1 ≠ H. | ||
Exploring the scope of substitution revealed that the C3 substituent could include electron-rich benzene rings 4d, electron-deficient benzene rings 4e, and heteroaryl rings 4f (Fig. 1). It should be noted that cross-couplings between two heteroarenes in the ortho-position to both heteroatoms are often difficult to carry out and so this is an important strength of this route.20 Deoxybenzoin 2f could also be employed as the ketone partner allowing a phenyl substituent to be installed at the C4 position in 4h. The successful coupling of deoxybenzoin prompted investigation as to whether more acidic carbonyl derivatives could be employed with heteroatom functionality attached to the carbonyl α-position. 2-Methoxyacetophenone could be successfully coupled to give 4i providing a mild base such as K3PO4 was employed in the α-arylation step. However, other heteroatom-functionalized enolates (such as that from 2-(methylthio)acetophenone, 2h), decomposed under the action of base or failed to couple.
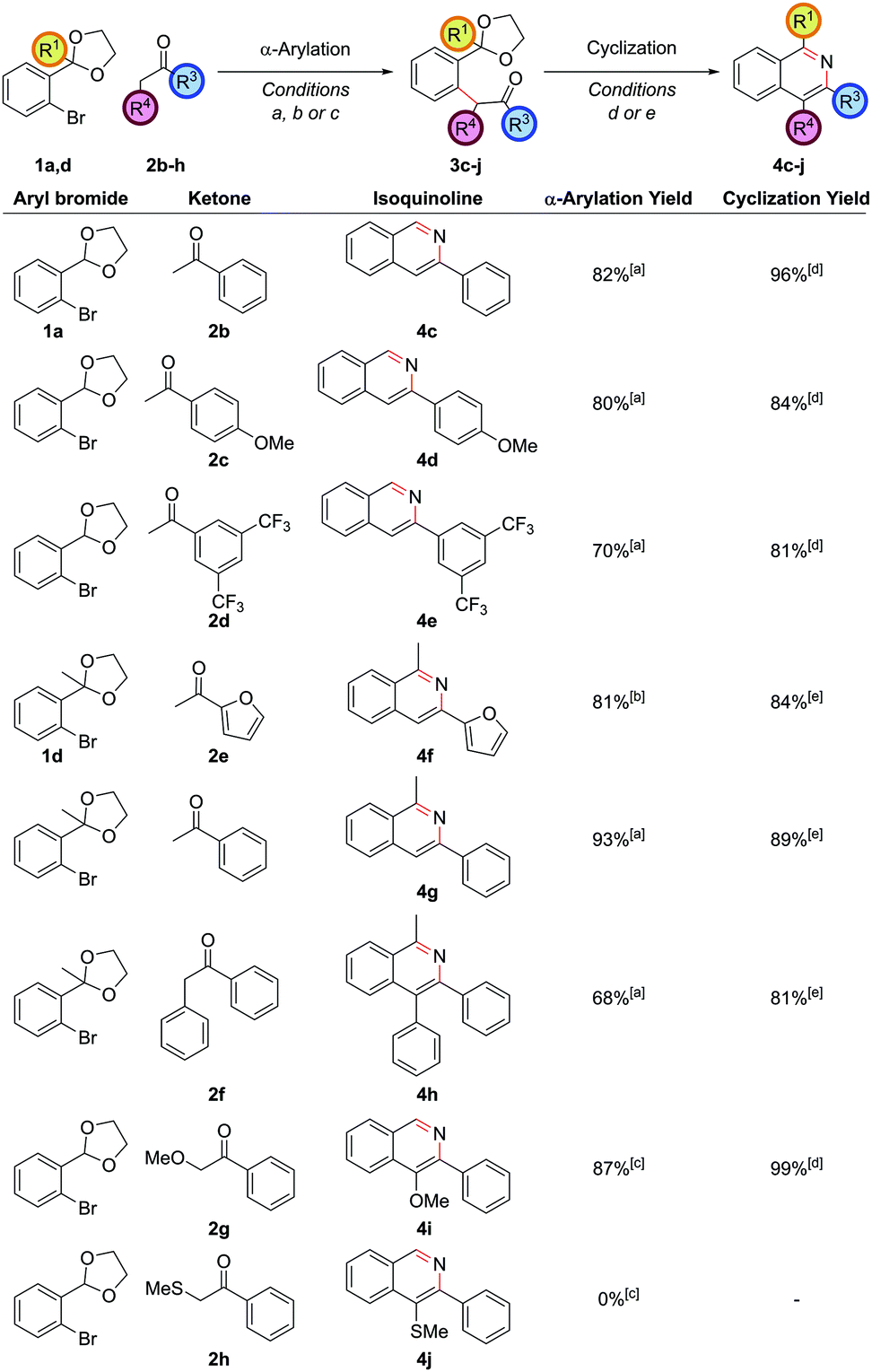 | ||
| Fig. 1 Exploring the scope with aryl ketones. [a] (DtBPF)PdCl2 (2.0 mol%), NaOtBu (250 mol%), ketone (120 mol%), THF, reflux, 18 h; [b] (DtBPF)PdCl2 (5.0 mol%), NaOtBu (250 mol%), ketone (200 mol%), THF, reflux, 18 h; [c] (DtBPF)PdCl2 (5.0 mol%), K3PO4 (250 mol%), ketone (120 mol%), THF, reflux, 18 h; [d] NH4Cl (1 M in 3:1 EtOH/H2O), 90 °C, 24 h; [e] NH4Cl (1 M in 3:1 EtOH/H2O), 90 °C, 18 h, then NH4HCO3 (2 M in H2O), 90 °C, 24 h. | ||
The α-arylation of cyclic ketones and subsequent cyclization furnished tricyclic isoquinoline moieties (i.e. with alkyl substitution at both the C3 and C4 positions) – these are difficult substituent patterns to access by most modern methods. Cycloheptanone was successfully arylated to give isoquinolines 4k and 4l (Fig. 2).
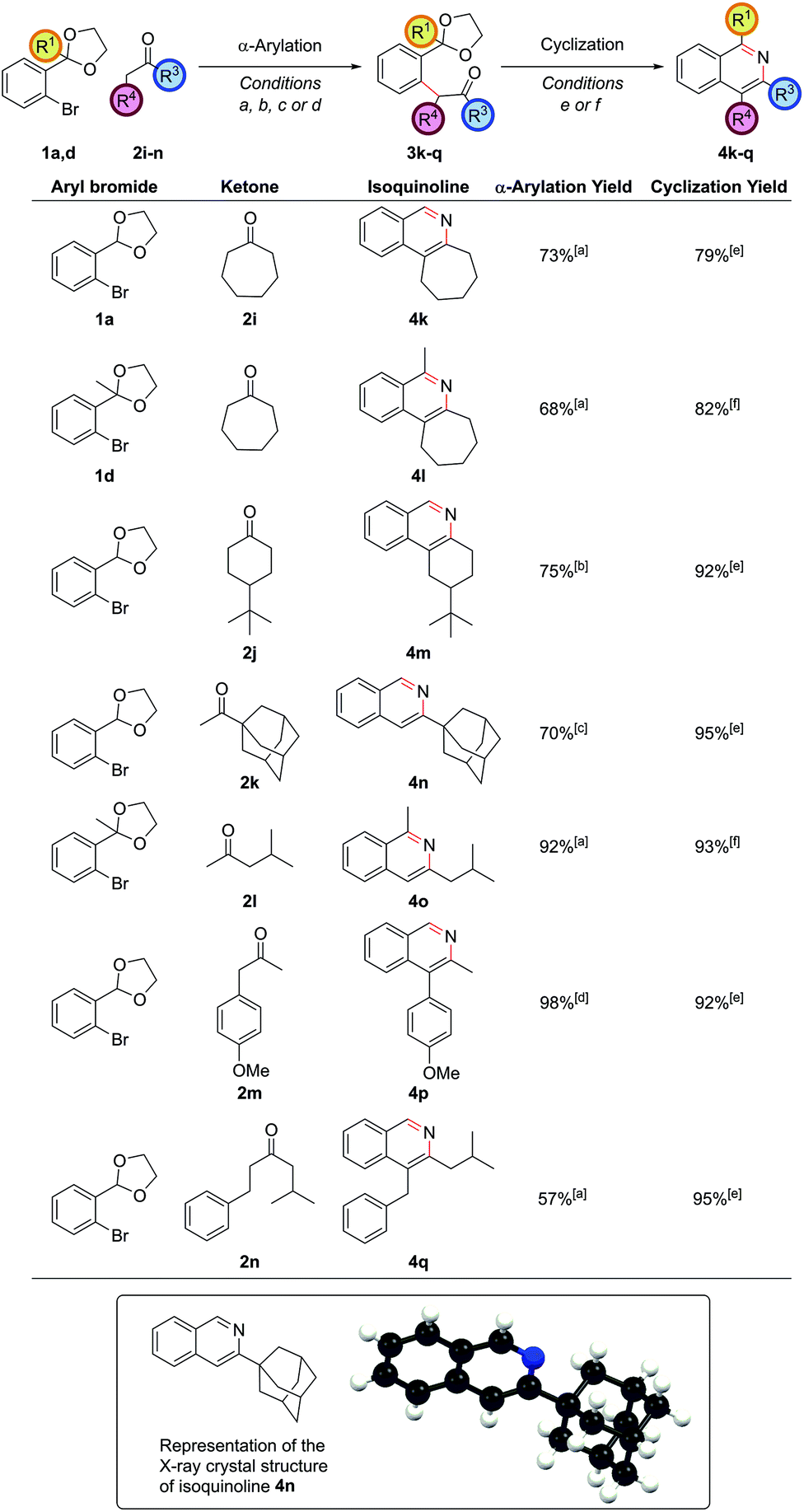 | ||
| Fig. 2 Exploring the scope with alkyl ketones. [a] (DtBPF)PdCl2 (5.0 mol%), NaOtBu (250 mol%), ketone (200 mol%), THF, reflux, 18 h; [b] (DtBPF)PdCl2 (5.0 mol%), nBuLi (250 mol%), TMP (270 mol%), ketone (200 mol%), THF, −78 °C to reflux,18 h; [c] (DtBPF)PdCl2 (2.0 mol%), NaOtBu (250 mol%), ketone (120 mol%), THF, reflux, 18 h; [d] (DtBPF)PdCl2 (5.0 mol%), Cs2CO3 (250 mol%), ketone (200 mol%), THF, reflux, 18 h; [e] NH4Cl (1 M in 3:1 EtOH/H2O), 90 °C, 24 h; [f] NH4Cl (1 M in 3:1 EtOH/H2O), 90 °C, 18 h, then NH4HCO3 (2 M in H2O), 90 °C, 24 h. | ||
Cyclic ketones with smaller rings than cycloheptanone proved more troublesome with NaOtBu, as had been noted in previous studies,21 due to increased likelihood for aldol condensations. With cyclohexanone-based ketones such as 2j, an α-arylation yield of 75% could be obtained by using the strong base lithium tetramethylpiperidide to fully deprotonate the ketone and form the enolate at −78 °C, before adding the aryl bromide and heating at reflux as before. The propensity for aldol condensations is also a problem for linear dialkyl ketones, making successful α-arylation of these substrates a challenging problem.22 Superior partners are usually ketones bearing a bulky group on one side of the carbonyl so as to ensure a regioselective arylation reaction. The α-arylation of adamantyl methyl ketone, 2k, enabled synthesis of isoquinoline 4n where the bulky adamantyl group is directly attached to the ring, as evidenced in the crystal structure. Attaching an adamantyl group on a preformed arene ring is very difficult, confirming the power of installing substituents pre-cyclization. Where the two alkyl groups differed substantially, such as in isobutyl methyl ketone, 2l, α-arylation could be accomplished exclusively on the methyl as opposed to the methylene position en route to isoquinoline 4o. In cases where the methylene carbon was more acidic, such as 4′-methoxyphenyl acetone, 2m, the strength of base became important in determining the outcome of the reaction. Utilizing NaOtBu as the base gave a 1:5 mixture of arylation at the methylene to methyl positions respectively. However, with the weaker base Cs2CO3 complete regioselectivity for arylation at the methylene position could be obtained. The use of stronger bases to achieve arylation exclusively at the methyl position led solely to decomposition of starting material in this case. On the more similarly substituted isobutyl 2-phenylethyl ketone, 2n, selectivity could even be achieved for α-arylation at the less hindered and slightly more activated methylene position on the side of the phenyl group, highlighting how subtle steric and electronic factors can give rise to useful regioselectivity.
On the aryl bromide partner, a variety of substitution could be tolerated. Methoxy-substituted partners worked well, allowing access to isoquinolines 4r, 4s and 4t (Fig. 3). The successful coupling to synthesize 4t is particularly noteworthy, as this α-arylation reaction features a bromide flanked by two ortho-substituents, both of which contain oxygen atoms that could possibly chelate to palladium. The electron-rich bromides could also include heteroaryl moieties such as a thiophene, leading to thienopyridine 4u.
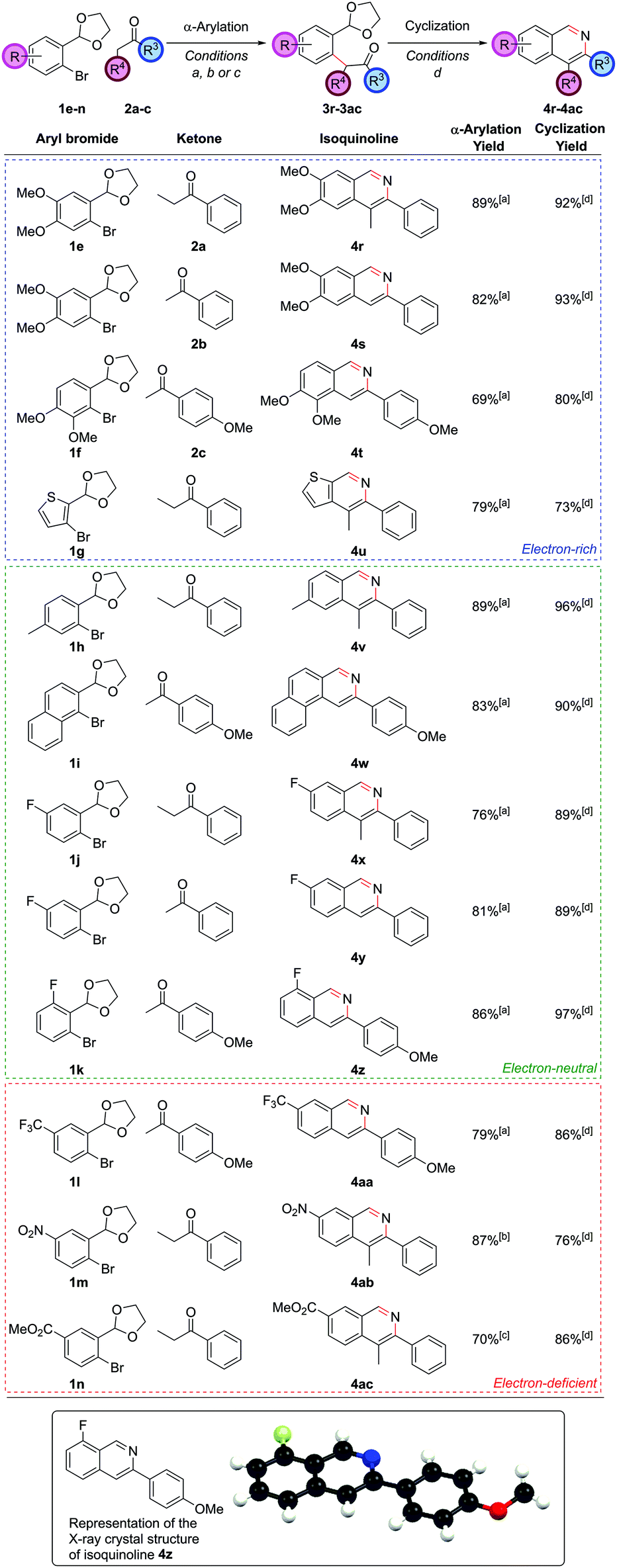 | ||
| Fig. 3 Exploring the scope of the aryl bromide partner. [a] (DtBPF)PdCl2 (2.0 mol%), NaOtBu (250 mol%), ketone (120 mol%), THF, reflux, 18 h; [b] PdCl2(Amphos)2 (5.0 mol%), Cs2CO3 (250 mol%), ketone (200 mol%), THF, reflux 18 h; [c] PdCl2(Amphos)2 (5.0 mol%), K3PO4 (250 mol%), ketone (120 mol%), THF, reflux, 18 h; [d] NH4Cl (1 M in 3:1 EtOH/H2O), 90 °C, 24 h. | ||
Of the electron-neutral aryl bromides, alkyl and aryl substituents could be readily incorporated to give isoquinolines 4v and 4w. Fluorinated aryl bromides were also competent substrates giving fluorinated isoquinolines 4x, 4y and 4z (of which a crystal structure of isoquinoline 4z was obtained) – the ability to install fluorinated motifs being a desirable property in medicinal chemistry to protect against metabolic instability. The trifluoromethyl group is also prevalent in medicinal chemistry and could be successfully installed at the C7 position in isoquinoline 4aa, giving the first truly electron-deficient isoquinoline accessed via this methodology. Other electron-poor systems could be accessed too. Nitro groups could be incorporated (4ab with the use of the weaker base Cs2CO3 and PdCl2(Amphos)2), and methyl esters could also be incorporated (4ac with the use of K3PO4 and PdCl2(Amphos)2); both of these are important functional groups for further synthetic manipulation post-heterocycle construction. The ready access to electron-deficient heterocyclic frameworks is a strength of this route over traditional methods. As well as demonstrating the success of the procedure with electron-rich through to electron-deficient aryl bromide partners, we had now shown that substitution could be incorporated at all positions on the carbocyclic ring of the isoquinolines, with no problems over regiocontrol.
This synthetic procedure enabled the synthesis of a range of substituted isoquinolines in two or three steps from commercial chemicals. However, in order to increase the synthetic practicality of the procedure it was thought that a one-pot procedure could be developed. This would be aided by the fact that the THF solvent for the α-arylation reaction and the EtOH/H2O solvent for the cyclization were miscible, allowing both reactions to be performed sequentially in one vessel. Therefore, after TLC analysis indicated that the α-arylation was complete, the reaction mixture was adjusted to pH 5 by the dropwise addition of aqueous 1 M HCl, an NH4Cl solution was added and the mixture heated to ensure cyclization. This procedural change eliminated one step and purification from the reaction sequence. Seven isoquinolines were synthesized via this one-pot protocol (Fig. 4) and the one-pot yield was only marginally less than the overall yield of the stepwise procedure. This more practical synthetic procedure represents a three-component (aryl bromide, ketone, ammonia source), one-pot coupling, where three of the six heteroarene ring bonds were made in a single synthetic operation.
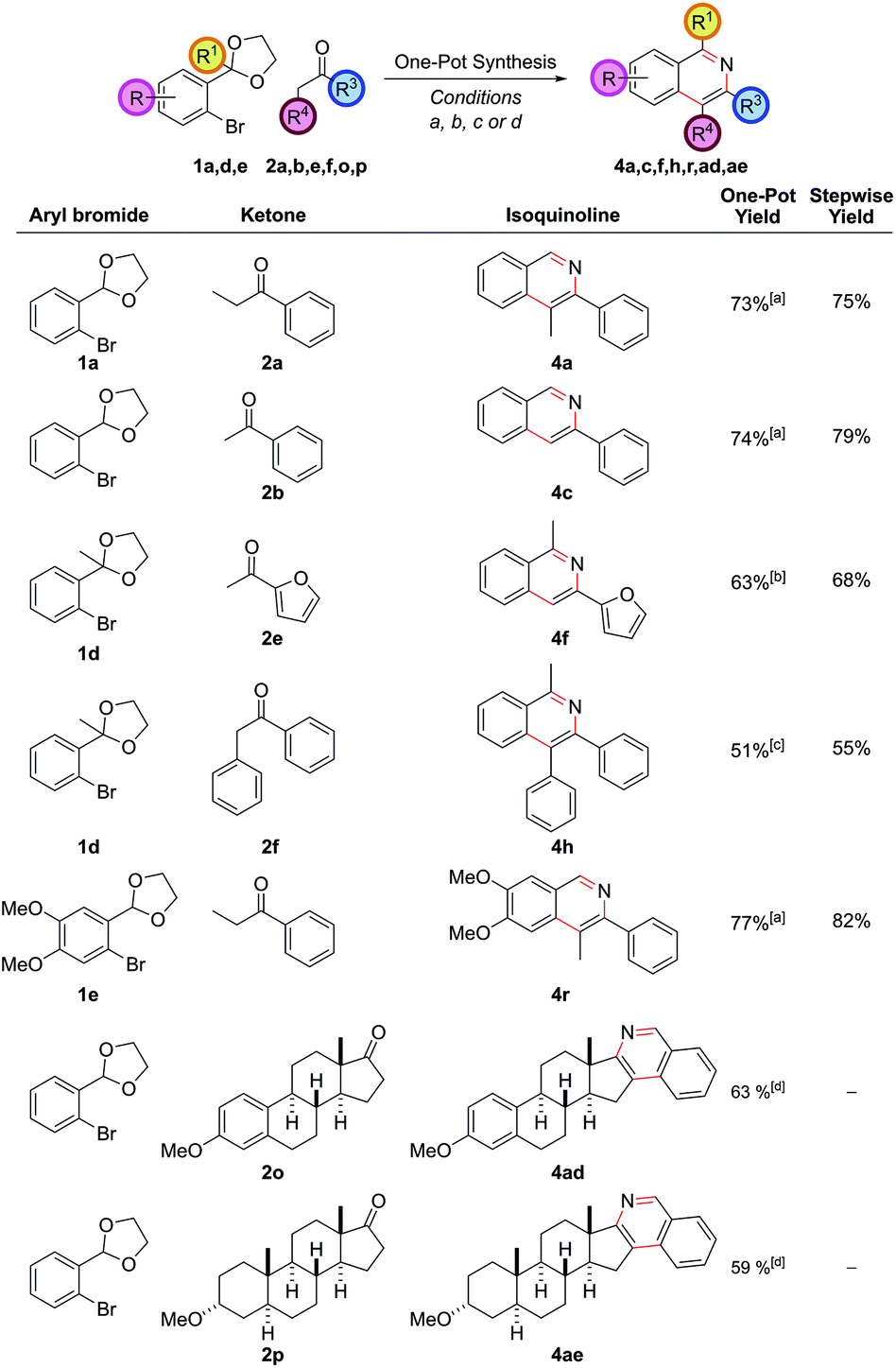 | ||
| Fig. 4 One-pot synthesis of isoquinolines. [a] (DtBPF)PdCl2 (2.0 mol%), NaOtBu (250 mol%), ketone (120 mol%), THF, reflux, 18 h, then HCl(aq), NH4Cl (1 M in 3:1 EtOH/H2O), 90 °C, 24 h; [b] (DtBPF)PdCl2 (5.0 mol%), NaOtBu (250 mol%), ketone (200 mol%), THF, reflux, 18 h, then HCl(aq), NH4Cl (1 M in 3:1 EtOH/H2O), 90 °C, 18 h, then NH4HCO3 (2 M in H2O), 90 °C, 24 h; [c] (DtBPF)PdCl2 (2.0 mol%), NaOtBu (250 mol%), ketone (120 mol%), THF, reflux, 18 h, then HCl(aq), NH4Cl (1 M in 3:1 EtOH/H2O), 90 °C, 18 h, then NH4HCO3 (2 M in H2O), 90 °C, 24 h; [d] (DtBPF)PdCl2 (5.0 mol%), NaOtBu (250 mol%), aryl bromide (150 mol%), THF, reflux, 18 h, then HCl(aq), NH4Cl (1 M in 3:1 EtOH/H2O), 90 °C, 24 h. | ||
Included in the one-pot examples were two ketones derived from steroidal hormones which could be converted into hexacyclic isoquinoline skeletons (with the steroidal ketone, now the expensive coupling partner, used as the limiting reagent). A large number of pharmaceutical drugs are constructed upon a steroid core, often significantly modified on the five-membered D-ring with the addition of extra heteroatom or heterocyclic functionality. The one-pot protocol hence allows expedient access to an array of modified steroids by variation in the aryl bromide partner which may be of value in target screening.
Change of nitrogen oxidation state: isoquinoline N-oxides
Isoquinoline N-oxides are also high value targets, and are particularly useful if further substitution on the arene ring is desired after the ring construction. As they are more electron-rich than the corresponding isoquinoline they undergo electrophilic aromatic substitution with much greater ease and they also show propensity for C–H functionalization reactions.23 Direct access to isoquinoline N-oxides avoids the need for oxidation of the parent isoquinoline – a reaction which often requires conditions that do not tolerate sensitive functionality. Limited literature precedent revealed that analogous 1,5-dicarbonyls had been converted into isoquinoline N-oxides directly by treatment with hydroxylamine, however, no systematic study had been undertaken.24 A number of intermediates obtained in this work were treated with a solution of hydroxylamine hydrochloride (which had a pH of around 4.5). This leads to rapid and clean deprotection, and cyclization to the corresponding isoquinoline N-oxide (Fig. 5). Single crystal X-ray structures were also obtained of isoquinoline N-oxides 5a and 5b.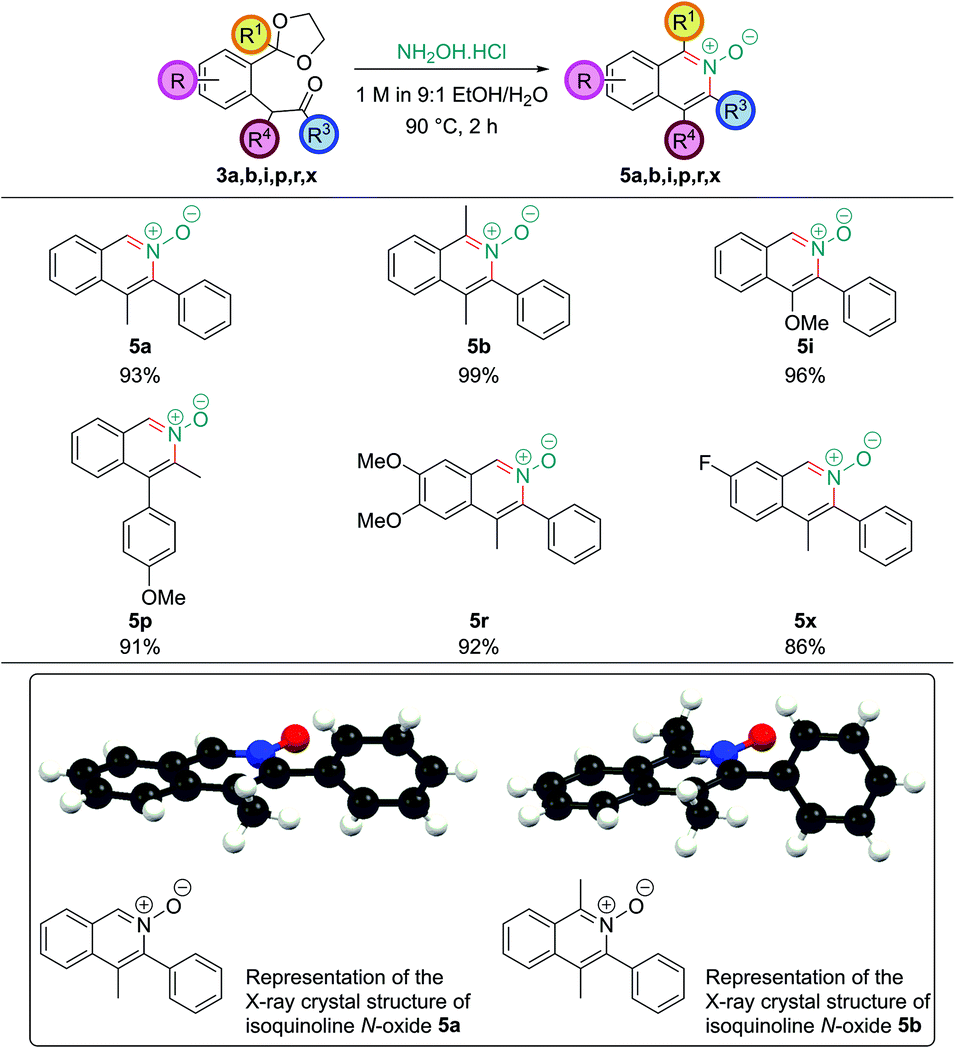 | ||
| Fig. 5 Synthesis of isoquinoline N-oxides. | ||
Isoquinoline N-oxides bearing C1 substitution, such as 5b, unlike their parent isoquinolines, could be synthesized without needing to basify the reaction conditions after acetal hydrolysis; this is likely due to the increased nucleophilicity of the hydroxylamine nitrogen atom.
Isoquinolinium salts
The successful cyclization to synthesize N-oxides raised the possibility as to whether the dicarbonyl intermediates could be treated with primary amines to also allow access to isoquinolinium salts directly. Pleasingly, intermediate 3a underwent clean conversion to isoquinolinium salt 6a upon treatment with methylamine hydrochloride (Scheme 4).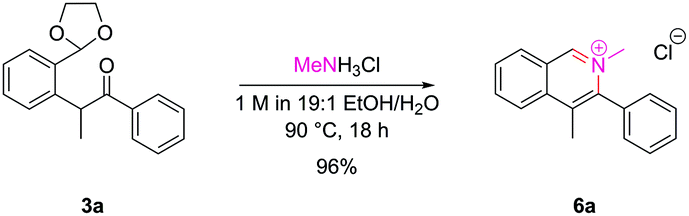 | ||
| Scheme 4 Synthesis of isoquinolinium salt 6a. | ||
With bulkier primary amines, higher temperatures were required to encourage cyclization, and significant amounts of the dealkylated isoquinoline were observed, which unfortunately limited this approach to sterically-unhindered primary amines. However, the inherent nucleophilicity of the isoquinoline nitrogen atom can be used to alkylate post-cyclization with a tethered electrophile which we employed en route to several natural products within our laboratory.16
Change of carbon oxidation state: 3-amino and 3-hydroxyisoquinolines
By employing hydroxylamine hydrochloride instead of ammonium chloride in the cyclization step, the resulting heterocycle was formed one oxidation level higher. Alternatively, we can utilize a higher oxidation level reagent in the α-arylation step, leading to the synthesis of 3-aminoisoquinolines or 3-hydroxyisoquinolines, derivatives of which are known to display significant biological activity.25 This tactic would require the arylation of the enolate of a nitrile, ester or similar compound rather than a ketone. The α-arylation of nitriles is significantly more challenging than ketones for a variety of reasons; for example, the proton to be removed is less acidic, meaning stronger bases are often required and there are problems of diarylation due to the less sterically-hindered nature of the nitrile product. However, recently some advances have been made in this area.26 Applying the α-arylation conditions that we had used successfully with ketones to nitriles such as propionitrile were unsuccessful, but as we desired an operationally simple procedure that could directly employ commercially available nitriles, we examined nitriles bearing an additional acidifying substituent such that their pKa was similar to the ketones that had been successfully coupled (2-phenylacetonitrile has a pKa of 21.9 in DMSO,27 compared to 24.4 for propiophenone28 for example). It was found that 2-phenylacetonitrile, 7a, could be successfully coupled with aryl bromide 1a with 5.0 mol% PdCl2(Amphos)2 and Cs2CO3 as the base (Fig. 6). Cyclization of intermediate 8a required basification and furnished the 3-aminoisoquinoline 9a (as opposed to the 3-hydroxyisoquinoline or its isoquinolone tautomer), clearly evidenced by the broad singlet at 4.42 ppm (relative integral 2H) from the NH2 group in the 1H NMR spectrum. This matched the literature data for this compound29 and was expected as isoquinolines substituted in the 3-position are known to favour the amino/hydroxy forms over the imine/oxo forms.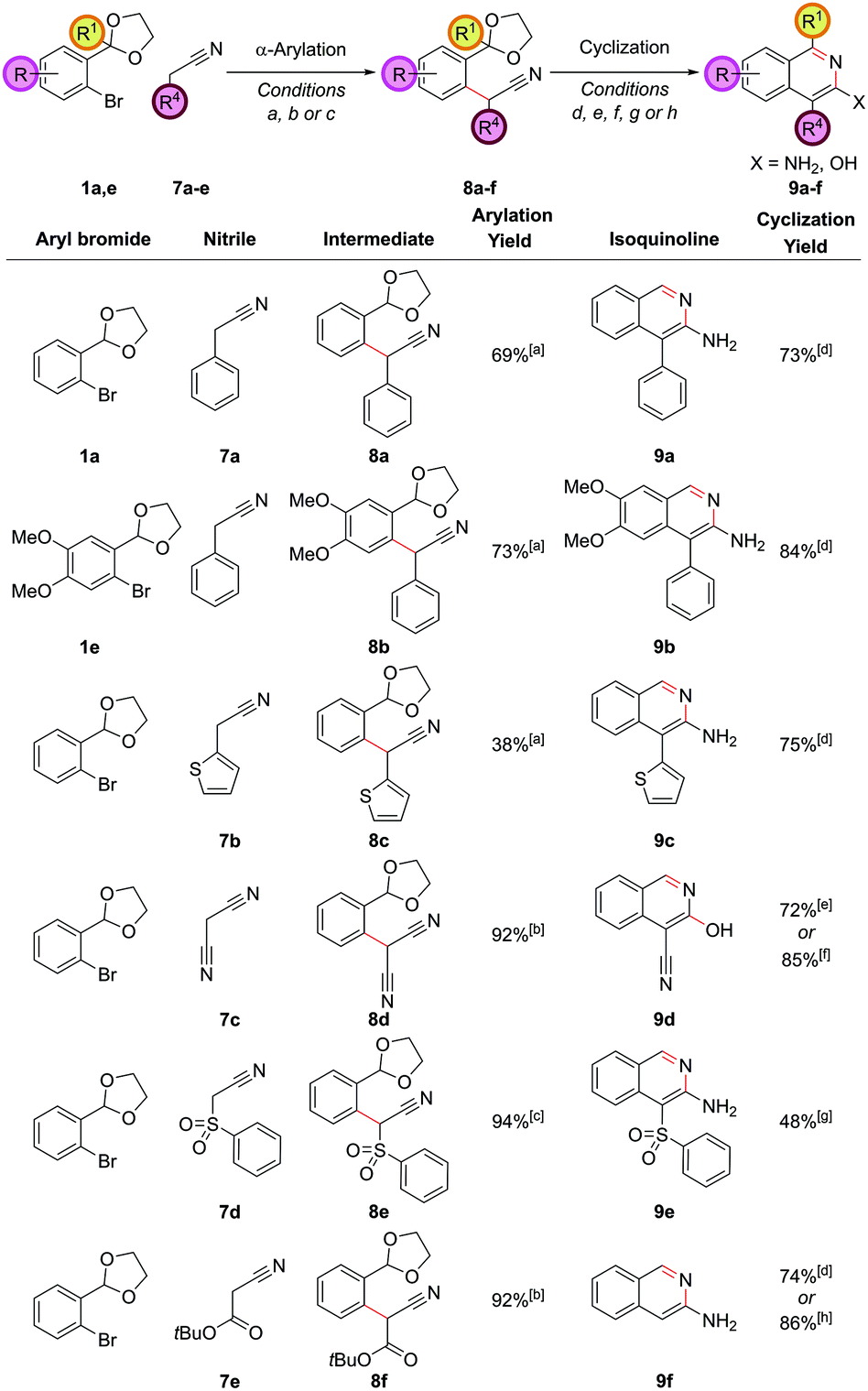 | ||
| Fig. 6 Access to 3-amino and 3-hydroxyisoquinolines. [a] PdCI2(Amphos)2 (5.0 mol%), Cs2CO3 (250 mol%), nitrile (200 mol%), THF, reflux, 18 h; [b] (DPPF)PdCl2 (2.0 mol%), NaOtBu (250 mol%), nitrile (100 mol%), aryl bromide (120 mol%), 1,4-dioxane, 70 °C, 4 h; [c] (DPPF)PdCl2 (2.0 mol%), NaOtBu (250 mol%), nitrile (100 mol%), aryl bromide (120 mol%), 1,4-dioxane, 70 °C, 18 h; [d] NH4Cl (1 M in 3:1 EtOH/H2O), 90 °C, 18 h, then NH4HCO3 (2 M in H2O), 90 °C, 24 h; [e] NH4Cl (1 M in 3:1 EtOH/H2O), 90 °C, 18 h; [f] pTsOH (10 mol%), THF/H2O (1:1), 50 °C, 18 h; [g] NH4Cl (1 M in 3:1 EtOH/H2O), 90 °C, 4 h, then NH4HCO3 (2 M in H2O), 65 °C, 1 h; [h] EtOH/H2O (3:2), 90 °C, 18 h, then NH4Cl (1000 mol%), 90 °C, 3 h, then NH4HCO3 (2 M in H2O), 90 °C, 3 h. | ||
Other classes of acidifying substituents could also be employed, but here again a slight change in the α-arylation conditions was required in order to get optimal yields. Utilizing (DPPF)PdCl2 as the catalyst30 in dioxane solvent led to a 92% yield in the coupling of malononitrile, 7c, with aryl bromide 1a. Interestingly, this intermediate showed a preference to cyclize to the 3-hydroxyisoquinoline 9d under the NH4Cl cyclization conditions, confirmed by high resolution mass spectrometry, the OH stretch in the IR, and by inference from the fact that this same cyclization could also be effected in higher yield in the absence of an external nitrogen source by the action of para-toluenesulfonic acid on intermediate 8d. (Phenylsulfonyl)acetonitrile, 7d, functioned well as the nitrile substrate, and gave intermediate 8e which could be cyclized to 3-aminoisoquinoline 9e. tert-Butyl cyanoacetate, 7e, was also proficient in the α-arylation reaction, but surprisingly after treatment with the cyclization conditions the unsubstituted 3-aminoisoquinoline, 9f, was produced, due to an ester hydrolysis and decarboxylation under the reaction conditions prior to cyclization. This meant the protocol had achieved the equivalent of α-arylating acetonitrile directly;31 this is an extremely challenging substrate in α-arylation methodology. This in situ decarboxylation also gave opportunities for further diversification which we sought to use to our advantage (vide infra).
Esters are also significantly less acidic than the corresponding ketones and have the potential to undergo Claisen condensations under basic conditions, complicating an attempted α-arylation.32 Again hoping to design a general procedure that didn't rely on specialized reagents, we proceeded with tert-butyl acetate, 10, LiHMDS as a base and PtBu3 (formed in situ from deprotonation of the air stable tetrafluoroborate salt) similar to conditions initially developed by Hartwig and coworkers.32b This delivered intermediate 11 in 68% yield, which could be cyclized to 3-hydroxyisoquinoline 12 in 70% yield, by hydrolysis of the acetal with pTsOH and then basification with NH4OH (Scheme 5); the data of this compound matched that from commercial suppliers. Unfortunately, the coupling of methyl and ethyl esters in this system was unsuccessful and tert-butyl esters of more sophisticated carboxylic acids coupled only in poor yields.
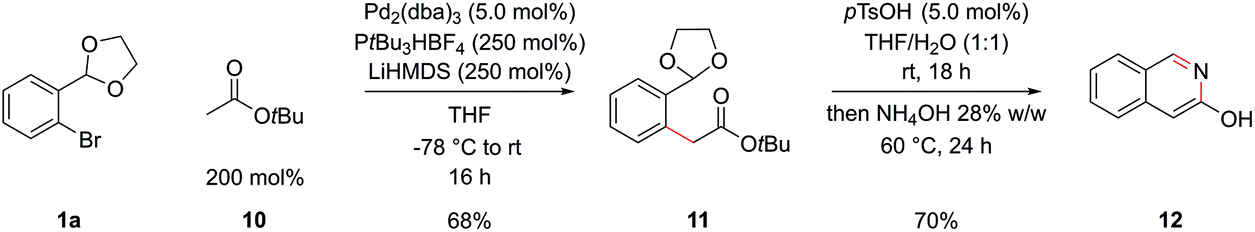 | ||
| Scheme 5 α-Arylation and cyclization of ester enolates. | ||
Functionalization at the C4 position
The isoquinoline C4 position is typically unreactive and difficult to selectively manipulate in preformed isoquinolines, since nucleophiles usually add to C1 or C3, and electrophiles to C5 or C8 (Scheme 6, part (a)).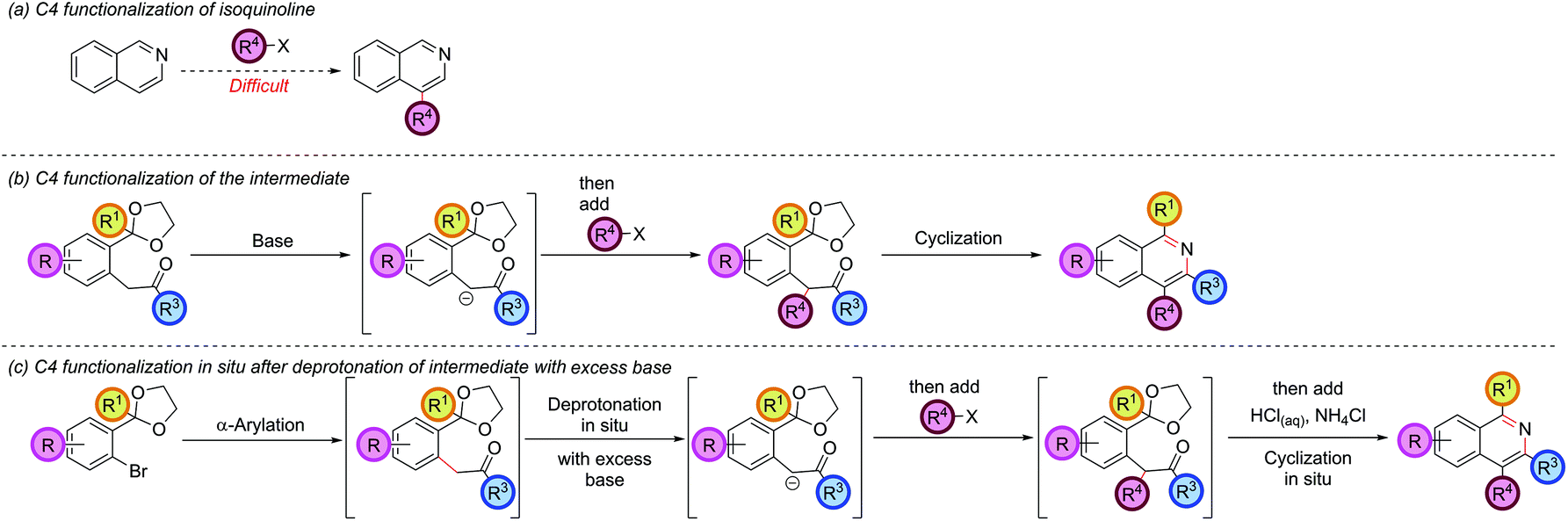 | ||
| Scheme 6 Functionalization of the C4 position. | ||
Functionalization at C4 typically requires reduction or addition across the C1–N bond to form a 1,2-dihydroisoquinoline, followed by reaction of the resulting enamine with an electrophile at C4, and a final elimination or reoxidation of the C1–N bond.33 Our strategy had thus far required the C4 substituent to be pre-installed on the ketone coupling partner and was limited by the accessibility of the requisite ketones and the compatibility of such ketones with the basic α-arylation conditions (such as the failure of 2h, Fig. 1). The regioselectivity of the α-arylation reaction for functionalization at the most acidic/least sterically-hindered position also imparted fundamental restrictions on the relative orientation of the groups present at C3 and C4 (vide supraFig. 2). However, it was thought that the enolate intermediate in our synthetic route was a prime candidate for further functionalization at the α-position via deprotonation and reaction with an electrophile. Subsequent cyclization would then install this group at the isoquinoline C4 position (Scheme 6, part (b)). In our earlier optimization, excess base was included in the α-arylation reaction to ensure complete reaction of the starting ketone, especially as the intermediate was preferentially deprotonated under the reaction conditions. Intercepting this intermediate in situ with an electrophile opened up the possibility of doing the α-arylation and C4-functionalization in one-pot (Scheme 6, part (c)). Combining this protocol with an in situ cyclization would transform the isoquinoline synthesis from a one-pot, three-component coupling (aryl bromide, ketone, and ammonia source) to a one-pot, four-component coupling protocol (aryl bromide, ketone, electrophile and ammonia source). Hartwig had previously employed iodomethane to trap the arylated enolate of diethylmalonate,34 and Wang also trapped the arylated enolate of tert-butylcyanoacetate,30 hinting at the possibility of a more general enolate trapping strategy being successful. Our envisaged approach also bore resemblance to Myers's trapping of eneamido anions with electrophiles en route to substituted isoquinolines.35
Resubjection of intermediate 3c to NaOtBu, followed by the addition of allyl bromide, 13a, furnished functionalized intermediate 14a in 68% yield. This could be cyclized to produce C4-functionalized isoquinoline 15a in 96% yield (Scheme 7). In order to combine all three steps in one-pot, the α-arylation was performed as previously, but the reaction was quenched first by addition of the electrophile. After stirring for a further period the reaction was acidified with HCl, the NH4Cl solution added, and cyclization ensued. This protocol gave isoquinoline 15a in 71% yield, compared to 75% overall yield for the isolated stepwise procedure. Hence a multi-component protocol had been developed which comprised three distinct chemical transformations, constructed four important skeletal bonds and coupled four different components in one-pot.
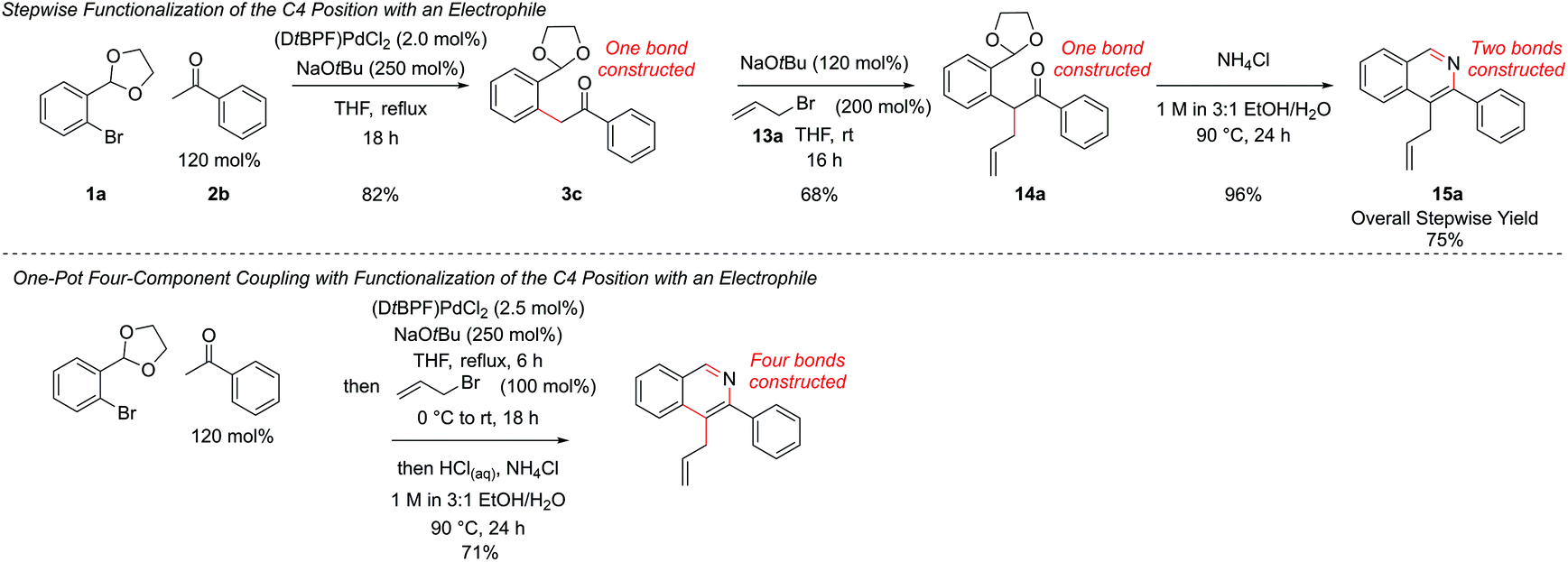 | ||
| Scheme 7 Development of four-component one-pot coupling. | ||
The method was broadly applicable to a range of carbon-based electrophiles (Fig. 7). Alkyl 15b and benzyl 15c groups could be readily installed. α-Bromoesters 13d and acrylates 13e were also reactive electrophiles and the cyclization conditions were sufficiently mild for the ester moieties to survive to furnish 15d and 15e. Vinyl bromides could also be introduced to form 15f, with no protodebromination observed, giving possibility for further synthetic modification via coupling reactions after isoquinoline formation. A single crystal X-ray structure was obtained for isoquinoline 15f. The use of Selectfluor® II, 13g, to give 4-fluoroisoquinoline 15g was also notable.
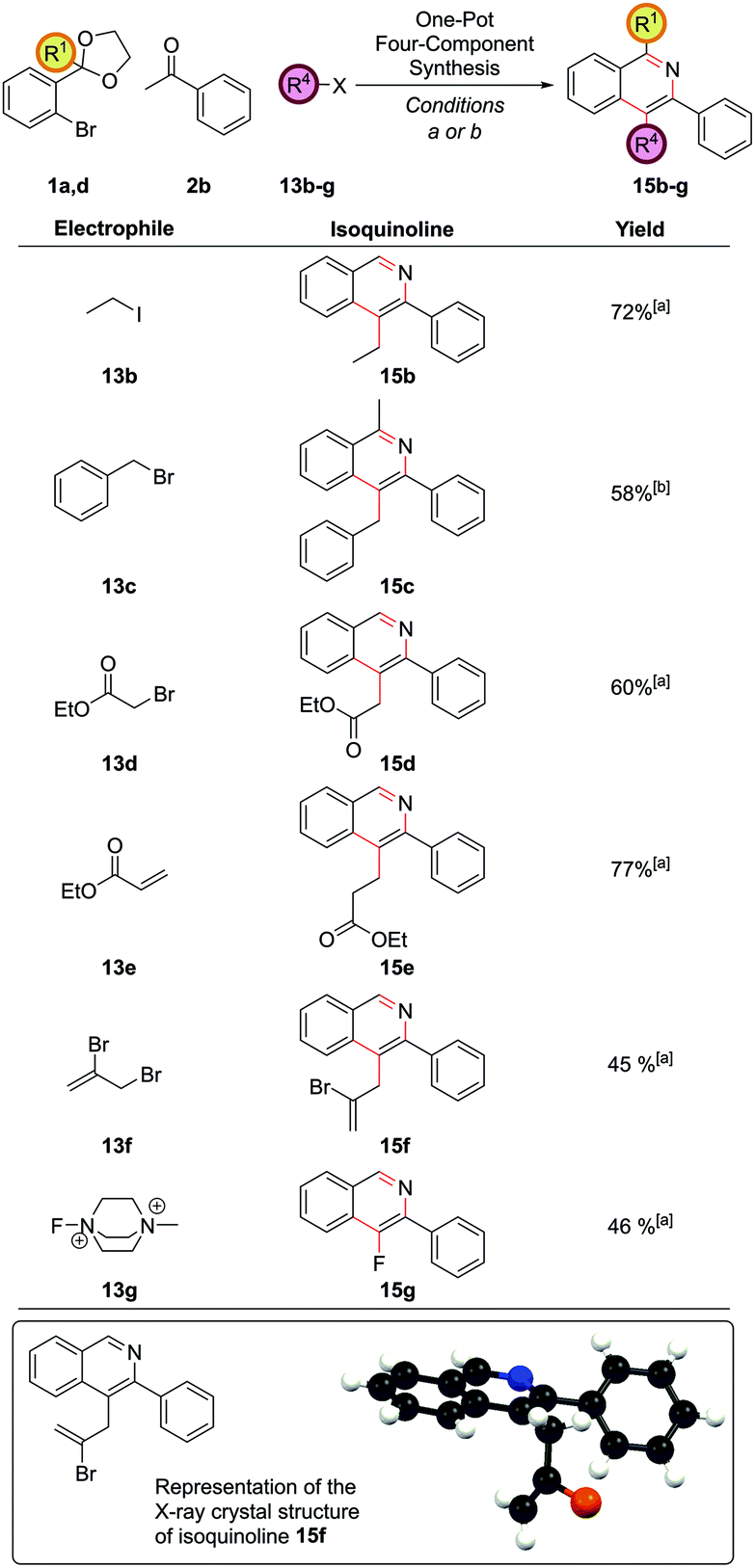 | ||
| Fig. 7 One-pot four-component coupling procedure. [a] (DtBPF)PdCl2 (2.5 mol%), NaOtBu (250 mol%), THF, reflux, 18 h, then electrophile, rt, 8 h, then HCl(aq), NH4Cl (1 M in 3:1 EtOH/H2O), 90 °C, 24 h; [b] (DtBPF)PdCl2 (5.0 mol%), NaOtBu (250 mol%), THF, reflux 18 h, then electrophile, rt, 8 h, then HCl(aq), NH4Cl (1 M in 3:1 EtOH/H2O), 90 °C, 24 h. | ||
Exploring the variation accessible with different aryl bromide and ketone partners illustrated that both electron-deficient 1l and electron-rich 1o aryl bromides could be employed as before to give 15h and 15i (Fig. 8). Trifunctionalized isoquinolines 15j and 15k could be accessed by the use of the sterically more hindered acetophenone-derived ortho-acetal 1d. Diphenyldisulfide, 13j, could also be employed as the electrophile, providing 4-thioether substituted isoquinoline 15l. Note that the direct arylation of a ketone bearing thioether functionality in the α-position to synthesize 4-thioether substituted isoquinolines could not be accomplished under our reaction conditions as the requisite ketone decomposed (vide supra ketone 2hFig. 1), highlighting a strength of this route. Isobutyl methyl ketone could also be employed to give a range of differently substituted dialkyl isoquinolines 15m, 15n and 4q (isoquinoline 4q also having been a target under our first generation approach vide supraFig. 2), which had required a more specialized ketone partner 2n, which had to be synthesized.
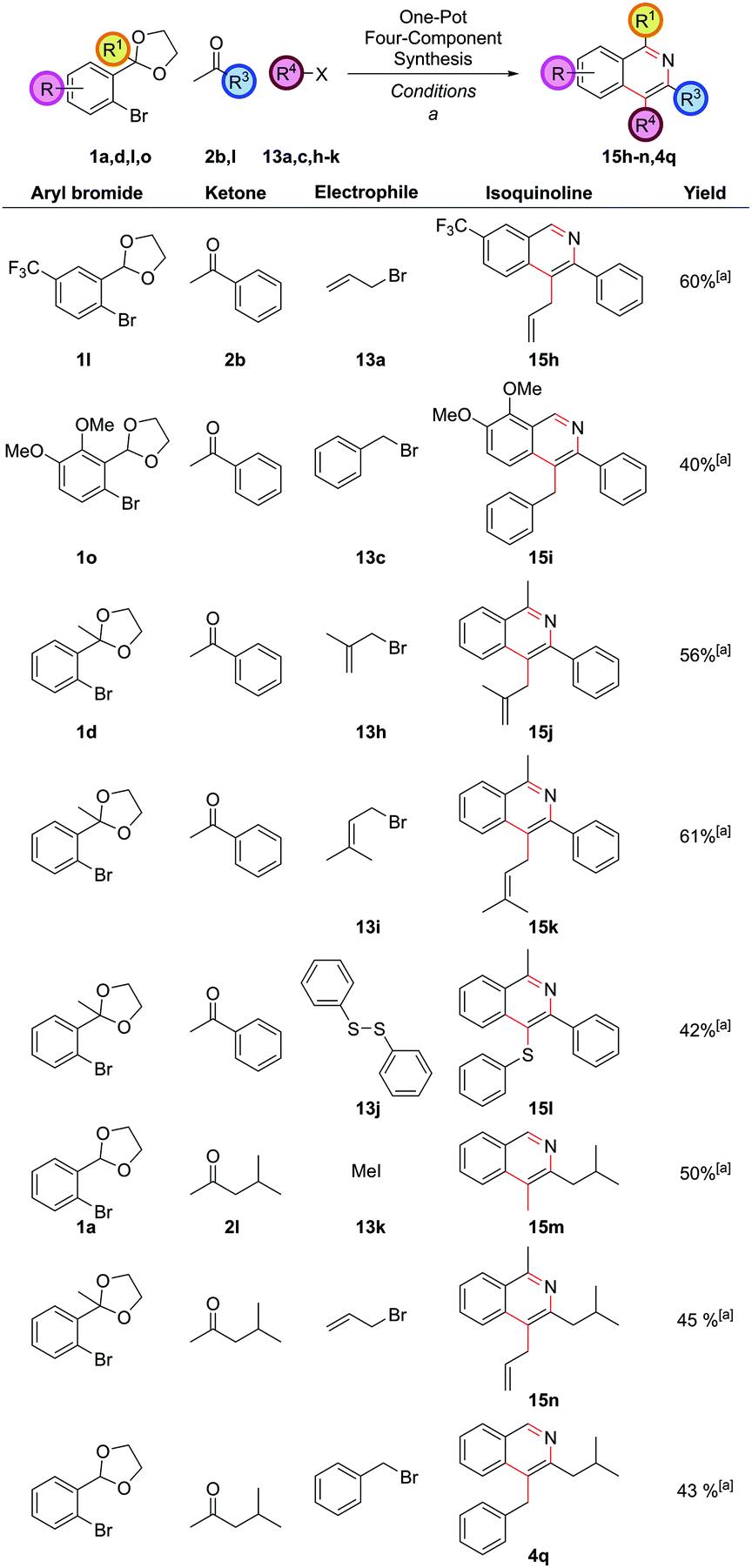 | ||
| Fig. 8 Further exemplification of the one-pot four-component coupling procedure. [a] (DtBPF)PdCl2 (2.5 mol%), NaOtBu (250 mol%), THF, reflux, 18 h, then electrophile, rt, 8 h, then HCl(aq), NH4Cl (1 M in 3:1 EtOH/H2O), 90 °C, 24 h. | ||
The incorporation of an aryl group at C4 was also challenging under the first generation route due to the limited commercial availability of benzyl ketones. It was envisaged that C4-aryl isoquinolines could be accessed by in situ functionalization as well, via the α,α-heterodiarylation of a methyl ketone, followed by acetal cleavage and cyclization. Although the palladium-catalyzed diarylation of methyl ketones had been previously reported, to the best of our knowledge this was limited to homodiarylation, achieved using an excess of one unhindered aryl halide36 or observed as an undesired over-reaction.12a,18e,37 Achieving an α,α-heterodiarylation is challenging. If two aryl bromides with similar reactivity were added simultaneously then a statistical mixture of homodiarylated and heterodiarylated products would be formed. If two aryl bromides with differing reactivity were added simultaneously then homodiarylation with the more reactive partner would occur first, followed by homodiarylation of the remaining ketone with the less reactive partner. There is little precedent for selective α,α-heterodiarylation in the literature, but it was thought this could be achieved by stepwise addition of the two aryl bromides, as long as the less reactive one was added first under conditions that did not induce diarylation before the more reactive one was added afterwards.
In our first generation isoquinoline synthesis,16 diarylation of the ketone enolates was not observed even when the aryl bromide was in excess, presumably due to the steric hindrance inhibiting the second coupling. Therefore, it was decided to attempt a second α-arylation in situ with a sufficiently unhindered aryl bromide, which would only be added after the first arylation was complete. Following the monoarylation of ketone 2b with aryl bromide 1a closely by TLC analysis indicated it was largely complete after 6 h, (Scheme 8) and so the second aryl bromide 16a was added at this point. This enabled the same palladium catalyst to promote both couplings; leaving the first reaction for longer times led to the need to add more palladium catalyst. The second arylation reaction was then run for 18 h to ensure complete conversion to intermediate 17a. No triarylated product was observed, presumably due to the now very high steric hindrance at this position. To the best of our knowledge, this transformation is the first reported one-pot palladium-catalyzed α,α-heterodiarylation reaction of a ketone, enabled by exploiting the steric dependence of the two aryl halides on the reaction. To fully generalise the procedure, increasing the temperature to 100 °C for the second α-arylation ensured it could always be driven to completion. This heterodiarylation could be incorporated seamlessly into the one-pot protocol to afford C4-aryl isoquinolines directly. In the case of isoquinoline 18a, the one-pot yield of 77% was comparable to the overall stepwise yield of 81%.
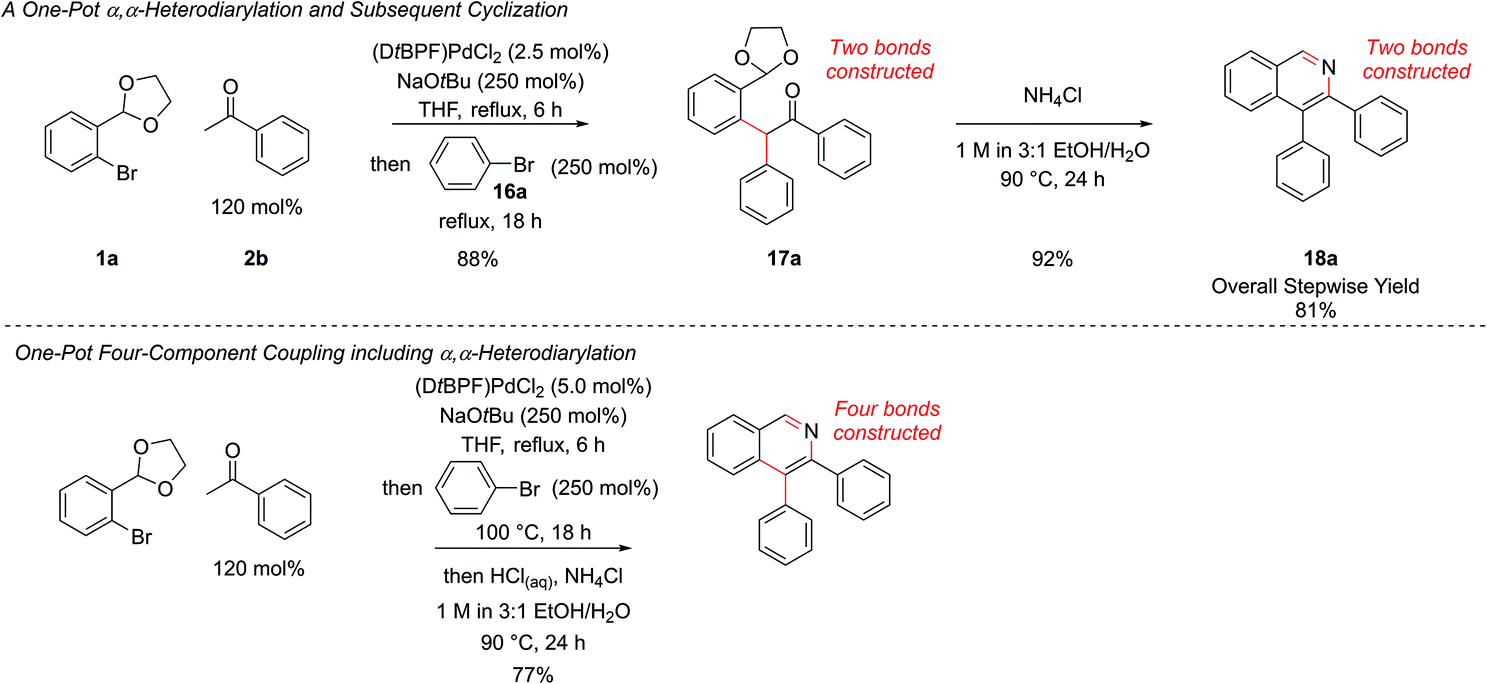 | ||
| Scheme 8 Development of one-pot diarylation. | ||
The reaction worked well with both electron-rich 16b and electron-deficient aryl bromides 16g (Fig. 9). Aryl bromides with significant steric hindrance such as ortho-methyl 16c and ortho-ethyl 16d groups could be employed, as could larger aromatic groups such as naphthalenes 16e and N-methyl indoles 16f. As before, a wide range of substitution was possible with the other reaction partners, including electron-rich 1o and electron-deficient 1l carbocyclic rings, C1-substitution 1d, and bulky alkyl substituents at C3 2k.
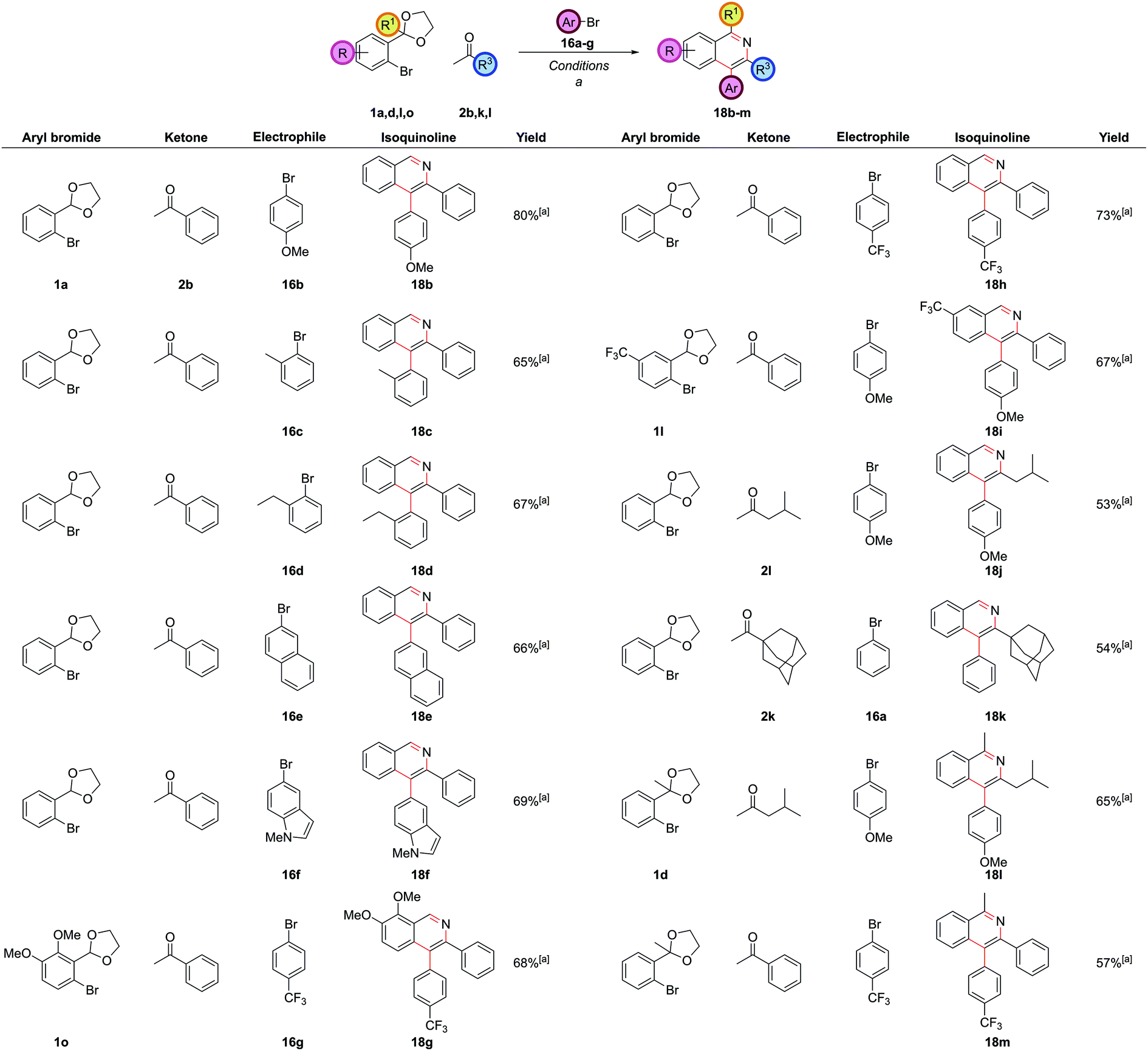 | ||
| Fig. 9 Scope of four-component coupling procedure with two aryl bromides. [a] (DtBPF)PdCl2 (5.0 mol%), NaOtBu (250 mol%), THF, reflux, 6 h, then aryl bromide, reflux, 18 h, then HCl(aq), NH4Cl (1 M in 3:1 EtOH/H2O), 90 °C, 24 h. | ||
Electrophile trapping and decarboxylation of tert-butyl cyanoacetate
Extending the four-component coupling procedure to nitriles would allow a greater variety of 3-amino isoquinolines to be synthesized, however this would require acetonitrile as one of the coupling partners, a difficult substrate to α-arylate. Our previous studies had shown that tert-butyl cyanoacetate could behave as a synthetic equivalent for acetonitrile due to the decarboxylation that could be encouraged after the α-arylation (vide supraFig. 6). Literature precedent also illustrated that ethyl cyanoacetate could be α-arylated and then alkylated in situ in a one-pot procedure,30 and so we investigated in situ trapping in this system. Addition of allyl bromide, 13a, and heating to 70 °C could be employed to trap the product of the α-arylation of tert-butyl cyanoacetate, 7e, to give intermediate 19a in 84% yield (Fig. 10). Attempted decarboxylation of 19a under mildly acidic conditions (NH4Cl) led to partial hydration of the nitrile and formation of a mixture of the 3-hydroxy and 3-aminoisoquinoline. The optimal procedure involved decarboxylation under neutral conditions, which occurred when intermediate 19a was heated in aqueous ethanol. Hence, the prime set of cyclization conditions involved first heating in a mixture of ethanol and water to effect decarboxylation, adding ammonium chloride to cleave the acetal (the prior removal of the ester stopped the hydration of the nitrile at this stage) and then basification with ammonium bicarbonate to cyclize to 3-aminoisoquinoline 20a. This occurred in a yield of 95%, and was also a better set of conditions for the cyclization of the unalkylated intermediate (86% vs. 74%, vide supraFig. 6). With such tert-butyl cyanoacetate couplings, the one-pot procedure gave lower yields, presumably due to the free amino group in the product reacting with an excess of one of the starting reagents. Hence, exemplification of this protocol was performed over two steps. Benzyl bromide, 13c, and iodomethane, 13k, could be employed as electrophiles to give isoquinolines 20b and 20c respectively, the presence of the amino group always characterized by the broad singlet (relative integral 2H) between 4.3 and 4.5 ppm in the 1H NMR spectra. With methyl bromoacetate, 13l, the 3-amino group underwent further cyclization onto the tethered ester furnishing lactam-fused isoquinoline 20d in 65% yield. | ||
| Fig. 10 α-Arylation and functionalization of tert-butyl cyanoacetate. [a] (DPPF)PdCl2 (2.0 mol%), NaOtBu (250 mol%), 1,4-dioxane, 70 °C, 4 h, then R4-X, 70 °C, 8 h; [b] EtOH/H2O (3:2), 90 °C, 18 h, then NH4Cl (1000 mol%), 90 °C, 3 h, then NH4HCO3 (2 M in H2O), 90 °C, 3 h. | ||
A C–H functionalization approach to isoquinolines
During the past two decades, chelation-assisted direct C–H functionalization has emerged as a powerful synthetic strategy due to its ability to bypass prefunctionalized starting materials. Imines,38 amines,39 oximes,40 amides41 and other nitrogen-centred directing groups42 have all been utilized for C–H functionalization en route to isoquinolines using transition metal-catalyzed annulation of alkynes or alkenes or allenes. However, these methods often suffer from regioselectivity issues when using unsymmetrical substrates and are limited by the commercial availability of the requisite starting materials. Our α-arylation methodology offered broad functional group tolerance and reliable regioselectivity but required the difunctionalized ortho-halobenzaldehydes or ortho-halo phenyl ketones as precursors. We therefore set out to adapt this methodology to incorporate a C–H functionalization reaction, thus allowing the inexpensive and commercially more abundant benzaldehydes to be integrated into this route. At the outset of this project, the direct arylation of nucleophilic carbonyl derivatives via an intermolecular C–H functionalization was a mode of reactivity with little precedent. Recently, some groups have made efforts towards this, however, a general procedure remains elusive.9b,43 Whilst studies towards this endeavour are still underway in our laboratories, an alternative strategy was envisaged whereby known C–H halogenation chemistry could pave the way for the economical benzaldehydes to enter our synthetic route to isoquinolines.O-Methyl oximes have recently been reported as proficient directing groups for palladium-catalyzed C–H functionalization and it was thought that these could not only serve to direct the C–H halogenation but also to protect the aldehyde/ketone in the subsequent α-arylation reaction from aldol reactions with the enolate. The O-methyl oxime of benzaldehyde, 22a could be synthesized from benzaldehyde, 21a, in quantitative yield by treatment of 21a with methoxylamine hydrochloride under basic conditions (Scheme 9). The desired ortho-bromide 23a was obtained by using the conditions reported by Sanford and coworkers.44 NBS was used as the limiting reagent to minimise the formation of polybrominated products. The α-arylation reaction could be performed in a 91% yield to furnish intermediate 24a with 5.0 mol% PdCl2(Amphos)2 catalyst. Pleasingly, the O-methyl oxime survived the α-arylation reaction conditions when the weaker base Cs2CO3 was employed but was destroyed if NaOtBu was used.
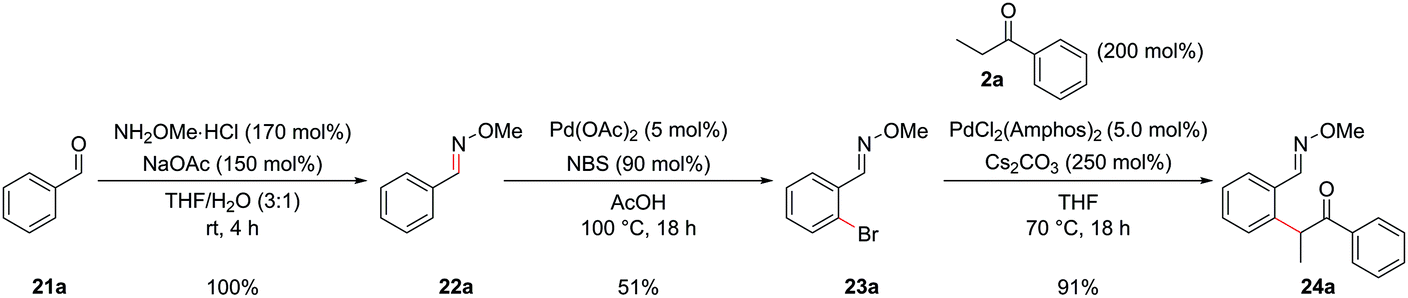 | ||
| Scheme 9 C–H functionalization allowing benzaldehyde derivatives to be incorporated into the synthetic route. | ||
Cyclization of oxime intermediate 24a under the conditions developed for the acetal-protected carbonyls gave a mixture of starting material, the desired isoquinoline 4a and the isoquinoline N-oxide 5a, the latter presumably formed by solvent-mediated demethylation of the oxygen from the O-methyl oxime after cyclization to the isoquinoline. The ratio between these was highly pH dependant. Full conversion to the isoquinoline could be achieved by cyclization with a mixture of ammonium chloride and ammonium hydroxide (at pH 10) at an elevated temperature of 110 °C in a yield of 88% (Scheme 10). It was also possible to exploit this pH dependence of the cyclization to selectively synthesize isoquinoline N-oxide 5a in quantitative yield by treating intermediate 24a with 1 M HCl. It is worth highlighting the atom-efficiency of this protocol since, in contrast to some C–H functionalization reactions, the directing group is incorporated into the desired product, acting as the N–O source, and is not cleaved in a subsequent step.
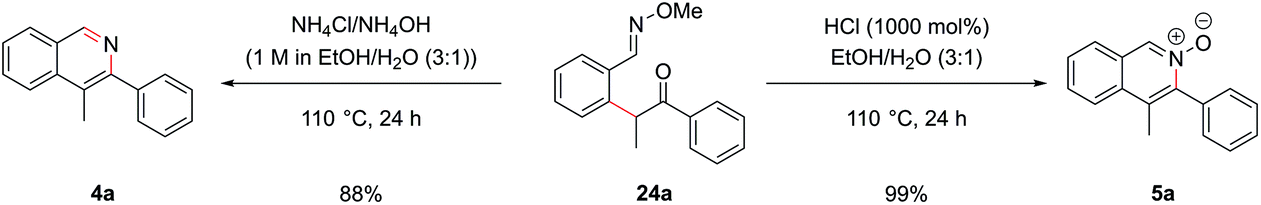 | ||
| Scheme 10 Cyclization of oxime 24a to both isoquinoline 4a and isoquinoline N-oxide 5a. | ||
The α-arylation and cyclization reactions could again be combined into a one-pot protocol, furnishing isoquinoline 4a from bromo oxime 24a in 83% yield, or N-oxide 5a in 64% yield. This modified protocol sufficiently enhanced the scope available at the C1 position of the resulting isoquinoline, which was previously limited by the low commercial availability of ortho-bromophenyl ketones and the difficulty of forming cyclic acetals on such moieties. With O-methyl oximes of ketones there is the possibility of E/Z isomerism, with the E isomer generally predominating unless the R group is sterically bulky. It was envisaged that E geometry, where the lone pair on nitrogen is oriented syn to the aromatic ring, is required to direct in the palladium for C–H functionalization, as has been previously noted for rhodium-catalyzed alkyne annulation.40f The geometric isomers of ethyl oxime 22c could be separated via column chromatography but equilibrated back to the thermodynamic ratio after standing in CDCl3 and hence it was postulated that the unreactive Z oximes could isomerize under the acidic bromination conditions and thus also be reacted. Methyl oxime 22b and ethyl oxime 22c were both converted through to the corresponding isoquinolines 4b and 25a respectively (Fig. 11), with only the α-arylation step occurring in lower yields than the benzaldehyde case. Significant unreacted starting material was recovered from these reactions and it was thought that deprotonation α to the oxime under the basic reaction conditions might be retarding the reaction. This notion was supported by the failure of the benzyl-substituted variant to undergo arylation. In the case of the ethyl variant, both E and Z isomers of intermediate 24d could be separately carried through the sequence successfully. Phenyl oxime 22d was a particularly efficient substrate and this was attributed to the conformational rigidity of the intermediates, placing the nitrogen lone pair close to the C–H bond to be functionalized, hence increasing the scope of substitution available at the C1 position (isoquinoline 25b).
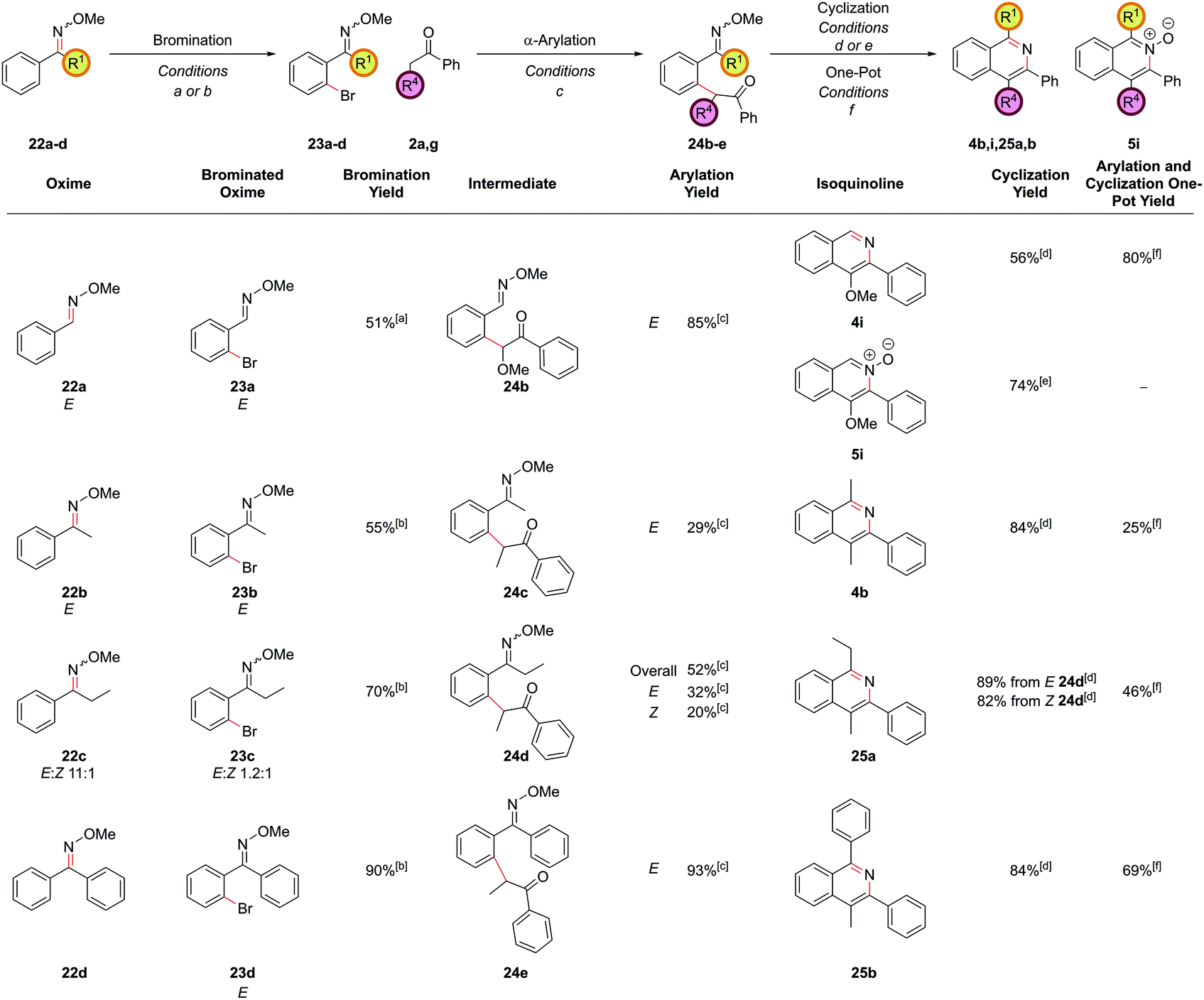 | ||
| Fig. 11 C–H bromination route to isoquinolines. [a] Pd(OAc)2 (5.0 mol%), NBS (90 mol%), AcOH, 100 °C, 18 h; [b] Pd(OAc)2 (5.0 mol%), NBS (110 mol%), AcOH, 100 °C, 18 h; [c] (PdCl2(Amphos)2 (5.0 mol%), Cs2CO3 (250 mol%), ketone (200 mol%), THF, 70 °C, 18 h; [d] NH4Cl/NH4OH (1 M in 3:1 EtOH/H2O), 110 °C, 24 h; [e] HCl (1000 mol%), EtOH/H2O (3:1), 110 °C, 24 h; [f] (PdCl2(Amphos)2 (5.0 mol%), Cs2CO3 (250 mol%), THF, 70 °C, 18 h, then NH4Cl/NH4OH (1 M in 3:1 EtOH/H2O), 110 °C, 24 h. | ||
Conclusions
The advent of innovative transition metal-catalyzed reactions enables aromatic heterocycles to be constructed in ever more adventurous ways – the palladium-catalyzed α-arylation of enolates in the synthesis of isoquinolines being a prime example of this retrosynthetic strategy. Throughout this project we sought to keep the ideals of modularity and practicality at the forefront. The three-component or four-component coupling procedures herein developed, involving multiple bond construction processes, provide access to highly complex isoquinoline frameworks in one-pot from largely commerically available starting materials, with only one work up and purification procedure required. Versatility of a synthetic route is also highly prized and demonstrated here by the utilization of ketone, ester or nitrile enolates, electron-rich, electron-deficient or sterically-hindered aryl halides, the direct access to 3-aminoisoquinolines, 3-hydroxyisoquinolines, isoquinoline N-oxides and isoquinolinium salts and the in situ functionalization of intermediates. The robustness of the process has been successfully proven in the arena of natural product synthesis,45 and also in the creation of a library of novel compounds charting new areas of chemical space giving chemists much greater flexibility when constructing their desired isoquinoline skeleton.Experimental
General experimental details
1H NMR and 13C NMR spectra were recorded on a 300 MHz or 400 MHz spectrometer in CDCl3, CD2Cl2, CD3OD or acetone-d6 and referenced to residual solvent peaks. Chemical shifts are quoted in ppm (parts per million) to the nearest 0.01 ppm with signal splitting recorded as singlet (s), doublet (d), triplet (t), quartet (q), septet (sept.), multiplet (m) and broad singlet (br. s). Coupling constants, J, are measured in Hz to the nearest 0.1 Hz. 1H and 13C NMR spectra were recorded at room temperature. Infrared spectra were recorded as thin films of neat samples on a Bruker Tensor 27 FT-IR spectrometer equipped with Attenuated Total Reflectance sampling accessories. High resolution mass spectra are given to four decimal places and were recorded on a Bruker MicroTof (resolution = 10000 FWHM) under conditions of electrospray ionization (ESI). Melting points (m.p.) were obtained from recrystallized samples using a Lecia VMTG heated-stage microscope and are uncorrected. The solvent systems used for recrystallization are quoted in parentheses. Flash column chromatography was performed using silica gel (60 Å, 0.033–0.070 mm, BDH) or using basic alumina (pH 9.5, 58 Å, 150 mesh, Sigma-Aldrich). TLC analyses were performed on Merck Kiesegel 60 F254 0.25 mm precoated silica plates or Macherey-Nagel Alugram Alox N/UV254 0.20 mm precoated alumina plates. Reagents obtained from Sigma-Aldrich, Alfa, Fluorochem and TCI suppliers were used directly as supplied other than alkyl bromides which were first purified by being passed through a short plug of K2CO3. All palladium catalysts were obtained from Johnson Matthey. The palladium catalysts and bases were stored in a desiccator. Compounds that contained acetals of electron-rich benzaldehydes were found to undergo slow hydrolysis (over a period of weeks) due to atmospheric moisture and hence were also stored in a desiccator. This increased their lifetime to many months. All anhydrous reactions were carried out in flame dried glassware and under an inert atmosphere of argon provided by a balloon. All reactions were stirred with magnetic followers. Petrol refers to petroleum ether in the boiling range 40–60 °C. THF, toluene and CH2Cl2 were dried by purification through two activated alumina purification columns. Brine refers to a saturated aqueous solution of NaCl. Crystal structure determination was carried out using X-ray diffraction data measured on an Enraf-Nonius Kappa CCD diffractometer. Intensity data were processed using the DENZO SMN package and further processing was carried out using the Crystals, Cameron and Mercury software. Experimental procedures and characterization data for new compounds or compounds made by a new method are listed below; for the experimental procedures and characterization data of the remaining compounds, see ref. 10, 15 and 16.
General procedure A
A resealable reaction tube, containing a magnetic follower, was sealed with a rubber septum and flame dried under a flow of argon. (DtBPF)PdCl2 (2.0 mol%) and NaOtBu (250 mol%) were added to the tube. The aryl bromide (100 mol%) was dissolved in dry THF (5 mL mmol−1) and the resulting solution was added via syringe to the tube. The ketone (120 mol%) was then added to the tube. The rubber septum was replaced with a screw cap and the tube was heated at 70 °C for 18 h. The reaction was then cooled to room temperature and quenched by the addition of H2O (25 mL). The aqueous layer was extracted with Et2O (3 × 25 mL) and the combined organics were dried over Na2SO4, filtered, and the solvent removed in vacuo.General procedure B
A solution of NH4Cl (1000 mol%, 1.0 M in 3:1 EtOH/H2O) was added to the cyclization substrate (100 mol%) in a resealable reaction tube containing a magnetic follower. The tube was sealed with a screw cap and heated at 90 °C for 24 h. The reaction was then cooled to room temperature and quenched by the addition of saturated aqueous NaHCO3 (25 mL). The aqueous layer was extracted with Et2O (3 × 25 mL) and the combined organics were dried over Na2SO4, filtered, and the solvent removed in vacuo.
General procedure C
A solution of NH4Cl (1000 mol%, 1.0 M in 3:1 EtOH/H2O) was added to the cyclization substrate (100 mol%) in a resealable reaction tube containing a magnetic follower. The tube was sealed with a screw cap and heated at 90 °C for 18 h. A solution of NH4HCO3 (2.0 M in H2O) was then added until the pH of the reaction mixture had been adjusted to approximately pH 9. The tube was resealed and heated for a further 24 h at 90 °C. The reaction was then cooled to room temperature and quenched by the addition of H2O (25 mL). The aqueous layer was extracted with Et2O (3 × 25 mL) and the combined organics were dried over Na2SO4, filtered, and the solvent removed in vacuo.
General procedure D
A solution of hydroxylamine hydrochloride (1000 mol%, 1.0 M in 9:1 EtOH/H2O) was added to the cyclization substrate (100 mol%) in a resealable reaction tube containing a magnetic follower. The tube was sealed with a screw cap and heated at 90 °C for 2 h. The reaction was then cooled to room temperature and the solvent removed in vacuo using a toluene azeotrope.
General procedure E
A resealable reaction tube, containing a magnetic follower, was sealed with a rubber septum and flame dried under a flow of argon. PdCl2(Amphos)2 (5.0 mol%) and Cs2CO3 (250 mol%) were added to the tube. The aryl bromide (100 mol%) was dissolved in dry THF (5 mL mmol−1) and the resulting solution was added via syringe to the tube. The nitrile (200 mol%) was then added to the tube. The rubber septum was replaced with a screw cap and the tube was heated at 70 °C for 18 h. The reaction was then cooled to room temperature and quenched by the addition of H2O (25 mL). The aqueous layer was extracted with EtOAc (3 × 25 mL) and the combined organics were dried over Na2SO4, filtered, and the solvent removed in vacuo.General procedure F
A microwave vial, containing a magnetic follower, was sealed with a rubber septum and flame dried under vacuum. (DPPF)PdCl2 (5.0 mol%), NaOtBu (250 mol%) and the nitrile (100 mol%) were added to the vial. The septum was replaced with a microwave cap and the vial was evacuated and backfilled with argon before a solution of the aryl bromide (120 mol%) in anhydrous 1,4-dioxane (4 mL mmol−1) was added via syringe to the vial and the reaction was heated at 70 °C for a specified time. The reaction was then cooled to room temperature and quenched by the addition of saturated aqueous NH4Cl (25 mL). The aqueous layer was extracted with EtOAc (3 × 25 mL) and the combined organics were dried over Na2SO4, filtered, and the solvent removed in vacuo.General procedure G
Methoxylamine hydrochloride (160 mol%) was added to a solution of the ketone (100 mol%) and pyridine (1.2 mL mmol−1) in EtOH (10 mL mmol−1) and the reaction was stirred at a specified temperature for a specified time. The solvent was removed in vacuo and then the remaining residue redissolved in EtOAc (10 mL mmol−1), washed with H2O (50 mL) and brine (50 mL), dried over Na2SO4, filtered, and the solvent removed in vacuo.General procedure H44
Palladium(II) acetate (5.0 mol%) was added to a solution of the O-alkyl oxime (100 mol%) and N-bromosuccinimide (110 mol%) in acetic acid (8.0 mL mmol−1) in a resealable reaction tube containing a magnetic follower. The reaction was heated at 100 °C for a specified time then cooled to room temperature and the solvent removed in vacuo.General procedure I
A resealable reaction tube, containing a magnetic follower, was sealed with a rubber septum and flame dried under a flow of argon. PdCl2(Amphos)2 (5.0 mol%) and Cs2CO3 (250 mol%) were added to the tube. A solution of the aryl bromide (100 mol%) in anhydrous THF (5.0 mL mmol−1) and the ketone (200 mol%) were then added via syringe. The septum was replaced with a screw cap and the reaction was heated at 70 °C for 18 h. The reaction was cooled to room temperature, quenched with H2O (25 mL), the aqueous layer extracted with Et2O (3 × 25 mL) and the combined organics were dried over Na2SO4, filtered, and the solvent removed in vacuo.General procedure J
A solution of NH4Cl (1000 mol%, 1.0 M in 3:1 EtOH/H2O) which had been basified to ∼pH 9 by the dropwise addition of 25% aqueous NH4OH was added to the cyclization substrate (100 mol%) in a resealable reaction tube containing a magnetic follower. The tube was sealed with a screw cap and heated at 110 °C for 24 h. The reaction was then cooled to room temperature and quenched by the addition of saturated aqueous NH4Cl (25 mL). The aqueous layer was extracted with Et2O (3 × 25 mL) and the combined organics were dried over Na2SO4, filtered, and the solvent removed in vacuo.
General procedure K
A resealable reaction tube, containing a magnetic follower, was sealed with a rubber septum and flame dried under a flow of argon. PdCl2(Amphos)2 (5.0 mol%) and Cs2CO3 (250 mol%) were added to the tube. A solution of the aryl bromide (100 mol%) in anhydrous THF (5.0 mL mmol−1) and the ketone (200 mol%) were then added via syringe. The septum was replaced with a screw cap and the reaction was heated at 70 °C for 18 h. The reaction was cooled to room temperature, then a solution of NH4Cl (1000 mol%, 1.0 M in 3:1 EtOH/H2O) which had been basified to ∼pH 9 by the dropwise addition of 25% aqueous NH4OH was added, the tube resealed with a screw cap and then heated at 110 °C for 24 h. The reaction was cooled to room temperature, quenched with saturated aqueous NH4Cl (25 mL), the aqueous layer extracted with Et2O (3 × 25 mL) and the combined organics were dried over Na2SO4, filtered, and the solvent removed in vacuo.
General procedure L
A solution of HCl (1000 mol%, 1.0 M in 9:1 EtOH/H2O) was added to the cyclization substrate (100 mol%) in a resealable reaction tube containing a magnetic follower. The tube was sealed with a screw cap and heated at 110 °C for 24 h. The reaction was then cooled to room temperature and the solvent removed in vacuo using a toluene azeotrope.
:1 grading to 9:1] furnished ketone 3d (53.8 mg, 0.180 mmol, 80%) as rods. M.p. 131–133 °C (petrol/CH2Cl2); 1H NMR (400 MHz, CDCl3) δH: 8.04 (2H, d, J 8.9), 7.59 (1H, dd, J 7.1, 1.8), 7.34–7.28 (2H, m), 7.18 (1H, dd, J 6.6, 2.0), 6.95 (2H, d, J 8.9), 5.90 (1H, s), 4.45 (2H, s), 4.06–3.94 (4H, m), 3.87 (3H, s); 13C[1H] NMR (100 MHz, CDCl3) δC: 196.2, 163.4, 135.5, 134.0, 131.3, 130.7, 130.0, 129.2, 126.9, 126.9, 113.8, 102.7, 65.0, 55.5, 42.3; IRνmax (thin film)/cm−1 2894, 2839, 1679, 1600, 1575, 1510, 1420, 1329, 1258, 1220, 1170, 1112, 1074, 1027, 992, 963, 833; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C18H18NaO4 321.1097; Found 321.1097.
:1 grading to 19:1] furnished ketone 3e (191 mg, 0.472 mmol, 70%) as prisms. M.p. 88–90 °C (petrol/CH2Cl2); 1H NMR (400 MHz, CDCl3) δH: 8.51 (2H, s), 8.06 (1H, s), 7.61–7.59 (1H, m), 7.38–7.31 (2H, m), 7.21–7.18 (1H, m), 5.91 (1H, s), 4.52 (2H, s), 4.04–4.01 (2H, m), 3.99–3.96 (2H, m); 13C NMR[1H] (100 MHz, CDCl3) δC: 195.1, 138.4, 135.4, 132.3 (q, 2J 33.9), 132.1, 131.1, 129.4, 128.6 (q, 3J 3.2), 127.5, 127.5, 126.1 (sept., 3J 3.6), 122.9 (q, 1J 272.9), 102.9, 64.9, 42.9; 19F[1H] NMR (377 MHz, CDCl3) δF: −62.9; IRνmax (thin film)/cm−1 2895, 1699, 1617, 1381, 1322, 1277, 1249, 1173, 1129, 1077, 1046, 968, 943, 914, 842; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C19H14F6NaO3 427.0739; Found 427.0732.
:1 grading to 24:1] furnished ketone 3g (189 mg, 0.670 mmol, 93%) as plates. M.p. 55–57 °C (petrol/CH2Cl2); 1H NMR (400 MHz, CDCl3) δH: 8.08 (2H, dd, J 6.9, 1.5), 7.65–7.63 (1H, m), 7.61–7.58 (1H, m), 7.51 (2H, t, J 7.5), 7.35–7.28 (2H, m), 7.18–7.16 (1H, m), 4.55 (2H, s), 3.89–3.85 (2H, m), 3.66–3.63 (2H, m), 1.69 (3H, s); 13C NMR[1H] (100 MHz, CDCl3) δC: 197.8, 141.2, 137.4, 133.0, 132.8, 132.3, 128.6, 128.1, 128.1, 127.1, 126.4, 109.0, 64.2, 43.8, 27.7; IRνmax (thin film)/cm−1 2988, 2892, 1690, 1484, 1447, 1374, 1331, 1238, 1214, 1193, 1124, 1066, 1034, 995, 951, 868; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C18H18NaO3 305.1148; Found 305.1140.
:1 grading to 5:1] furnished ketone 3m (59.3 mg, 0.241 mmol, 75%) as a pale yellow oil. 1H NMR (400 MHz, CDCl3) δH: 7.48 (1H, d, J 7.6), 7.29 (1H, m), 7.19 (1H, m), 7.14 (1H, d, J 7.8), 5.79 (1H, s), 4.06 (1H, m), 4.00–3.95 (2H, m), 3.93–3.88 (2H, m), 2.49–2.39 (2H, m), 2.26–2.20 (1H, m), 2.14–2.08 (1H, m), 1.77–1.64 (2H, m), 1.60–1.47 (1H, m), 0.86 (9H, s); 13C NMR[1H] (100 MHz, CDCl3) δC: 210.5, 138.0, 135.3, 129.0, 128.8, 126.7, 126.3, 102.4, 65.1, 65.0, 52.2, 47.7, 41.8, 36.6, 32.6, 28.6, 27.7; IRνmax (thin film)/cm−1 2954, 2869, 2360, 1715, 1477, 1366, 1227; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C19H26NaO3 325.1762; Found 325.1774.
:1 grading to 14:1] furnished ketone 3p (301 mg, 0.965 mmol, 98%) as an oil. 1H NMR (400 MHz, acetone-d6) δH: 7.57 (1H, d, J 7.3), 7.33 (1H, t, J 7.4), 7.28 (1H, t, J 7.3), 7.18 (2H, d, J 8.6), 7.15 (1H, d, J 7.8), 6.90 (2H, d, J 8.6), 5.98 (1H, s), 5.79 (1H, s), 4.19–4.03 (4H, m), 3.78 (3H, s), 2.18 (3H, s); 13C[1H] NMR (100 MHz, acetone-d6) δC: 206.0, 159.6, 139.0, 136.5, 131.5, 131.2, 130.6, 129.6, 127.8, 127.4, 114.7, 103.3, 65.8, 65.6, 59.3, 55.5, 30.0; IRνmax (thin film)/cm−1 2956, 2891, 2837, 1714, 1609, 1582, 1510, 1456, 1408, 1353, 1303, 1250, 1181, 1155, 1109, 1071, 1031, 970, 945; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C19H20NaO4 335.1254; Found 335.1238.
:1] furnished alcohol S1 (1.35 g, 7.02 mmol, 47%) as an oil. 1H NMR (400 MHz, CDCl3) δH: 7.32–7.26 (2H, m), 7.25–7.16 (3H, m), 3.71 (1H, tt, J 8.6, 4.4), 2.82 (1H, ddd, J 13.7, 9.9, 5.9), 2.68 (1H, ddd, J 13.7, 9.9, 6.5), 2.12 (1H, s), 1.86–1.67 (3H, m), 1.44 (1H, ddd, J 14.1, 8.8, 5.4), 1.29 (1H, ddd, J 13.7, 8.8, 4.4), 0.93 (6H, t, J 6.1); 13C NMR[1H] (100 MHz, CDCl3) δC: 142.3, 128.4 (2 signals), 125.7, 69.3, 46.8, 39.7, 32.1, 24.6, 23.9, 22.2. Data were consistent with those previously reported.46
:1] furnished ketone 2n (680 mg, 3.57 mmol, 95%) as an oil. 1H NMR (400 MHz, CDCl3) δH: 7.31–7.25 (2H, m), 7.22–7.16 (3H, m), 2.89 (2H, t, J 8.0), 2.71 (2H, t, J 8.0), 2.27 (2H, d, J 6.9), 2.20–2.07 (1H, m), 0.89 (6H, d, J 6.6); 13C NMR[1H] (100 MHz, CDCl3) δC: 210.1, 141.3, 128.6, 128.5, 126.2, 52.2, 44.9, 29.8, 24.7, 22.7; IRνmax (thin film)/cm−1 3028, 2871, 2360, 2341, 1711; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C13H19O 191.14325; Found 191.14304.
:1] furnished ketone 3q (84.2 mg, 0.249 mmol, 57%) as an oil. 1H NMR (400 MHz, CDCl3) δH: 7.50–7.44 (1H, m), 7.30–7.00 (8H, m), 5.62 (1H, s), 4.38 (1H, dd, J 8.4, 5.8), 4.06–3.91 (4H, m), 3.33 (1H, dd, J 13.5, 8.4), 2.75 (1H, dd, J 13.5, 5.8), 2.15–2.00 (2H, m), 1.99–1.87 (1H, m), 0.68 (3H, d, J 6.1), 0.57 (3H, d, J 6.1); 13C NMR[1H] (100 MHz, CDCl3) δC: 209.7, 140.3, 137.6, 135.4, 129.5, 129.1, 128.2, 128.0, 127.1, 126.9, 126.1, 102.4, 65.1, 65.0, 55.6, 51.2, 39.1, 24.4, 22.6, 22.1; IRνmax (thin film)/cm−1 3028, 2872, 2360, 2341, 1709; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C22H27O3 339.19562; Found 339.19547.
:1 grading to 4:1] furnished ketone 3s (79.9 mg, 0.243 mmol, 82%) as prisms. M.p. 97–99 °C (petrol/CH2Cl2); 1H NMR (400 MHz, CDCl3) δH: 8.05 (2H, d, J 7.4), 7.58 (1H, t, J 7.3), 7.48 (2H, t, J 7.6), 7.14 (1H, s), 6.68 (1H, s), 5.82 (1H, s), 4.43 (2H, s), 4.06–3.93 (4H, m), 3.90 (3H, s), 3.84 (3H, s); 13C NMR[1H] (100 MHz, CDCl3) δC: 197.8, 149.2, 147.7, 136.9, 133.1, 128.6, 128.4, 127.7, 125.9, 114.3, 110.1, 102.4, 65.0, 55.9, 55.9, 42.1; IRνmax (thin film)/cm−1 2944, 1684, 1596, 1573, 1525, 1466, 1447, 1349, 1331, 1273, 1242, 1215, 1199, 1115, 1002, 887, 861; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C19H20NaO5 predicted 351.1203, Found 351.1198.
:1 grading to 14:1] furnished ketone 3y (95.0 mg, 0.332 mmol, 81%) as prisms. M.p. 63–65 °C (petrol/CH2Cl2); 1H NMR (400 MHz, CDCl3) δH: 8.05 (2H, dd, J 8.0, 1.3), 7.60 (1H, t, J 7.5), 7.50 (2H, t, J 7.6), 7.34 (1H, dd, J 9.7, 2.7), 7.15 (1H, dd, J 7.9, 5.6), 7.03 (1H, td, J 8.3, 2.8), 5.86 (1H, s), 4.46 (2H, s), 4.03–3.98 (2H, m), 3.97–3.93 (2H, m); 13C[1H] NMR (100 MHz, CDCl3) δC: 197.3, 161.8 (d, 1J 245.2), 138.4 (d, 3J 7.2), 136.8, 133.2, 133.0 (d, 3J 8.0), 129.0 (d, 4J 3.2), 128.7, 128.3, 115.7 (d, 2J 20.7), 113.8 (d, 2J 23.1), 101.6 (d, 4J 1.6), 65.0, 41.8; 19F[1H] NMR (377 MHz, CDCl3) δF: −115.2; IRνmax (thin film)/cm−1 3064, 2929, 1689, 1639, 1597, 1499, 1448, 1382, 1333, 1257, 1215, 1159, 1113, 1066, 1036, 1014, 962, 907, 873, 834, 812; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C17H15FNaO3 309.0897; Found 309.0897.
:3] furnished isoquinoline 4a (46.3 mg, 0.211 mmol, 88%) as a solid. Method B: Oxime 23a (67.0 mg, 0.313 mmol) was subjected to General procedure K with propiophenone, 2a. Purification by flash column chromatography [petrol/EtOAc 17:3] furnished isoquinoline 4a (56.7 mg, 0.258 mmol, 83%) as a solid. M.p. 97–99 °C; 1H NMR (400 MHz, CDCl3) δH: 9.22 (1H, s), 8.07 (1H, d, J 8.3), 8.01 (1H, d, J 8.1), 7.77 (1H, ddd, J 8.4, 7.0, 1.3), 7.66–7.58 (3H, m), 7.54–7.46 (2H, m), 7.46–7.38 (1H, m), 2.67 (3H, s); 13C[1H] NMR (100 MHz, CDCl3) δC: 151.8, 150.2, 141.3, 136.2, 130.4, 129.9, 128.1, 128.1, 127.6, 127.3, 126.6, 124.0, 123.6, 15.5. Data were consistent with those previously reported.10
:3] furnished isoquinoline 4b (39.8 mg, 0.171 mmol, 84%) as a solid. Method B: Aryl bromide 23b (240 mg, 1.05 mmol) was subjected to General procedure K with propiophenone, 2a. Purification by flash column chromatography [CH2Cl2] furnished isoquinoline 4b (61.2 mg, 0.263 mmol, 25%) as a solid. M.p. 96–98 °C; 1H NMR (400 MHz, CDCl3) δH: 8.18 (1H, d, J 8.3), 8.07 (1H, d, J 8.3), 7.76 (1H, t, J 7.7), 7.67–7.55 (3H, m), 7.49 (2H, t, J 7.5), 7.44–7.36 (1H, m), 3.01 (3H, s), 2.62 (3H, s); 13C[1H] NMR (100 MHz, CDCl3) δC: 155.9, 150.6, 141.5, 136.3, 129.9, 129.9, 128.1, 127.5, 126.3, 126.2, 126.1, 124.2, 122.3, 22.5, 15.4. Data were consistent with those previously reported.10
:1 grading to 9:1] furnished isoquinoline 4d (79.8 mg, 0.339 mmol, 84%) as prisms. M.p. 99–101 °C (petrol/CH2Cl2); 1H NMR (400 MHz, CDCl3) δH: 9.31 (1H, s), 8.10 (2H, dt, J 8.9, 2.4), 7.99 (1H, s), 7.96 (1H, d, J 8.3), 7.83 (1H, d, J 8.1), 7.67 (1H, td, J 7.6, 1.1), 7.55 (1H, td, J 7.5, 0.9), 7.05 (2H, dt, J 8.9, 2.5), 3.88 (3H, s); 13C[1H] NMR (100 MHz, CDCl3) δC: 160.1, 152.3, 151.0, 136.8, 132.2, 130.4, 128.2, 127.5, 127.4, 126.7, 126.7, 115.4, 114.2, 55.4. Data were consistent with those previously reported.47
:1 grading to 19:1] furnished isoquinoline 4e (26.9 mg, 0.0788 mmol, 81%) as needles. M.p. 121–122 °C (petrol/CH2Cl2); 1H NMR (400 MHz, CDCl3) δH: 9.36 (1H, s), 8.62 (2H, s), 8.16 (1H, s), 8.04 (1H, d, J 8.1), 7.95–7.92 (2H, m), 7.77 (1H, ddd, J 8.2, 6.9, 1.3), 7.68 (1H, ddd, J 8.1, 7.0, 1.1); 13C NMR[1H] (100 MHz, CDCl3) δC: 152.9, 147.8, 141.6, 136.4, 132.1 (q, 2J 33.1), 131.1, 128.3, 128.2, 127.7, 127.1, 126.9 (q, 3J 3.1), 123.5 (q, 1J 272.6), 121.9 (sept., 3J 3.8), 117.3; 19F[1H] NMR (377 MHz, CDCl3) δF: −62.8; IRνmax (thin film)/cm−1 2925, 1628, 1495, 1391, 1383, 1336, 1288, 1212, 1167, 1125, 1058, 895, 882, 845; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C17H9F6NNa 364.0531; Found 364.0523.
:1 grading to 19:1] furnished isoquinoline 4g (42.3 mg, 0.193 mmol, 89%) as an oil. 1H NMR (400 MHz, CDCl3) δH: 8.17–8.13 (3H, m), 7.94 (1H, s), 7.87 (1H, d, J 8.3), 7.68 (1H, ddd, J 8.1, 7.1, 1.2), 7.58 (1H, ddd, J 7.9, 6.9, 1.3), 7.52 (2H, t, J 7.6), 7.41 (1H, tt, J 7.4, 1.2), 3.06 (3H, s); 13C NMR[1H] (100 MHz, CDCl3) δC: 158.6, 150.0, 139.9, 136.8, 130.0, 128.7, 128.3, 127.6, 127.0, 126.8, 126.6, 125.7, 115.2, 22.7. Data were consistent with those previously reported.48
:1] furnished isoquinoline 4i (25.3 mg, 0.108 mmol, 56%) as an oil. Method B: Oxime 23a (102 mg, 0.477 mmol) was subjected to General procedure K with 2-methoxyacetophenone, 2g. Purifcation by flash column chromatography [petrol/EtOAc 97:3] furnished isoquinoline 4i (89.6 mg, 0.381 mmol, 80%) as an oil. 1H NMR (400 MHz, CDCl3) δH: 9.16 (1H, s), 8.23 (1H, d, J 8.3), 8.10 (2H, d, J 7.6), 8.02 (1H, d, J 8.3), 7.76 (1H, t, J 7.6), 7.63 (1H, t, J 7.8), 7.52 (2H, t, J 7.7), 7.42 (1H, t, J 7.3), 3.70 (3H, s); 13C[1H] NMR (100 MHz, CDCl3) δC: 149.0, 147.8, 143.1, 138.0, 132.0, 130.4, 129.3, 129.2, 128.4, 128.1, 127.4, 127.3, 121.6, 61.2. Data were consistent with those previously reported.10
:1 grading to 3:1] furnished isoquinoline 4m (38.4 mg, 0.160 mmol, 92%) as yellow prisms. M.p. 300 °C (decomp.) (petrol/CH2Cl2); 1H NMR (400 MHz, CDCl3) δH: 9.03 (1H, s), 7.92–7.90 (1H, m), 7.90–7.87 (1H, m), 7.67 (1H, dd, J 7.1, 1.5), 7.50 (1H, t, J 7.3), 3.23–3.13 (2H, m), 3.11–3.01 (1H, m), 2.75–2.65 (1H, m), 2.18–2.11 (1H, m), 1.62–1.53 (2H, m), 1.04 (9H, s); 13C[1H] NMR (100 MHz, CDCl3) δC: 150.0, 135.6, 130.2, 128.2, 126.8, 125.8, 125.8, 124.6, 121.9, 44.2, 33.8, 32.6, 27.4, 26.5, 24.3; IRνmax (thin film)/cm−1 2959, 2361, 2342, 1623, 1584, 1454, 1366, 1226; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C17H22N 240.1747, Found 240.1754.
:1 grading to 14:1] furnished isoquinoline 4p (30.4 mg, 0.122 mmol, 92%) as prisms. M.p. 96–98 °C (petrol/CH2Cl2); 1H NMR (400 MHz, CDCl3) δH: 9.21 (1H, s), 7.97 (1H, d, J 7.3), 7.58–7.50 (2H, m), 7.46 (1H, d, J 8.6), 7.22 (2H, d, J 8.6), 7.06 (2H, d, J 8.5), 3.91 (3H, s), 2.51 (3H, s); 13C NMR[1H] (100 MHz, CDCl3) δC: 159.0, 150.9, 149.2, 136.2, 131.1, 130.6, 129.7, 130.3, 126.0, 127.5, 126.8, 125.0, 114.0, 55.3, 23.0; IRνmax (thin film)/cm−1 3000, 2956, 2929, 2836, 1779, 1723, 1611, 1573, 1513, 1456, 1441, 1379, 1286, 1242, 1175, 1106, 1029, 964, 922, 830; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C17H16NO 250.1226; Found 250.1220.
:1 pentane/EtOAc] furnished isoquinoline 4q (31.1 mg, 0.113 mmol, 95%) as a solid. M.p. 63–64 °C (petrol/CH2Cl2); 1H NMR (400 MHz, CDCl3) δH: 9.10 (1H, s), 7.85 (1H, d, J 7.9), 7.76 (1H, d, J 8.5), 7.49 (1H, t, J 8.5), 7.40 (1H, t, J 7.9), 7.21–7.04 (3H, m), 6.97 (2H, d, J 7.3), 4.39 (2H, s), 2.80 (2H, d, J 7.2), 2.24–2.10 (1H, m), 0.86 (6H, d, J 6.6); 13C NMR[1H] (100 MHz, CDCl3) δC: 153.6, 150.9, 140.0, 135.9, 130.3, 128.5, 128.1, 128.1, 127.3, 126.1, 125.9, 125.9, 123.7, 44.3, 33.4, 29.5, 22.6. Data were consistent with those previously reported.15
:1 grading to 4:1] furnished isoquinoline 4s (39.3 mg, 0.148 mmol, 93%) as prisms. M.p. 126–127 °C (petrol/CH2Cl2); 1H NMR (400 MHz, CDCl3) δH: 9.11 (1H, s), 8.08 (2H, dt, J 7.4, 1.6), 7.91 (1H, s), 7.49 (2H, t, J 7.6), 7.39 (1H, tt, J 7.4, 1.3), 7.19 (1H, s), 7.09 (1H, s), 4.02 (3H, s), 4.02 (3H, s); 13C NMR[1H] (100 MHz, CDCl3) δC: 153.2, 150.3, 150.2, 149.8, 139.9, 133.3, 128.8, 128.2, 126.8, 123.8, 115.5, 105.2, 105.0, 56.1, 56.1. Data were consistent with those previously reported.49
:1 grading to 14:1] furnished isoquinoline 4y (40.4 mg, 0.181 mmol, 89%) as prisms. M.p. 132–134 °C (petrol/CH2Cl2); 1H NMR (400 MHz, CDCl3) δH: 9.30 (1H, s), 8.12 (2H, dd, J 8.0, 1.6), 8.06 (1H, s), 7.89 (1H, dd, J 9.0, 5.2), 7.60 (1H, dd, J 8.5, 2.3), 7.54–7.41 (4H, m); 13C[1H] NMR (100 MHz, CDCl3) δC: 160.8 (d, 1J 249.3), 151.6 (d, 4J 5.6), 151.0 (d, 6J 3.2), 139.3, 133.7, 129.6 (d, 3J 8.8), 128.8, 128.6, 128.2 (d, 3J 8.0), 126.9, 121.2 (d, 2J 25.6), 116.2 (d, 5J 1.6), 110.6 (d, 2J 20.7); 19F[1H] NMR (377 MHz, CDCl3) δF: −111.3. Data were consistent with those previously reported.47
:1] furnished isoquinoline N-oxide 5a (22.2 mg, 0.0943 mmol, 99%) as a solid. Method B: A resealable reaction tube, containing a magnetic follower, was sealed with a rubber septum and flame dried under a flow of argon. PdCl2(Amphos)2 (22.8 mg, 0.0322 mmol) and Cs2CO3 (525 mg, 1.61 mmol) were added to the tube. A solution of the aryl bromide 23a (138 mg, 0.645 mmol) in anhydrous THF (3.2 mL) and propiophenone, 2a, (174 mg, 1.29 mmol) were then added via syringe. The septum was replaced with a screw cap and the reaction was heated at 70 °C for 18 h. The reaction was cooled to room temperature, then a solution of HCl (12.9 mL, 1.0 M in 9:1 EtOH/H2O) was added, the tube resealed with a screw cap and then heated at 110 °C for 24 h. The reaction was cooled to room temperature and the solvent removed in vacuo using a toluene azeotrope. Purification by flash column chromatography on alumina [EtOAc/MeOH 19:1] furnished isoquinoline N-oxide 5a (97.4 mg, 0.413 mmol, 64%) as a solid. M.p. 180–181 °C; 1H NMR (400 MHz, CDCl3) δH: 8.87 (1H, s), 8.02–7.89 (1H, m), 7.79–7.70 (1H, m), 7.69–7.58 (2H, m), 7.57–7.45 (3H, m), 7.44–7.36 (2H, m), 2.42 (3H, s); 13C[1H] NMR (100 MHz, CDCl3) δC: 146.1, 135.1, 132.9, 130.9, 130.0, 129.5, 128.9, 128.9, 128.6, 128.5, 128.5, 125.4, 124.0, 16.1. Data were consistent with those previously reported.10
:1 grading to 3:1] furnished isoquinoline N-oxide 5i (13.8 mg, 0.0549 mmol, 96%) as a solid. Method B: Ketone24b (104 mg, 0.367 mmol) was subjected to General procedure L. Purification by flash column chromatography on alumina [EtOAc] furnished isoquinoline N-oxide 5i (67.9 mg, 0.270 mmol, 74%) as a solid. M.p. 175–178 °C; 1H NMR (400 MHz, CDCl3) δH: 8.81 (1H, s), 8.10–8.01 (1H, m), 7.78–7.69 (1H, m), 7.67–7.58 (4H, m), 7.57–7.45 (3H, m), 3.55 (3H, s); 13C[1H] NMR (100 MHz, CDCl3) δC: 152.1, 140.3, 133.4, 130.6, 129.5, 129.2 (2 signals), 128.7, 128.5, 128.3, 125.8, 124.7, 122.0, 61.6; IRνmax (thin film)/cm−1 3062, 2941, 2852, 1733, 1659, 1624, 1588, 1490, 1465, 1445, 1427, 1365, 1312, 1223, 1179, 1098, 1045, 1025, 962, 909, 894, 862; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C16H14NO2 252.1019; Found 252.1024.
:1 grading to 39:1] furnished isoquinoline N-oxide 5p (37.7 mg, 0.142 mmol, 91%) as prisms. M.p. 237–238 °C (MeOH/EtOAc); 1H NMR (400 MHz, CD2Cl2) δH: 8.88 (1H, s), 7.75 (1H, d, J 7.6), 7.55 (1H, t, J 7.5), 7.45 (1H, t, J 7.6), 7.35 (1H, d, J 8.6), 7.24 (2H, d, J 8.6), 7.10 (2H, d, J 8.6), 3.92 (3H, s), 2.40 (3H, s); 13C[1H] NMR (100 MHz, CD2Cl2) δC: 160.0, 145.1, 135.7, 135.4, 131.1, 129.7, 128.6, 128.6, 128.4, 128.3, 126.0, 124.8, 114.6, 55.8, 15.7; IRνmax (thin film)/cm−1 2992, 2955, 2930, 2832, 1607, 1593, 1513, 1490, 1462, 1423, 1384, 1315, 1288, 1243, 1203, 1164, 1146, 1107, 1029, 1002, 970, 943, 894, 872, 850; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C17H16NO2 266.1176; Found 266.1166.
:1 grading to 3:1] furnished isoquinoline-N-oxide 5r (41.3 mg, 0.188 mmol, 92%) as prisms. M.p. 193–195 °C (petrol/CH2Cl2); 1H NMR (400 MHz, CDCl3) δH: 8.72 (1H, s), 7.54–7.50 (2H, m), 7.48–7.44 (1H, m), 7.40–7.38 (2H, m), 7.11 (1H, s), 6.98 (1H, s), 4.04 (3H, s), 4.03 (3H, s), 2.35 (3H, s); 13C[1H] NMR (75 MHz, CDCl3) δC: 151.8, 151.6, 144.5, 133.8, 133.3, 130.1, 129.1, 128.7, 128.6, 125.7, 124.6, 103.5, 102.8, 56.2, 56.1, 16.3; IRνmax (thin film)/cm−1 2926, 1622, 1506, 1465, 1423, 1388, 1364, 1312, 1254, 1208, 1167, 1072, 1030, 990; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C18H17NNaO3 318.1101; Found 318.1101.
:1 EtOH/H2O) was added to ketone 3a (69.7 mg, 0.247 mmol) (synthesis10) in a resealable reaction tube containing a magnetic follower. The tube was sealed with a screw cap and heated at 90 °C for 18 h. The reaction was then cooled to room temperature and quenched by the addition of saturated aqueous NaHCO3 (25 mL). The aqueous layer was extracted with EtOAc (3 × 25 mL) and the combined organics were dried over Na2SO4, filtered and the solvent removed in vacuo. Purification by flash column chromatography on alumina [EtOAc/MeOH 4:1 grading to MeOH] furnished isoquinolinium salt 6a (63.6 mg, 0.237 mmol, 96%) as prisms. M.p. 280 °C (decomposed) (MeOH/EtOAc); 1H NMR (400 MHz, CDCl3) δH: 11.49 (1H, s), 8.81 (1H, d, J 8.3), 8.18–8.12 (2H, m), 7.90 (1H, td, J 7.3, 1.7), 7.63–7.61 (3H, m), 7.35 (2H, dd, J 6.5, 3.0), 4.31 (3H, s) 2.46 (3H, s); 13C[1H] NMR (100 MHz, CDCl3) δC: 151.9, 143.5, 137.4, 137.1, 133.1, 132.6, 131.4, 130.9, 130.7, 130.0, 129.1, 126.9, 123.9, 48.2, 16.1; IRνmax (thin film)/cm−1 1633, 1597, 1500, 1481, 1445, 1376, 1343, 1269, 1189, 1077, 1007, 811; HRMS (ESI-TOF) m/z: M+ Calcd for C17H16N 234.1278, Found 234.1281.
:1 grading to 9:1] furnished nitrile 8a (65.6 mg, 0.247 mmol, 69%) as an oil. 1H NMR (400 MHz, CDCl3) δH: 7.62 (1H, d, J 7.7), 7.45 (1H, d, J 8.2), 7.42–7.32 (7H, m,), 5.95 (1H, s), 5.90 (1H, s), 4.16–4.03 (4H, m); 13C NMR[1H] (100 MHz, CDCl3) δC: 135.8, 134.9, 134.7, 130.0, 129.7, 129.1, 128.3, 128.1, 127.9, 127.6, 120.0, 102.3, 65.2, 65.1, 37.8; IRνmax (thin film)/cm−1 2892, 2244, 1601, 1494, 1452, 1406, 1217, 1112, 1075, 1043, 970, 943, 894; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C17H15NNaO2 288.0995; Found 288.0992.
:1 grading to 1:1] furnished nitrile 8b (72.6 mg, 0.223 mmol, 73%) as prisms. M.p. 57–59 °C (petrol/CH2Cl2); 1H NMR (400 MHz, CDCl3) δH: 7.37–7.36 (4H, m), 7.34–7.31 (1H, m), 7.11 (1H, s), 6.84 (1H, s), 5.82 (1H, s), 5.82 (1H, s), 4.17–4.02 (4H, m), 3.91 (3H, s), 3.81 (3H, s); 13C[1H] NMR (100 MHz, CDCl3) δC: 149.9, 148.6, 135.9, 129.0, 128.0, 127.6, 127.4, 126.7, 120.0, 112.2, 110.1, 101.8, 65.1, 65.0, 56.0, 56.0, 37.4; IRνmax (thin film)/cm−1 2960, 2893, 2243, 1609, 1518, 1494, 1453, 1402, 1351, 1290, 1268, 1201, 1180, 1114, 1066, 1030, 1003, 959, 940, 913, 868; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C19H19NNaO4 348.1206; Found 348.1203.
:1 then petrol/CH2Cl2 1:1] furnished nitrile 8c (48.1 mg, 0.177 mmol, 38%) as an oil. 1H NMR (400 MHz, CDCl3) δH: 7.59 (2H, m), 7.45 (1H, td, J 7.5, 1.6), 7.39 (1H, td, J 7.5, 1.3), 7.27 (1H, dd, J 5.2, 1.2), 7.11 (1H, dt, J 3.5, 1.1), 6.97 (1H, dd, J 5.1, 3.6), 6.09 (1H, s), 5.90 (1H, s), 4.20–4.05 (4H, m); 13C[1H] NMR (100 MHz, CDCl3) δC: 138.5, 134.5, 134.2, 130.0, 129.2, 128.6, 127.6, 126.9, 126.8, 126.4, 119.3, 102.2, 65.1, 65.0, 33.6; IRνmax (thin film)/cm−1 2959, 2925, 2891, 2855, 1738, 1601, 1470, 1389, 1259, 1216, 1074, 1043, 1023, 969, 942, 910; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C15H13NNaO2S 294.0559; Found 294.0554.
:3] furnished nitrile 8d (72.5 mg, 0.338 mmol, 85%) as an oil. 1H NMR (400 MHz, CDCl3) δH: 7.75 (1H, dd, J 7.5, 1.3), 7.60–7.48 (3H, m), 5.89 (1H, s), 5.87 (1H, s), 4.21–4.14 (2H, m), 4.14–4.07 (2H, m); 13C[1H] NMR (100 MHz, CDCl3) δC: 134.6, 130.7, 130.3, 129.3, 129.2, 125.5, 112.3, 102.8, 65.0, 24.9; IRνmax (thin film)/cm−1 2899, 2257, 1459, 1405, 1289, 1228, 1114, 1075, 1043; HRMS (ESI-TOF) m/z: [M − H]− Calcd for C12H9N2O2 213.0670; Found 213.0662.
:3] furnished nitrile 8e (158 mg, 0.480 mmol, 94%) as a solid. M.p. 99–101 °C; 1H NMR (400 MHz, CDCl3) δH: 7.83 (2H, dd, J 8.3, 1.2), 7.75 (1H, td, J 7.5, 1.1), 7.68 (1H, d, J 7.6), 7.60–7.56 (2H, m), 7.47 (1H, ddd, J 7.9, 6.3, 2.5), 7.37–7.31 (2H, m), 6.38 (1H, s), 6.18 (1H, s), 4.10–3.94 (4H, m); 13C[1H] NMR (100 MHz, CDCl3) δC 138.1, 135.2, 135.0, 130.6, 130.4, 129.9, 129.2, 129.1, 127.0, 123.7, 114.1, 101.4, 65.0, 64.9, 58.9; IRνmax (powder)/cm−1 2936, 2891, 1691, 1582, 1447, 1391, 1327, 1310, 1154, 1104, 1064, 1027; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C17H15NNaO4S 352.0614; Found 352.0605.
:3] furnished nitrile 8f (140 mg, 0.484 mmol, 92%) as an oil. 1H NMR (400 MHz, CDCl3) δH: 7.61 (1H, dd, J 7.6, 1.3), 7.55 (1H, dd, J 7.5, 1.5), 7.50–7.36 (2H, m), 5.93 (1H, s), 5.34 (1H, s), 4.17–4.00 (4H, m), 1.46 (9H, s); 13C[1H] NMR (100 MHz, CDCl3) δC: 164.0, 135.0, 129.9, 129.7, 129.5, 128.9, 128.0, 116.6, 102.7, 84.2, 65.1, 64.9, 41.0, 27.7; IRνmax (neat)/cm−1 2980, 1739, 1456, 1395, 1370, 1259, 1145, 1109, 1076, 1045; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C16H19NNaO4 312.1206; Found 312.1193.
:1 grading to 2:1] furnished isoquinoline 9a (32.1 mg, 0.146 mmol, 73%) as prisms. M.p. 145–147 °C (CH2Cl2/EtOAc); 1H NMR (400 MHz, CDCl3) δH: 8.91 (1H, s), 7.83 (1H, d, J 8.1), 7.56 (2H, t, J 7.3), 7.47 (1H, t, J 7.5), 7.42–7.40 (3H, m), 7.29 (1H, d, J 9.7), 7.25 (1H, d, J 7.1), 4.42 (2H, br. s); 13C NMR[1H] (100 MHz, CDCl3) δC: 151.7, 151.2, 137.3, 135.7, 130.6, 130.5, 129.4, 127.9, 127.9, 123.8, 123.0, 122.7, 111.6. Data were consistent with those previously reported.29
:1 grading to EtOAc/MeOH 39:1] furnished isoquinoline 9b (29.5 mg, 0.105 mmol, 84%) as an oil. 1H NMR (400 MHz, CD3OD) δH: 8.59 (1H, s), 7.58 (2H, t, J 7.5), 7.48 (1H, t, J 7.1), 7.37 (2H, d, J 7.3), 7.23 (1H, s), 6.49 (1H, s), 3.91 (3H, s), 3.65 (3H, s); 13C[1H] NMR (100 MHz, CD3OD) δC: 154.3, 151.2, 148.0, 147.5, 136.2, 134.9, 130.6, 129.6, 128.1, 119.9, 112.3, 105.9, 101.2, 55.3, 54.9; IRνmax (thin film)/cm−1 3476, 3375, 3176, 3001, 2934, 2830, 2360, 1627, 1598, 1579, 1495, 1452, 1425, 1401, 1354, 1246, 1197, 1160, 1092, 1031, 1011, 927, 844; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C17H17N2O2 281.1285; Found 281.1292.
:1] furnished isoquinoline 9c (43.1 mg, 0.143 mmol, 75%) as an oil. 1H NMR (300 MHz, CDCl3) δH: 8.82 (1H, s), 7.72 (1H, dd, J 8.1, 0.7), 7.45 (1H, dt, J 5.2, 0.9), 7.38 (2H, d, J 3.6), 7.22–7.14 (2H, m), 7.03 (1H, dd, J 3.4, 0.9), 4.55 (2H, br. s); 13C[1H] NMR (75 MHz, CDCl3) δC: 153.2, 152.3, 138.5, 136.0, 130.9, 128.8, 127.9, 127.8, 127.2, 123.6, 122.9, 122.9, 103.4; IRνmax (neat)/cm−1 3301, 3170, 1620, 1578, 1492, 1455, 1426, 1373, 1346, 1264, 1217, 1147, 956, 925, 853, 816; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C13H11N2S 227.0638; Found 227.0633.
:1 EtOAc/nBuOH (5 × 10 mL). Purification by flash column chromatography [C18 reversed phase SiO2, H2O] furnished isoquinoline 9d (48.3 mg, 0.284 mmol, 72%) as a bright yellow solid. Method B:para-Toluenesulfonic acid monohydrate (9.5 mg, 0.050 mmol) was added to a solution of nitrile 8d (97.1 mg, 0.453 mmol) in 1:1 THF/H2O (2.5 mL) in a resealable reaction tube containing a magnetic follower. The tube was sealed with a screw cap and heated at 50 °C for 18 h. The reaction was then cooled to room temperature and quenched by the addition of saturated aqueous NaHCO3 (25 mL). The aqueous layer was extracted with 4:1 EtOAc/nBuOH (5 × 10 mL) and the combined organics were dried over Na2SO4, filtered, and the solvent removed in vacuo. Purification by flash column chromatography [C18 reversed phase SiO2, H2O] furnished isoquinoline 9d (65.4 mg, 0.384 mmol, 85%) as a bright yellow solid. M.p. >320 °C; 1H NMR (400 MHz, CD3OD) δH: 8.83 (1H, s), 7.75 (1H, d, J 8.1), 7.59–7.52 (2H, m), 7.12 (1H, ddd, J 8.1, 6.4, 1.5); 13C[1H] NMR (100 MHz, CD3OD) δC: 173.0, 158.1, 141.9, 133.0, 130.0, 122.4, 121.8, 121.7, 120.7, 83.2; IRνmax (powder)/cm−1 3263, 2581, 2214, 1622, 1580, 1555, 1467, 1438, 1221, 1179, 1047, 1014; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C10H6N2NaO 193.0372; Found 193.0369.
:1 EtOH/H2O) was added to nitrile 8e (53.8 mg, 0.163 mmol) in a resealable reaction tube containing a magnetic follower. The tube was sealed with a screw cap and heated at 90 °C for 4 h. A solution of NH4HCO3 (3.26 mL, 2.0 M in H2O) was then added. The tube was resealed and heated for a further 1 h at 65 °C. The reaction was then cooled to room temperature and quenched by the addition of H2O (25 mL). The aqueous layer was extracted with 4:1 EtOAc/nBuOH (3 × 10 mL) and the combined organics were dried over Na2SO4, filtered, and the solvent removed in vacuo. Purification by flash column chromatography [petrol/EtOAc 4:1] furnished isoquinoline 9e (22.2 mg, 0.0781 mmol, 48%) as a solid. M.p. 191–193 °C; 1H NMR (400 MHz, CDCl3) δH: 8.91 (1H, s), 8.44 (1H, dd, J 8.8, 0.7), 8.01–7.98 (2H, m), 7.74 (1H, dd, J 8.1, 0.8), 7.62–7.51 (2H, m), 7.49–7.45 (2H, m), 7.28 (1H, dd, J 7.0, 0.9), 6.66 (2H, br. s); 13C[1H] NMR (100 MHz, CDCl3) δC: 159.1, 155.3, 143.0, 134.6, 133.1, 133.0, 129.2, 129.0, 126.0, 123.6, 123.5, 122.0, 103.1; IRνmax (powder)/cm−1 3441, 3293, 3170, 1633, 1557, 1478, 1437, 1291, 1136, 1083; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C15H12N2NaO2S 307.0512; Found 307.0503.
:1] furnished isoquinoline 9f (25.8 mg, 0.179 mmol, 74%) as an oil. Method B: A solution of nitrile 8f (53.6 mg, 0.185 mmol) in 3:2 EtOH/H2O (1.85 mL) was added to a resealable reaction tube containing a magnetic follower. The tube was sealed with a screw cap and heated at 90 °C for 18 h. The reaction was cooled to room temperature, NH4Cl (99.0 mg, 1.85 mmol) was added and then the reaction was reheated at 90 °C for 3 h. The reaction was cooled to room temperature and basified by the addition of 2 M aqueous NH4HCO3 (1.85 mL). The reaction mixture was heated at 90 °C for 3 h and then cooled to room temperature. The aqueous layer was extracted with 4:1 EtOAc/nBuOH (3 × 10 mL) and the combined organics were dried over Na2SO4, filtered and the solvent removed in vacuo. Purification by flash column chromatography [petrol/EtOAc 4:1] furnished isoquinoline 9f as a yellow solid (22.8 mg, 0.158 mmol, 86%). M.p. 175–180 °C 1H NMR (400 MHz, CDCl3) δH: 8.87 (1H, s), 7.78 (1H, dd, J 8.2, 0.5), 7.58–7.46 (2H, m), 7.28–7.21 (1H, m), 6.74 (1H, s), 4.19 (2H, br. s); 13C[1H] NMR (100 MHz, CDCl3) δC: 154.5, 151.6, 138.9, 130.4, 127.8, 124.6, 123.8, 123.0, 99.4. Data were consistent with those previously reported.50
:1 grading to 5:1] furnished ester 11 (67.8 mg, 0.256 mmol, 68%) as a pale yellow oil. 1H NMR (400 MHz, CDCl3) δH: 7.46 (1H, dd, J 7.5, 1.7), 7.28–7.15 (3H, m), 5.89 (1H, s), 4.10–4.01 (2H, m), 4.00–3.93 (2H, m), 3.63 (2H, s), 1.37 (9H, s); 13C[1H] NMR (100 MHz, CDCl3) δC: 170.9, 135.6, 133.6, 131.4, 129.2, 127.0, 126.8, 102.6, 80.7, 65.1, 39.6, 28.0; IRνmax (neat)/cm−1 2360, 1731, 1455, 1393, 1368, 1334, 1147; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C15H20NaO4 287.1254; Found 287.1260.
:1 THF/H2O (1.6 mL) in a resealable reaction tube containing a magnetic follower. The tube was sealed with a screw cap and stirred at room temperature for 18 h. Aqueous NH4OH (28% w/w, 1.6 mL) was added and the reaction was heated at 60 °C for 24 h. The reaction was then cooled to room temperature, diluted with brine (25 mL) and extracted with EtOAc (2 × 25 mL). The aqueous layer was then neutralised to pH 7 by the addition of 2 M HCl and again extracted with EtOAc (2 × 25 mL). The combined organics were then washed with 2 M HCl (3 × 25 mL) and the organic phase discarded. The aqueous phase was then neutralised to pH 7 by the addition of 2 M NaOH and then extracted with EtOAc (3 × 25 mL). The combined organics were then dried over Na2SO4, filtered, and the solvent removed in vacuo. Purification by flash column chromatography [EtOAc/MeOH 100:1 grading to 3:1] furnished isoquinoline 12 (40.2 mg, 0.275 mmol, 70%) as bright yellow prisms. 1H NMR (400 MHz, CD3OD) δH: 8.66 (1H, s), 7.81 (1H, dd, J 8.6, 1.0), 7.59–7.49 (2H, m), 7.23 (1H, dd, J 6.4, 1.2), 6.87 (1H, s); 13C[1H] NMR (100 MHz, CD3OD) δC: 161.0, 144.1, 142.2, 131.7, 127.8, 124.7, 123.3, 121.3, 104.9. Data were consistent with a commercially available sample from Sigma Aldrich.
:1 EtOH/H2O (5.24 mL) was added and the reaction was heated at 90 °C for 24 h. The reaction was cooled to room temperature and quenched by the addition of saturated aqueous NaHCO3 (25 mL). The aqueous layer was extracted with EtOAc (3 × 15 mL) and the combined organics were then dried over Na2SO4, filtered, and the solvent removed in vacuo. Purification by flash column chromatography [CH2Cl2] furnished isoquinoline 18c (100 mg, 0.339 mmol, 65%) as a solid. M.p. 125–130 °C; 1H NMR (400 MHz, CDCl3) δH: 9.42 (1H, d, J 0.7), 8.10–8.04 (1H, m), 7.64–7.58 (2H, m), 7.47–7.39 (3H, m), 7.33–7.28 (1H, m), 7.26–7.16 (6H, m), 1.90 (3H, s); 13C[1H] NMR (100 MHz, CDCl3) δC: 151.8, 150.3, 140.6, 137.0, 136.5, 135.8, 131.3, 130.5, 130.1, 129.7, 129.8, 129.7, 127.8, 127.6, 127.2, 127.2, 126.8, 125.7, 125.5, 19.9; IRνmax (neat)/cm−1 3058, 1617, 1558, 1498, 1449, 1370, 1247, 1029; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C22H18N 296.1434; Found 296.1434.
:1 H2O/THF (18.9 mL). The reaction was stirred at room temperature for 4 h and then diluted with Et2O (50 mL), washed with brine (100 mL), then dried over Na2SO4, filtered, and the solvent removed in vacuo to furnish (E)-O-methyl oxime 22a (1.23 g, 9.41 mmol, 100%) as a colourless oil. 1H NMR (400 MHz, CDCl3) δH: 8.10 (1H, s), 7.67–7.55 (2H, m), 7.43–7.35 (3H, m), 4.01 (3H, s); 13C NMR[1H] (100 MHz, CDCl3) δC: 148.6, 132.2, 129.8, 128.7, 127.0, 62.0. Data were consistent with those previously reported.51
:1 mixture of (E)- and (Z)-O-methyl oxime 22c (1.42 g, 8.70 mmol, 91%) as a colourless oil. (E)-22c: 1H NMR (400 MHz, CDCl3) δH: 7.72–7.62 (2H, m), 7.46–7.33 (3H, m), 4.01 (3H, s), 2.78 (2H, q, J 7.6), 1.16 (3H, t, J 7.6); 13C[1H] NMR (100 MHz, CDCl3) δC: 159.8, 135.6, 129.0, 128.5, 126.3, 61.9, 20.1, 11.2. (Z)-22c: 1H NMR (400 MHz, CDCl3) δH: 7.74–7.61 (2H, m), 7.46–7.32 (3H, m), 3.86 (3H, s), 2.58 (2H, q, J 7.6), 1.09 (3H, t, J 7.5); 13C[1H] NMR (100 MHz, CDCl3) δC: 159.0, 133.9, 128.6, 128.1, 127.8, 61.6, 28.9, 11.6. Data were consistent with those previously reported.53
:1] furnished (E)-oxime 23a (44.4 mg, 0.208 mmol, 51%) as a colourless oil. 1H NMR (400 MHz, CDCl3) δH: 8.46 (1H, s), 7.88 (1H, d, J 7.8), 7.57 (1H, d, J 8.1), 7.35–7.18 (2H, m), 4.01 (3H, s); 13C[1H] NMR (100 MHz, CDCl3) δC: 147.9, 133.1, 131.5, 131.0, 127.5, 127.5, 123.8, 62.3. Data were consistent with those previously reported.51
:1] furnished (E)-oxime 23b (669 mg, 2.93 mmol, 55%) as a colourless oil. 1H NMR (400 MHz, CDCl3) δH: 7.64–7.53 (1H, m), 7.37–7.17 (3H, m), 4.00 (3H, s), 2.22 (3H, s); 13C[1H] NMR (100 MHz, CDCl3) δC: 157.0, 138.9, 133.1, 130.3, 130.0, 127.4, 121.8, 61.9, 16.5; IRνmax (thin film)/cm−1 2937, 1471, 1427, 1100, 1048; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C9H1179BrNO 228.0019; Found 228.0023.
:1] furnished a 1.2:1 mixture of stereoisomers of oxime 23c (517 mg, 2.14 mmol, 70%) as a colourless oil. Major isomer:1H NMR (400 MHz, CDCl3) δH: 7.65–7.54 (1H, m), 7.39–7.30 (1H, m), 7.29–7.16 (2H, m), 3.97 (3H, s), 2.75 (2H, q, J 7.7), 1.00 (3H, t, J 7.6); 13C[1H] NMR (100 MHz, CDCl3) δC: 161.9, 137.5, 132.9, 130.8, 129.9, 127.3, 122.3, 61.9, 22.7, 10.0. Minor isomer:1H NMR (400 MHz, CDCl3) δH: 7.64–7.54 (1H, m), 7.39–7.29 (1H, m), 7.29–7.17 (1H, m), 7.07 (1 H, dd, J 7.6, 1.3), 3.83 (3H, s), 2.54 (2H, q, J 7.4), 1.12 (3H, t, J 7.5); 13C[1H] NMR (100 MHz, CDCl3) δC: 158.4, 136.9, 132.6, 129.5, 128.5, 127.1, 120.5, 61.8, 28.4, 10.8; IRνmax (thin film)/cm−1 2972, 2937, 1463, 1432, 1048, 1026; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C10H1379BrNO 242.0175; Found 242.0176.
:1] furnished (E)-oxime 23d (1.24 g, 4.27 mmol, 90%) as an oil. 1H NMR (400 MHz, CDCl3) δH: 7.65–7.53 (3H, m), 7.49 (1H, dd, J 7.6, 1.8), 7.44–7.34 (4H, m), 7.32–7.23 (1H, m), 4.07 (3H, s); 13C[1H] NMR (100 MHz, CDCl3) δC: 155.8, 138.0, 133.4, 132.4, 131.9, 130.3, 130.1, 129.5, 128.0, 127.4, 123.5, 62.6; IRνmax (thin film)/cm−1 2936, 1468, 1445, 1328, 1059, 1036; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C14H1379BrNO 290.0175; Found 290.0176.
:1] furnished (E)-ketone 24a (227 mg, 0.850 mmol, 91%) as an oil. 1H NMR (400 MHz, CDCl3) δH: 8.45 (1H, s), 7.93–7.86 (2H, m), 7.62–7.55 (1H, m), 7.50–7.43 (1H, m), 7.40–7.32 (2H, m), 7.30–7.20 (2H, m), 7.18–7.11 (1H, m), 5.34 (1H, q, J 6.8), 3.96 (3H, s), 1.52 (3H, d, J 6.8); 13C[1H] NMR (100 MHz, CDCl3) δC: 200.5, 148.1, 140.3, 136.3, 132.8, 130.2, 129.7, 129.1, 128.7, 128.5, 128.1, 127.1, 62.1, 44.2, 18.6; IRνmax (thin film)/cm−1 2953, 1684, 1597, 1448, 1222, 1048; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C17H17NNaO2 290.1151; Found 290.1151.
:1 petrol] furnished (E)-ketone 24b (118 mg, 0.417 mmol, 85%) as an oil. 1H NMR (400 MHz, CDCl3) δH: 8.36 (1H, s), 7.99 (2H, d, J 7.3), 7.59 (1H, d, J 7.1), 7.55–7.48 (2H, m), 7.47–7.31 (4H, m), 6.23 (1H, s), 3.76 (3H, s), 3.51 (3H, s); 13C[1H] NMR (100 MHz, CDCl3) δC: 195.8, 148.3, 135.6, 134.8, 133.2, 130.7, 129.8, 129.8, 128.8, 128.6, 128.5, 82.5, 61.9, 58.0; IRνmax (thin film)/cm−1 2936, 1693, 1597, 1448, 1210, 1088, 1043, 1003; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C17H17NNaO3 306.1101; Found 306.1097.
:1] furnished (E)-ketone 24c (69.5 mg, 0.247 mmol, 29%) as an oil. 1H NMR (400 MHz, CDCl3) δH: 8.06–7.98 (2H, m), 7.51–7.43 (1H, m), 7.42–7.34 (2H, m), 7.32–7.18 (4H, m), 5.17 (1H, q, J 6.8), 3.94 (3H, s), 2.27 (3H, s), 1.53 (3H, d, J 6.8); 13C[1H] NMR (100 MHz, CDCl3) δC: 200.9, 155.8, 139.2, 136.3, 132.7, 129.0, 128.9, 128.8, 128.4, 127.9, 126.9, 61.8, 44.1, 19.4, 16.6; IRνmax (thin film)/cm−1 2935, 1684, 1448, 1220, 1048; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C18H19NNaO2 304.1308; Found 304.1300.
:1] furnished (E)-ketone (E)-24d (54.9 mg, 0.186 mmol, 32%) as an oil. 1H NMR (400 MHz, CDCl3) δH: 8.11–8.00 (2H, m), 7.51–7.43 (1H, m), 7.42–7.33 (2H, m), 7.31–7.18 (4H, m) 5.13 (1H, q, J 6.8), 3.91 (3H, s), 2.87–2.66 (2H, m), 1.52 (3H, d, J 7.1), 1.13 (3H, t, J 7.6,); 13C[1H] NMR (100 MHz, CDCl3) δC: 201.0, 160.8, 139.6, 136.4, 135.1, 132.7, 129.0, 128.9, 128.9, 128.3, 128.0, 126.7, 61.7, 44.0, 23.4, 19.6, 10.6; IRνmax (thin film)/cm−1 2965, 1683, 1449, 1260, 1221, 1046; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C19H21NNaO2 318.1465; Found 318.1460.
:3] furnished (Z)-ketone (Z)-24d (34.7 mg, 0.115 mmol, 20%) as an oil. Major rotamer:1H NMR (400 MHz, CDCl3) δH: 8.01 (2H, d, J 7.6), 7.59–7.23 (6H, m), 7.11–6.96 (1H, m), 4.74–4.59 (1H, m), 3.44 (3H, s), 2.70–2.51 (2H, m), 1.51 (3H, d, J 7.1), 1.18 (3H, t, J 7.5); 13C[1H] NMR (100 MHz, CDCl3) δC: 200.5, 159.4, 138.0, 136.3, 134.6, 132.7, 128.8, 128.6, 128.5, 126.9, 126.7, 61.2, 43.7, 29.8, 20.1, 11.4. Minor rotamer:1H NMR (400 MHz, CDCl3) δH: 7.90 (2H, d, J 7.6), 7.58–7.23 (6H, m), 7.11–6.96 (1H, m), 4.75–4.57 (1H, m), 3.90 (3H, s), 2.46–2.21 (2H, m), 1.57 (3H, d, J 6.8), 1.01 (3H, t, J 7.5); 13C[1H] NMR (100 MHz, CDCl3) δC: 201.8, 159.2, 137.2, 137.1, 135.1, 132.9, 128.7, 128.5 (2 carbons), 128.4, 128.4, 128.2, 126.3, 61.6, 43.7, 29.7, 19.2, 10.6; IRνmax (thin film)/cm−1 2935, 1684, 1449, 1252, 1221, 1057, 1030; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C19H21NNaO2 318.1465; Found 318.1458.
:3] furnished (E)-ketone 24e (110 mg, 0.320 mmol, 93%) as a solid. M.p. 84–86 °C; 1H NMR (400 MHz, CDCl3) δH: 7.88 (2H, d, J 7.3), 7.65–7.55 (2H, m), 7.49–7.37 (4H, m), 7.36–7.20 (6H, m), 5.02 (1H, q, J 6.7), 4.04 (3H, s), 1.35 (3H, d, J 6.8); 13C[1H] NMR (100 MHz, CDCl3) δC: 200.8, 155.8, 140.4, 136.3, 135.7, 133.4, 132.7, 131.1, 130.1, 129.7, 129.5, 128.8, 128.4, 128.2, 128.0, 126.8, 62.5, 44.3, 18.9; IRνmax (powder)/cm−1 2934, 1683, 1447, 1220, 1042; HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C23H21NNaO2 366.1465; Found 366.1453.
:1] furnished isoquinoline 25a (37.2 mg, 0.150 mmol, 89%) as a solid. Method B: Ketone (Z)-24d (45.0 mg, 0.152 mmol) was subjected to General procedure J. Purification by flash column chromatography [petrol/EtOAc 49:1] furnished isoquinoline 25a (30.8 mg, 0.125 mmol, 82%) as a solid. Method C: Oxime 23c (73.1 mg, 0.302 mmol) was subjected to General procedure K with propiophenone, 2a. Purification by flash column chromatography [petrol/EtOAc 49:1] furnished isoquinoline 25a (34.7 mg, 0.140 mmol, 46%) as a solid. M.p. 58–61 °C; 1H NMR (400 MHz, CDCl3) δH: 8.23 (1H, d, J 8.3), 8.08 (1H, d, J 8.6), 7.75 (1H, td, J 7.6, 1.1), 7.67–7.57 (3H, m), 7.50 (2H, t, J 7.5), 7.45–7.36 (1H, m), 3.39 (2H, q, J 7.6), 2.63 (3H, s), 1.48 (3H, t, J 7.6); 13C[1H] NMR (100 MHz, CDCl3) δC: 160.7, 150.6, 141.7, 136.6, 130.0, 129.7, 128.1, 127.4, 126.2, 125.8, 125.3, 124.3, 122.1, 28.7, 15.5, 14.3. Data were consistent with those previously reported.54
:1] furnished isoquinoline 25b (36.4 mg, 0.123 mmol, 84%) as a solid. Method B: Oxime 23d (105 mg, 0.362 mmol) was subjected to General procedure K with propiophenone, 2a, (95.4 mg, 0.724 mmol). Purification by flash column chromatography [petrol/EtOAc 99:1] furnished isoquinoline 25b (74.0 mg, 0.251 mmol, 69%) as a solid. M.p. 78–83 °C; 1H NMR (400 MHz, CDCl3) δH: 8.15 (2H, dd, J 8.5, 3.9), 7.84–7.71 (3H, m), 7.67 (2H, d, J 7.3), 7.62–7.38 (7H, m), 2.73 (3H, s); 13C[1H] NMR (100 MHz, CDCl3) δC: 158.3, 151.0, 141.4, 139.8, 137.1, 130.2, 130.1, 130.0, 128.3, 128.2, 128.1, 128.1, 127.5, 126.4, 125.4, 123.9, 123.2, 15.7. Data were consistent with those previously reported.38c
Acknowledgements
B.S.P. would like to thank St. John's College Oxford for financial support. A.E.G. and T.J.D. would like to thank the EPSRC and GlaxoSmithKline for financial support. C.H.A.E. would like to thank CAPES, Coordenação de Aperfeiçoamento de Pessoal de Nível Superior, Brazil for financial support.References
- A. Bischler and B. Napieralski, Ber. Dtsch. Chem. Ges., 1893, 26, 1903–1908 CrossRef.
- A. Pictet and T. Spengler, Ber. Dtsch. Chem. Ges., 1911, 44, 2030–2036 CrossRef CAS.
- (a) C. Pomeranz, Monatsh., 1893, 14, 116–119 CrossRef; (b) P. Fritsch, Ber. Dtsch. Chem. Ges., 1893, 26, 419–422 CrossRef.
- K. W. Bentley, Nat. Prod. Rep., 2006, 23, 444–463 RSC.
- V. W. Pike, C. Halldin, C. Crouzel, L. Barré, D. J. Nutt, S. Osman, F. Shah, D. R. Turton and S. L. Waters, Nucl. Med. Biol., 1993, 20, 503–525 CrossRef CAS PubMed.
- A. Tsuboyama, H. Iwawaki, M. Furugori, T. Mukaide, J. Kamatani, S. Igawa, T. Moriyama, S. Miura, T. Takiguchi, S. Okada, M. Hoshino and K. Ueno, J. Am. Chem. Soc., 2003, 125, 12971–12979 CrossRef CAS PubMed.
- C. W. Lim, O. Tissot, A. Mattison, M. W. Hooper, J. M. Brown, A. R. Cowley, D. I. Hulmes and A. J. Blacker, Org. Process Res. Dev., 2003, 7, 379–384 CrossRef CAS.
- (a) A. Coppola, D. Sucunza, C. Burgos and J. J. Vaquero, Org. Lett., 2015, 17, 78–81 CrossRef CAS PubMed; (b) T. Ikawa, H. Urata, Y. Fukumoto, Y. Sumii, T. Nishiyama and S. Akai, Chem. – Eur. J., 2014, 20, 16228–16232 CrossRef CAS PubMed; (c) B. Zhang and A. Studer, Org. Biomol. Chem., 2014, 12, 9895–9898 RSC; (d) H. Jiang, Y. Cheng, R. Wang, Y. Zhang and S. Yu, Chem. Commun., 2014, 50, 6164–6167 RSC; (e) D. Fischer, H. Tomeba, N. K. Pahadi, N. T. Patil, Z. Huo and Y. Yamamoto, J. Am. Chem. Soc., 2008, 130, 15720–15725 CrossRef CAS PubMed; (f) Y.-Y. Yang, W.-G. Shou, Z.-B. Chen, D. Hong and Y.-G. Wang, J. Org. Chem., 2008, 73, 3928–3930 CrossRef CAS PubMed.
- (a) D. Zhao, F. Lied and F. Glorius, Chem. Sci., 2014, 5, 2869–2873 RSC; (b) Z. Shi, D. C. Koester, M. Boultadakis-Arapinis and F. Glorius, J. Am. Chem. Soc., 2013, 135, 12204–12207 CrossRef CAS PubMed; (c) X. Wang and Z. Wang, Tetrahedron, 2014, 70, 6728–6732 CrossRef CAS; (d) D. Yang, S. Burugupalli, D. Daniel and Y. Chen, J. Org. Chem., 2012, 77, 4466–4472 CrossRef CAS PubMed; (e) Y.-N. Niu, Z.-Y. Yan, G.-L. Gao, H.-L. Wang, X.-Z. Shu, K.-G. Ji and Y.-M. Liang, J. Org. Chem., 2009, 74, 2893–2896 CrossRef CAS PubMed; (f) R. He, Z.-T. Huang, Q.-Y. Zheng and C. Wang, Tetrahedron Lett., 2014, 55, 5705–5713 CrossRef CAS.
- T. J. Donohoe, B. S. Pilgrim, G. R. Jones and J. A. Bassuto, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11605–11608 CrossRef CAS PubMed.
- (a) F. Bellina and R. Rossi, Chem. Rev., 2010, 110, 1082–1146 CrossRef CAS PubMed; (b) C. C. C. Johansson and T. J. Colacot, Angew. Chem., Int. Ed., 2010, 49, 676–707 CrossRef CAS PubMed.
- (a) M. Palucki and S. L. Buchwald, J. Am. Chem. Soc., 1997, 119, 11108–11109 CrossRef CAS; (b) B. C. Hamann and J. F. Hartwig, J. Am. Chem. Soc., 1997, 119, 12382–12383 CrossRef CAS; (c) T. Satoh, Y. Kawamura, M. Miura and M. Nomura, Angew. Chem., Int. Ed. Engl., 1997, 36, 1740–1742 CrossRef CAS.
- (a) J. Barluenga, A. Jiménez-Aquino, C. Valdés and F. Aznar, Angew. Chem., Int. Ed., 2007, 46, 1529–1532 CrossRef CAS PubMed; (b) J. Barluenga, A. Jiménez-Aquino, F. Aznar and C. Valdés, J. Am. Chem. Soc., 2009, 131, 4031–4041 CrossRef CAS PubMed; (c) J. L. Rutherford, M. P. Rainka and S. L. Buchwald, J. Am. Chem. Soc., 2002, 124, 15168–15169 CrossRef CAS PubMed; (d) S. H. Spergel, D. R. Okoro and W. Pitts, J. Org. Chem., 2010, 75, 5316–5319 CrossRef CAS PubMed; (e) Z. Jin, S.-X. Guo, L.-L. Qiu, G.-P. Wu and J.-X. Fang, Appl. Organomet. Chem., 2011, 25, 502–507 CrossRef CAS; (f) J. M. Knapp, J. S. Zhu, D. J. Tantillo and M. J. Kurth, Angew. Chem., Int. Ed., 2012, 51, 10588–10591 CrossRef CAS PubMed.
- (a) C. Eidamshaus and J. D. Burch, Org. Lett., 2008, 10, 4211–4214 CrossRef CAS PubMed; (b) M. C. Willis, D. Taylor and A. T. Gillmore, Tetrahedron, 2006, 62, 11513–11520 CrossRef CAS; (c) M. C. Willis, D. Taylor and A. T. Gillmore, Org. Lett., 2004, 6, 4755–4757 CrossRef CAS PubMed; (d) F. Churruca, R. SanMartin, I. Tellitu and E. Domínguez, Eur. J. Org. Chem., 2005, 2481–2490 CrossRef CAS.
- B. S. Pilgrim, A. E. Gatland, C. T. McTernan, P. A. Procopiou and T. J. Donohoe, Org. Lett., 2013, 15, 6190–6193 CrossRef CAS PubMed.
- A. E. Gatland, B. S. Pilgrim, P. A. Procopiou and T. J. Donohoe, Angew. Chem., Int. Ed., 2014, 53, 14555–14558 CrossRef CAS PubMed.
- M. Prashad, Y. Liu and O. Repič, Adv. Synth. Catal., 2003, 345, 533–536 CrossRef CAS.
- (a) B. C. Hamann and J. F. Hartwig, J. Am. Chem. Soc., 1998, 120, 7369–7370 CrossRef CAS; (b) M. Kawatsura and J. F. Hartwig, J. Am. Chem. Soc., 1999, 121, 1473–1478 CrossRef CAS; (c) D. A. Culkin and J. F. Hartwig, Acc. Chem. Res., 2003, 36, 234–245 CrossRef CAS PubMed; (d) T. J. Colacot and H. A. Shea, Org. Lett., 2004, 6, 3731–3734 CrossRef CAS PubMed; (e) G. A. Grasa and T. J. Colacot, Org. Lett., 2007, 9, 5489–5492 CrossRef CAS PubMed.
- A. S. Guram, A. O. King, J. G. Allen, X. Wang, L. B. Schenkel, J. Chan, E. E. Bunel, M. M. Faul, R. D. Larsen, M. J. Martinelli and P. J. Reider, Org. Lett., 2006, 8, 1787–1789 CrossRef CAS PubMed.
- M. Hapke, L. Brandt and A. Lutzen, Chem. Soc. Rev., 2008, 37, 2782–2797 RSC.
- J. M. Fox, X. Huang, A. Chieffi and S. L. Buchwald, J. Am. Chem. Soc., 2000, 122, 1360–1370 CrossRef CAS.
- K. D. Hesp, R. J. Lundgren and M. Stradiotto, J. Am. Chem. Soc., 2011, 133, 5194–5197 CrossRef CAS PubMed.
- (a) L.-C. Campeau, D. R. Stuart, J.-P. Leclerc, M. Bertrand-Laperle, E. Villemure, H.-Y. Sun, S. Lasserre, N. Guimond, M. Lecavallier and K. Fagnou, J. Am. Chem. Soc., 2009, 131, 3291–3306 CrossRef CAS PubMed; (b) K. S. Kanyiva, Y. Nakao and T. Hiyama, Angew. Chem., Int. Ed., 2007, 46, 8872–8874 CrossRef CAS PubMed; (c) B. Xiao, Z.-J. Liu, L. Liu and Y. Fu, J. Am. Chem. Soc., 2013, 135, 616–619 CrossRef CAS PubMed.
- (a) J. Dusemund and E. Kröger, Arch. Pharm., 1987, 320, 617–620 CrossRef CAS; (b) L. Pongó, J. Reiter, G. Simig, B. ágai and F. Faigl, J. Heterocycl. Chem., 2006, 43, 1539–1547 CrossRef.
- P. W. Manley, P. Furet, G. Bold, J. Brüggen, J. Mestan, T. Meyer, C. R. Schnell, J. Wood, M. Haberey, A. Huth, M. Krüger, A. Menrad, E. Ottow, D. Seidelmann, G. Siemeister and K.-H. Thierauch, J. Med. Chem., 2002, 45, 5687–5693 CrossRef CAS PubMed.
- (a) J. You and J. G. Verkade, J. Org. Chem., 2003, 68, 8003–8007 CrossRef CAS PubMed; (b) L. Wu and J. F. Hartwig, J. Am. Chem. Soc., 2005, 127, 15824–15832 CrossRef CAS PubMed.
- F. G. Bordwell, J. E. Bares, J. E. Bartmess, G. J. McCollum, M. Van der Puy, N. R. Vanier and W. S. Matthews, J. Org. Chem., 1977, 42, 321–325 CrossRef CAS.
- F. G. Bordwell and J. A. Harrelson Jr., Can. J. Chem., 1990, 68, 1714–1718 CrossRef CAS.
- Y. Wan, W. Niu, W. J. Behof, Y. Wang, P. Boyle and C. B. Gorman, Tetrahedron, 2009, 65, 4293–4297 CrossRef CAS.
- X. Wang, A. Guram, E. Bunel, G.-Q. Cao, J. R. Allen and M. M. Faul, J. Org. Chem., 2008, 73, 1643–1645 CrossRef CAS PubMed.
- R. Shang, D.-S. Ji, L. Chu, Y. Fu and L. Liu, Angew. Chem., Int. Ed., 2011, 50, 4470–4474 CrossRef CAS PubMed.
- (a) W. A. Moradi and S. L. Buchwald, J. Am. Chem. Soc., 2001, 123, 7996–8002 CrossRef CAS PubMed; (b) M. Jørgensen, S. Lee, X. Liu, J. P. Wolkowski and J. F. Hartwig, J. Am. Chem. Soc., 2002, 124, 12557–12565 CrossRef; (c) T. Hama, X. Liu, D. A. Culkin and J. F. Hartwig, J. Am. Chem. Soc., 2003, 125, 11176–11177 CrossRef CAS PubMed.
- T. J. Kress and S. M. Costantino, J. Heterocycl. Chem., 1973, 10, 409–410 CrossRef CAS.
- N. A. Beare and J. F. Hartwig, J. Org. Chem., 2002, 67, 541–555 CrossRef CAS PubMed.
- C. Si and A. G. Myers, Angew. Chem., Int. Ed., 2011, 50, 10409–10413 CrossRef CAS PubMed.
- (a) D. W. Old, J. P. Wolfe and S. L. Buchwald, J. Am. Chem. Soc., 1998, 120, 9722–9723 CrossRef CAS; (b) T. Satoh, Y. Kametani, Y. Terao, M. Miura and M. Nomura, Tetrahedron Lett., 1999, 40, 5345–5348 CrossRef CAS; (c) F. Churruca, R. SanMartin, M. Carril, I. Tellitu and E. Domínguez, Tetrahedron, 2004, 60, 2393–2408 CrossRef CAS; (d) J. R. Schmink and N. E. Leadbeater, Org. Lett., 2009, 11, 2575–2578 CrossRef CAS PubMed.
- (a) A. Ehrentraut, A. Zapf and M. Beller, Adv. Synth. Catal., 2002, 344, 209–217 CrossRef CAS; (b) L. V. Desai, D. T. Ren and T. Rosner, Org. Lett., 2010, 12, 1032–1035 CrossRef CAS PubMed.
- (a) S.-G. Lim, J. H. Lee, C. W. Moon, J.-B. Hong and C.-H. Jun, Org. Lett., 2003, 5, 2759–2761 CrossRef CAS PubMed; (b) N. Guimond and K. Fagnou, J. Am. Chem. Soc., 2009, 131, 12050–12051 CrossRef CAS PubMed; (c) T. Fukutani, N. Umeda, K. Hirano, T. Satoh and M. Miura, Chem. Commun., 2009, 5141–5143 RSC; (d) R. He, Z.-T. Huang, Q.-Y. Zheng and C. Wang, Angew. Chem., Int. Ed., 2014, 53, 4950–4953 CrossRef CAS PubMed.
- (a) P. Villuendas and E. P. Urriolabeitia, J. Org. Chem., 2013, 78, 5254–5263 CrossRef CAS PubMed; (b) D.-S. Kim, J.-W. Park and C.-H. Jun, Adv. Synth. Catal., 2013, 355, 2667–2679 CrossRef CAS.
- (a) K. Parthasarathy and C.-H. Cheng, J. Org. Chem., 2009, 74, 9359–9364 CrossRef CAS PubMed; (b) X. Zhang, D. Chen, M. Zhao, J. Zhao, A. Jia and X. Li, Adv. Synth. Catal., 2011, 353, 719–723 CrossRef CAS; (c) L. Zheng, J. Ju, Y. Bin and R. Hua, J. Org. Chem., 2012, 77, 5794–5800 CrossRef CAS PubMed; (d) R. K. Chinnagolla, S. Pimparkar and M. Jeganmohan, Org. Lett., 2012, 14, 3032–3035 CrossRef CAS PubMed; (e) C. Kornhaaß, J. Li and L. Ackermann, J. Org. Chem., 2012, 77, 9190–9198 CrossRef PubMed; (f) P. C. Too, Y.-F. Wang and S. Chiba, Org. Lett., 2010, 12, 5688–5691 CrossRef CAS PubMed.
- (a) T. K. Hyster and T. Rovis, J. Am. Chem. Soc., 2010, 132, 10565–10569 CrossRef CAS PubMed; (b) T. K. Hyster, L. Knörr, T. R. Ward and T. Rovis, Science, 2012, 338, 500–503 CrossRef CAS PubMed.
- (a) N. Guimond, C. Gouliaras and K. Fagnou, J. Am. Chem. Soc., 2010, 132, 6908–6909 CrossRef CAS PubMed; (b) N. Guimond, S. I. Gorelsky and K. Fagnou, J. Am. Chem. Soc., 2011, 133, 6449–6457 CrossRef CAS PubMed; (c) B. Ye and N. Cramer, Science, 2012, 338, 504–506 CrossRef CAS PubMed; (d) S. Rakshit, C. Grohmann, T. Besset and F. Glorius, J. Am. Chem. Soc., 2011, 133, 2350–2353 CrossRef CAS PubMed; (e) H. Wang and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 7318–7322 CrossRef CAS PubMed; (f) H. Wang, C. Grohmann, C. Nimphius and F. Glorius, J. Am. Chem. Soc., 2012, 134, 19592–19595 CrossRef CAS PubMed.
- Z.-W. Zhang, A. Lin and J. Yang, J. Org. Chem., 2014, 79, 7041–7050 CrossRef CAS PubMed.
- D. Kalyani, A. R. Dick, W. Q. Anani and M. S. Sanford, Tetrahedron, 2006, 62, 11483–11498 CrossRef CAS.
- T. J. Donohoe and R. D. C. Pullin, Chem. Commun., 2012, 48, 11924–11938 RSC.
- J. M. Alderson, A. M. Phelps, R. J. Scamp, N. S. Dolan and J. M. Schomaker, J. Am. Chem. Soc., 2014, 136, 16720–16723 CrossRef CAS PubMed.
- S. Hwang, Y. Lee, P. H. Lee and S. Shin, Tetrahedron Lett., 2009, 50, 2305–2308 CrossRef CAS.
- M. Chaumontet, R. Piccardi and O. Baudoin, Angew. Chem., Int. Ed., 2009, 48, 179–182 CrossRef CAS PubMed.
- H. Gao and J. Zhang, Adv. Synth. Catal., 2009, 351, 85–88 CrossRef CAS.
- T. Zdrojewski and A. Jończyk, Tetrahedron, 1995, 51, 12439–12444 CrossRef CAS.
- E. Dubost, C. Fossey, T. Cailly, S. Rault and F. Fabis, J. Org. Chem., 2011, 76, 6414–6420 CrossRef CAS PubMed.
- S. Kawamorita, T. Miyazaki, H. Ohmiya, T. Iwai and M. Sawamura, J. Am. Chem. Soc., 2011, 133, 19310–19313 CrossRef CAS PubMed.
- Y. Chu, Z. Shan, D. Liu and N. Sun, J. Org. Chem., 2006, 71, 3998–4001 CrossRef CAS PubMed.
- Y.-F. Wang, K. K. Toh, J.-Y. Lee and S. Chiba, Angew. Chem., Int. Ed., 2011, 50, 5927–5931 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Copies of 1H and 13C NMR spectra for all novel compounds; crystallographic details for compound 4n, 4z, 5a, 5b and 15f. CCDC 1422535–1422539. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ob02320c |
| ‡ Current Address: Department of Chemistry, The University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, United Kingdom |
| This journal is © The Royal Society of Chemistry 2016 |