
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Efficient access to conjugated 4,4′-bipyridinium oligomers using the Zincke reaction: synthesis, spectroscopic and electrochemical properties†
Long
Chen
a,
Helen
Willcock
b,
Christopher J.
Wedge
 c,
František
Hartl
a,
Howard M.
Colquhoun
*a and
Barnaby W.
Greenland
*d
c,
František
Hartl
a,
Howard M.
Colquhoun
*a and
Barnaby W.
Greenland
*d
aDepartment of Chemistry, University of Reading, Whiteknights, Reading, RG6 6AD, UK. E-mail: h.m.colquhoun@reading.ac.uk
bDepartment of Materials, Loughborough University, Epinal Way, Loughborough, LE11 3TU, UK
cDepartment of Physics, University of Warwick, Coventry, CV4 7AL, UK
dSchool of Pharmacy, University of Reading, Whiteknights, Reading, RG6 6AH, UK. E-mail: b.w.greenland@reading.ac.uk
First published on 20th November 2015
Abstract
The cyclocondensation reaction between rigid, electron-rich aromatic diamines and 1,1′-bis(2,4-dinitrophenyl)-4,4′-bipyridinium (Zincke) salts has been harnessed to produce a series of conjugated oligomers containing up to twelve aromatic/heterocyclic residues. These oligomers exhibit discrete, multiple redox processes accompanied by dramatic changes in electronic absorption spectra.
Introduction
Bipyridinium residues are amongst the most intensively studied building blocks in supramolecular chemistry.1–3 They have found widespread application in molecular systems that exhibit controlled switching,4–8 rotational motion9–11 and potential for data storage.12–15 Many of these complex nanosystems harness the controllable electrochemical properties of 4,4′-bipyridinium ions (“viologens”) in either their synthesis or application.16 The reversible one- and two-electron reductions of 4,4′-bipyridinium derivatives (see ESI – Scheme 1†) are accompanied by a dramatic change in the UV-vis absorption spectra of the materials, an effect which has been widely studied in the context of electrochromic devices.4,17–19 The colour of cationic bipyridinium residues is highly dependent on the electronic ground state energy of the molecule, which can be finely tuned by varying the substituents.20–24There have been a number of studies concerning the synthesis of oligomers and polymeric materials containing 4,4′-bipyridine units in the backbone. Typically, the electroactive 4,4′-bipyridinium groups are separated by at least one methylene residue.25–28 This structural arrangement precludes electronic communication between the bipyridinium groups.29 In addition, there have been several reports of the synthesis of so-called “extended viologens” whereby two pyridinium residues are separated by increasing numbers of aromatic rings.30–40 These have rapidly found widespread application in the construction of supramolecular complexes.41–46 There are also examples of (di)phenyl viologen structures in which the conjugation length has been extended by the addition of an aromatic residue to the N-termini of the viologen.29,47–53 Small molecules containing 4,4′- or 3,3′-bipyridine units,36,54–57 and polymers58 with 3,3′-bipyridine residues have been shown to exhibit interesting conductivity properties in the solid state.
Our own work in this area has previously concentrated on the synthesis of 4,4′-bipyridinium-containing macrocycles such as 4 (Scheme 1). We synthesized a family of related macrocycles by exploiting the Zincke reaction,59 in which the condensation of nucleophilic amines with N-(2,4-dinitrophenyl)pyridinium salts resulted in efficient access to N-substituted pyridinium residues.60 These macrocycles underwent one-electron reduction both chemically [e.g., by triethylamine (TEA)] and electrochemically, to yield the corresponding radical cations such as 5.60 The unpaired spin density was found to be delocalized over four aromatic/heterocyclic rings, but was prevented from more extensive delocalization by the break in conjugation as a consequence of the ether linkages in these macrocyclic systems.
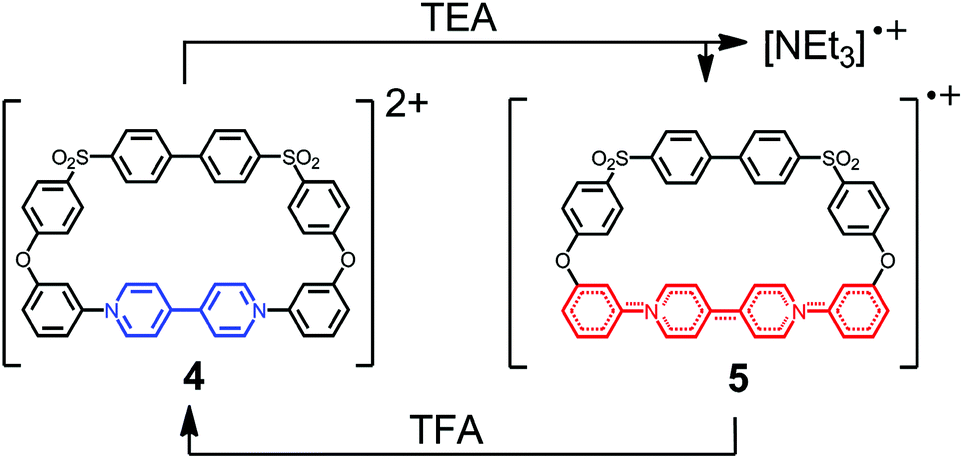 | ||
| Scheme 1 Reduction of dicationic macrocycle 4 to radical mono-cation 5 in which the unpaired electron is delocalized over the four aromatic rings highlighted in red. | ||
Here we report the syntheses and spectroelectrochemical analyses of a series of redox-active oligomers containing progressively increasing numbers of 4,4′-bipyridinium units, culminating in the synthesis of a hexa-cationic trimer containing twelve aromatic/heterocyclic residues.
Results and discussion
The syntheses of the new family of 4,4′-bipyridinium oligomers are shown in Scheme 2. The route to the targeted oligomers started from 1,1′-bis(2,4-dinitrophenyl)-[4,4′-bipyridin]-1,1′-diium dichloride (6), which can be readily prepared on a multigram scale.61 As an example of the efficiency of the Zincke reaction, the reaction of Zincke salt 6 with dimethyl 5-aminoisophthalate 7 afforded unimer 1 in 93% (isolated) yield. | ||
| Scheme 2 Synthesis of conjugated 4,4′-bipyridinium oligomers 1, 2 and 3. | ||
Higher molecular-weight oligomers in the series were accessed through intermediate 8 that was produced in 59% yield in two steps from known precursor 9,62 using simple extraction and crystallization procedures for purification. Condensation of two equivalents of 8 with the electron-rich aromatic diamine 10 resulted in the formation of dimer 2, in 77% yield. In contrast, reaction of three equivalents of 10 with 6 gave diamine 11 (95%) that was converted smoothly to trimer 3 by reaction with two equivalents of precursor 8 (72.5%), which was isolated as its hexafluorophosphate salt. Significant quantities of all three oligomers (e.g., ca. 1 g of trimer 3) could be obtained using these protocols, without recourse to column chromatography until the final step.
Electronic absorption spectra of 1, 2 and 3 in dimethylformamide (DMF) are presented in Fig. 1. The main difference between the spectra of 1 and 2 is the new absorption band of the dimer at ca. 380 nm, tailing into the visible region. Its intensity increases markedly with oligomer length. At the same time the dominant UV absorption at ca. 300 nm shifts slightly to higher energy.
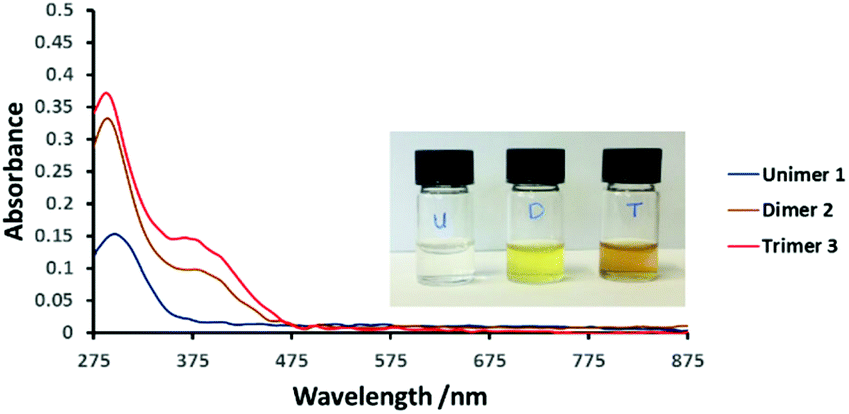 | ||
| Fig. 1 UV-Vis spectra of compounds 1, 2 and 3 in dimethylformamide (DMF, 0.2 mM) at 25 °C. Inset: Photograph of the compounds in DMF solution (0.2 mM). | ||
We next studied the chemically reversible redox process using TEA and trifluoroacetic acid (TFA), as described previously for the macrocyclic systems.60Fig. 2(A) shows the 1H NMR spectrum of an orange solution of trimer 3, which exhibits clearly resolved signals in the aromatic region (7.5 to 10.0 ppm), as well as two distinct resonances from the methoxy-ether and methoxy-ester residues (3.8 and 4.0 ppm).
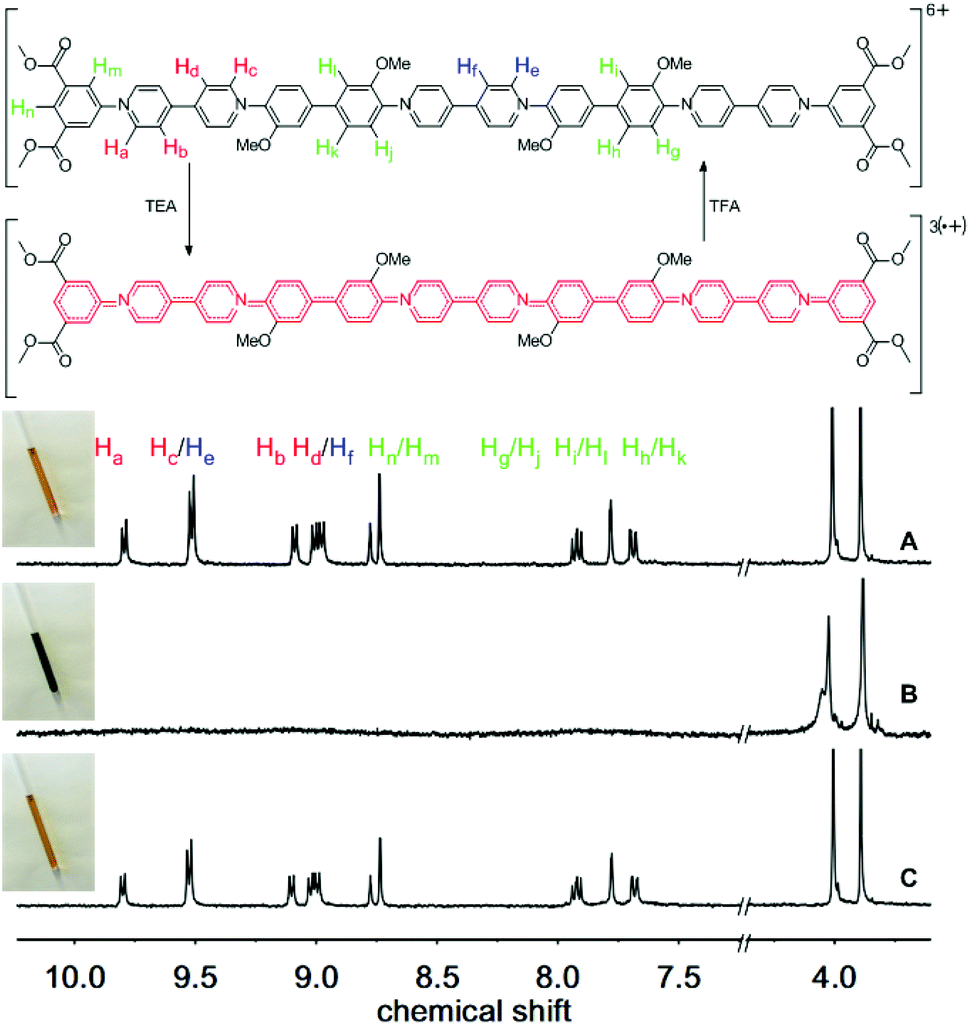 | ||
| Fig. 2 Chemical reduction (3× one-electron) and reoxidation of trimer 3 using TEA and TFA, respectively. The aromatic and heterocyclic rings highlighted in red indicate the region of unpaired spin density (not bond order). 1H NMR spectra and photographs of the samples: (A) trimer 3 in acetone-d6, (B) after addition of 20 equiv. of TEA and (C) subsequent to addition of an excess of TFA to B. | ||
The protons of the pyridinium residues at the termini of the oligomer, closest to the electron withdrawing ester groups, resonate at lower field (Ha at 9.80 ppm) compared to the signals from the bipyridinium residue in the central section (protons He, at 9.52 ppm). Thus, the three bipyridinium residues are in two distinct environments: two at the termini of the oligomer and one in the centre.
Upon addition of excess triethylamine, all the signals corresponding to the protons of the aromatic and heterocyclic rings disappear, as a consequence of the formation of a paramagnetic tris(radical cation) (Fig. 2B). This change in the 1H NMR spectrum is accompanied by a transformation in the color of the solution to a deep green, which is characteristic of the formation of radical cationic chromophores of this type.63 However, the 1H NMR signals corresponding to the non-conjugated methoxy groups remain visible at ca. 3.8 and 4.0 ppm. Addition of TFA to the tris(radical cation) not only regenerates the original color of the solution but also restores the missing signals in the aromatic region of the 1H NMR spectrum (Fig. 2C; see also the ESI – Fig. S1 and S2† for 1 and 2, respectively).
The radical cationic redox states of compounds 1, 2 and 3 were investigated by electron paramagnetic resonance (EPR) spectroscopy in acetone at room temperature. The EPR spectrum of 1˙+ centred at g = 2.0034(3) is characteristic of a viologen radical cation, showing well-resolved hyperfine splittings arising from a pair of equivalent 14N nuclei and multiple groups of equivalent 1H nuclei (Fig. 3).64,65 The bis(radical cationic) form of dimer 2 and the tris(radical cationic) form of trimer 3 are also EPR-active. This indicates that the electron spins of the dimer biradical do not exclusively pair to form a diamagnetic singlet state, though a half-field transition characteristic of a triplet state was not observed. The EPR signal of 22(˙+) exhibits the same broad envelope as 1˙+, but the hyperfine structure is apparent only through weak shoulders. The EPR spectrum of 33(˙+) also has unresolved hyperfine shoulders, though the peak-to-peak linewidth has narrowed. The changes in the spectral shape from unimer to dimer to trimer are characteristic of successive broadening of the hyperfine spectrum as may arise from increasingly rapid Heisenberg spin exchange or electron transfer processes.66
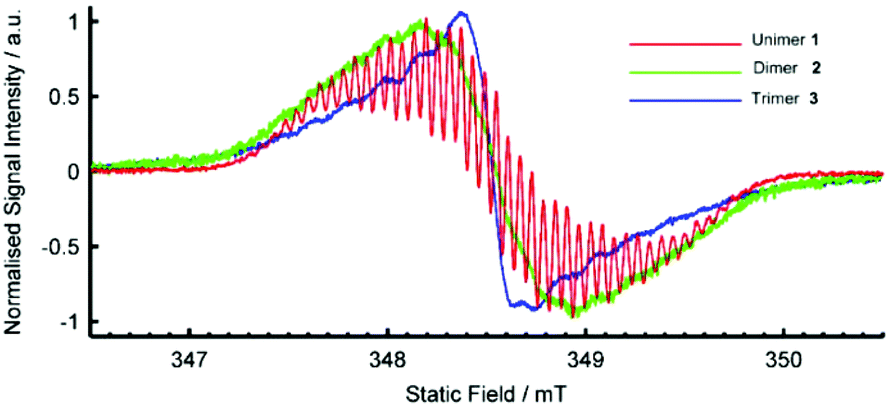 | ||
| Fig. 3 EPR Spectra of radical species generated from 1, 2 and 3 (1 mM) by addition of excess TEA in deaerated acetone solution at room temperature. | ||
The electrochemical reduction of 1, 2 and 3 was studied by cyclic voltammetry (CV) at a polished glassy carbon disc electrode, using anhydrous DMF as solvent, containing 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6) as supporting electrolyte. Ferrocene was used as internal reference. Cyclic voltammograms of compounds 1, 2 and 3 are shown in Fig. 4 and Table 1. Two reversible one-electron cathodic waves for unimer 1 occur at E1/2 = −0.63 and −0.84 V vs. Fc/Fc+. For dimer 2, these waves are found at E1/2 = −0.63 and −0.92 V. The data indicate that the two viologen moieties in tetracationic dimer 2 are electronically independent, as revealed by the identical first reduction potentials of 2 and dicationic compound 1, resulting from lack of conjugation through the twisted, central biphenyl rings. The second reduction potential of 2 is shifted to a more negative value compared to that of 1. This difference can be attributed to increased electronic conjugation in the more planar bis(radical cationic) form of 2 generated at the first cathodic wave.
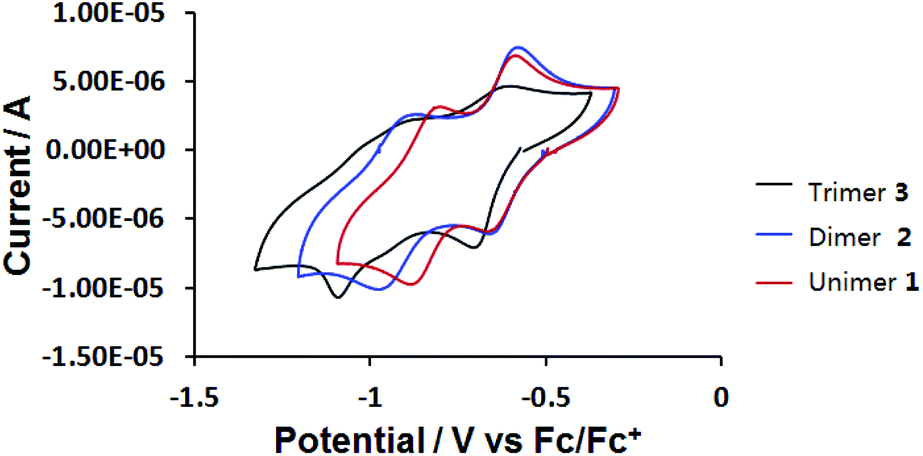 | ||
| Fig. 4 Cyclic voltammograms of 0.2 mM compounds 1, 2 and 3 at a glassy carbon disc (d = 2 mm) electrode in anhydrous DMF at v = 500 mV s−1. | ||
| CV = 0.5 V s−1 | CV = 0.5 V s−1 | |
|---|---|---|
| E 11/2 (V) vs. Fc/Fc+ | E 21/2 (V) vs. Fc/Fc+ | |
| Unimer 1 | −0.63 | −0.84 |
| Dimer 2 | −0.63 | −0.92 |
| Trimer 3 | −0.66 | −1.06 |
Hexacationic compound 3 is reduced at only a slightly more negative potential than 1 or 2 (E1/2 = −0.66 V, ΔEp = 100 mV). As observed for 2, all three viologen units in 3 are reduced to their corresponding radical cations at the same electrode potential, suggesting that the viologens are twisted with respect to the chain direction and are therefore electronically independent. The cyclic voltammogram of 3 indicates a strong influence of adsorption phenomena at this scan rate (resolved cathodic waves and diminished anodic counter-waves). Although the resolution of the CV improved slightly at higher scan rates (v = 3 V s−1; Fig. S8 in the ESI†), the rather unusual second cathodic step which ultimately converts the tris(radical cationic) form of 3 to the neutral species, remains indistinct.
Thus, to further investigate the electrochemical properties of 3, a square wave voltammogram was acquired. Under these conditions three distinct reduction events were resolved (signals A, B and C in Fig. 5 and Table 2). Although the peak current of square wave voltammetry (SWV) signals can be influenced by solubility effects and molecular re-organisation during reduction, it would appear that reduction steps for the single central (signal B, Fig. 5) and two terminal bipyridinium units (signal C, Fig. 5) to the neutral, quinoidal species can, nevertheless, be resolved during this experiment.
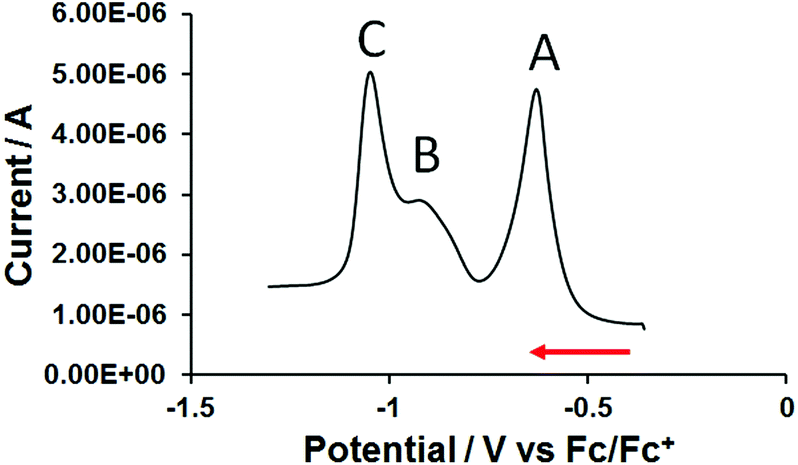 | ||
| Fig. 5 Square wave voltammogram of 0.2 mM trimer 3 on a glassy carbon disc electrode in anhydrous DMF/0.1 M TBAPF6. | ||
| SQW, f = 10 Hz, tp = 25 mV | CV, v = 0.5 V s−1 |
|---|---|
| E p (V) vs. Fc/Fc+ | E 1/2 (V) vs. Fc/Fc+ |
| −0.65 | −0.66 |
| −0.93 | Unresolved |
| −1.05 | −1.06 |
Infra-red spectroelectrochemistry (IR-SEC) was carried out on unimer 1 within an OTTLE cell67 (Fig. S6 in the ESI†) to gain insight into the changes in the conjugated structure throughout the redox cycle. Formation of the viologen radical cation was accompanied by the appearance of a strong ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) band at 1639 cm−1 associated with this species. There was, however, negligible change in the wavenumber of the ν(CO) band at 1734 cm−1 arising from the terminal ester groups. This observation suggests that the ester groups are not significantly conjugated with the aromatic system, so that their influence on the reduction potential will only be minor. Unfortunately, IR-SEC experiments with 2 and 3 were precluded by poor solubility of the reduced species in DMF at the high concentrations required to give reasonable absorbance values.
C) band at 1639 cm−1 associated with this species. There was, however, negligible change in the wavenumber of the ν(CO) band at 1734 cm−1 arising from the terminal ester groups. This observation suggests that the ester groups are not significantly conjugated with the aromatic system, so that their influence on the reduction potential will only be minor. Unfortunately, IR-SEC experiments with 2 and 3 were precluded by poor solubility of the reduced species in DMF at the high concentrations required to give reasonable absorbance values.
In order to study the redox-induced changes in electronic spectra of these systems, thin-layer ultraviolet-visible spectroelectrochemical (UV-vis SEC) measurements were carried out at 293 K with 0.2 mM 1, 2 or 3 in DMF/0.1 M TBAPF6 (Fig. S11 in ESI,†6 and 7). The observed spectral changes for all three species exhibit isosbestic points, excluding the possibility of side-reactions on the timescale of the experiment. All cathodic steps were fully reversible and parent electronic absorption spectra were recovered upon reoxidation. Fig. 6 show the UV-vis spectral monitoring of the electrochemical reduction of unimer 1 and dimer 2 at the two well-defined cathodic waves shown in Fig. 4. The tetracationic species shows two absorption bands at 290 and 375 nm (Fig. 6A). After completion of the first reduction step, the new absorption bands at 450, 600 and 715 nm are indicative of the bis(radical cation) (Fig. 6A and B).18 Continuation of the cathodic sweep results in a second transformation in the electronic absorption spectrum due to formation of the neutral redox form absorbing at 380 and 503 nm (Fig. 6B; see also the ESI – Fig. S11† for 1).
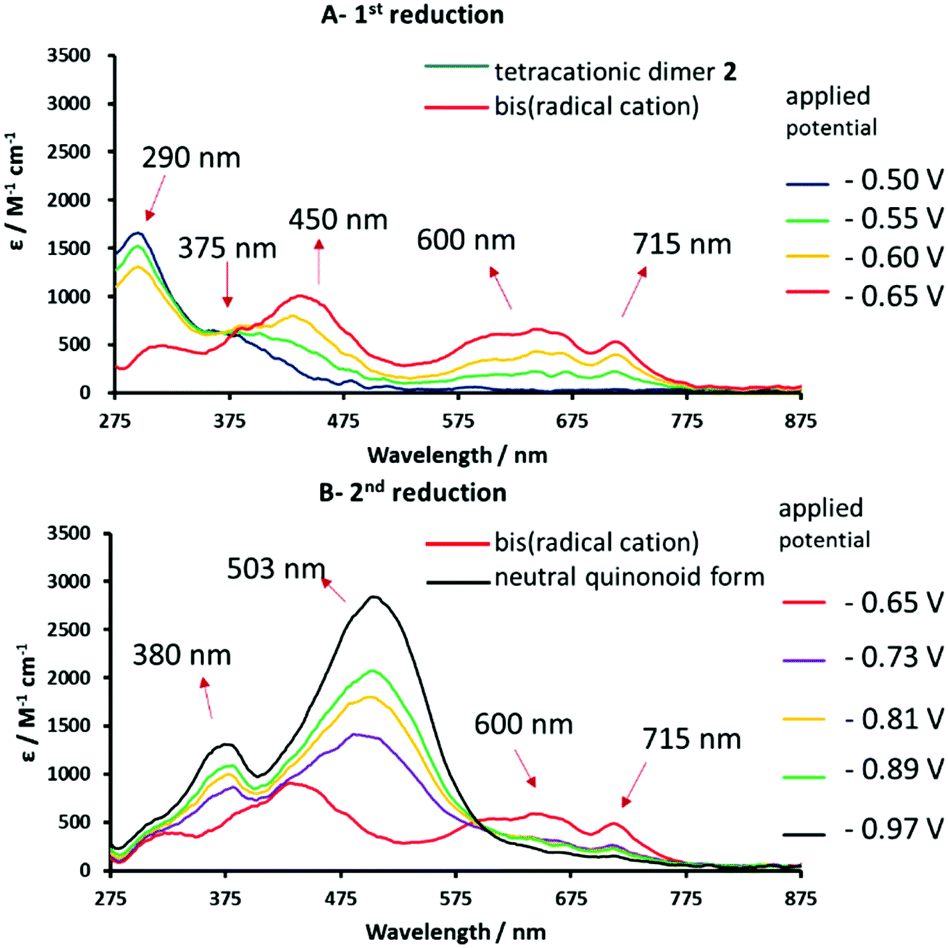 | ||
| Fig. 6 Reversible UV-vis spectral changes accompanying the stepwise 1e reduction of tetracationic dimer 2 to the corresponding bis(radical cation) (spectrum A) and the neutral quinonoid form (spectrum B). Spectra recorded in anhydrous DMF/0.1 M TABPF6, using an OTTLE cell.67 | ||
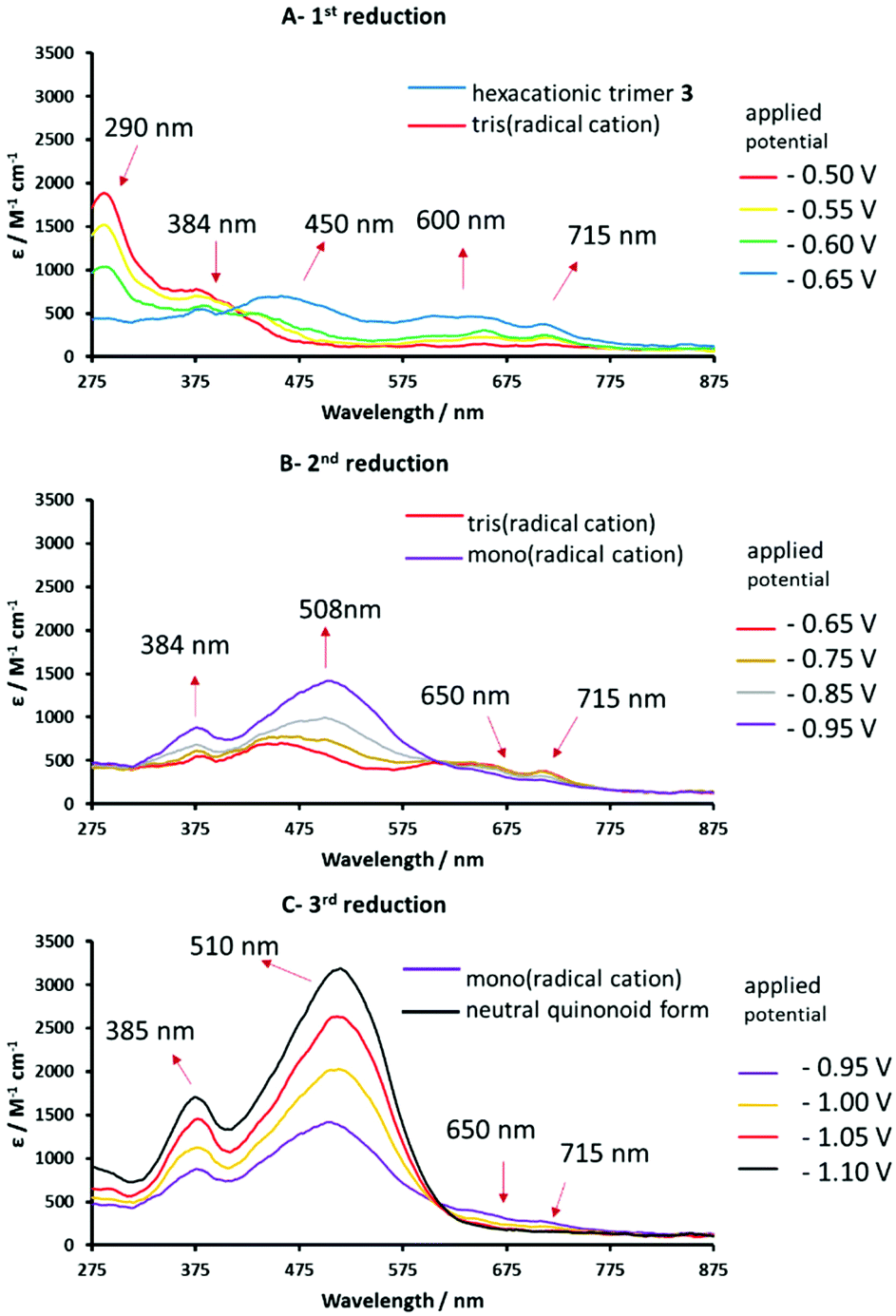 | ||
| Fig. 7 Reversible UV-vis spectral changes accompanying the stepwise reduction of hexacationic 3 to the corresponding tris(radical cation) (spectrum A) and the mono(radical cation) (spectrum B) and the neutral quinoidal form (spectrum C), recorded in anhydrous DMF/0.1 M TABPF6 using an OTTLE cell.67 | ||
Spectroelectrochemical (UV-vis) data for the four stable redox forms of trimeric species 3 were recorded at different reduction potentials corresponding to the three cathodic waves measured previously (Table 2). Conversion of the hexacationic species to the tris(radical cation) results in a spectrum similar to that observed for the analogous radical cation of 1 and bis(radical cation) of 2, with absorption maxima at 450, 600, 650 and 715 nm (Fig. 7A). Continued decrease in the applied cathodic potential results in the formation of a second stable species associated with absorption bands at 384 and 508 nm, which confirm the formation of fully reduced, quinoidal heterocycles. The absorption bands of the radical cation above 600 nm are diminished but still evident (Fig. 7B). This spectrum is consistent with a mixed oxidation state species containing both radical cationic and quinoidal heterocyclic species. Further lowering of the applied potential delivers the final species with intense absorption bands at 385 and 510 nm and no significant absorption bands above 600 nm (Fig. 7C), indicative of a fully reduced, neutral species.
Fig. 8 shows an overlaid plot of the UV-vis spectra of the four stable, spectroscopically distinct species in the redox cycle of 3. The increase in intensity of the absorption band of the quinoidal bipyridinium species at 515 nm as the molecule becomes reduced from the tris(radical cation) to the mixed oxidation state system is 870 au (Arrow A). This compares to a change in intensity of 1800 au when moving from the mixed oxidation state species to the fully reduced, neutral trimer (Arrow B). These data suggest that the intermediate species, which is formed at approximately −0.9 V (Table 2), contains a single quinoidal bipyridinium species compared to the three quinoidal species present in the ultimate, neutral form of 3. Data from the SWV (Fig. 5) and the UV-vis SEC (Fig. 7 and 8) of 3, supported by thin-layer cyclic voltammetry carried out during the latter experiment (see S14 in the ESI†), prove that the reduction of the three bipyridinium groups in the hexacationic trimer 3, giving a tris(radical cation), occurs by the addition of three electrons at the same potential (−0.65 V). However, the subsequent reduction of the tris(radical cation) to the neutral species occurs in a stepwise manner, by the addition of a single electron (at −0.93 V) followed by two electrons (at −1.05 V). This can be accounted for if the central bipyridinium radical cation is reduced first, followed by the two terminal radical cations (Scheme 3).
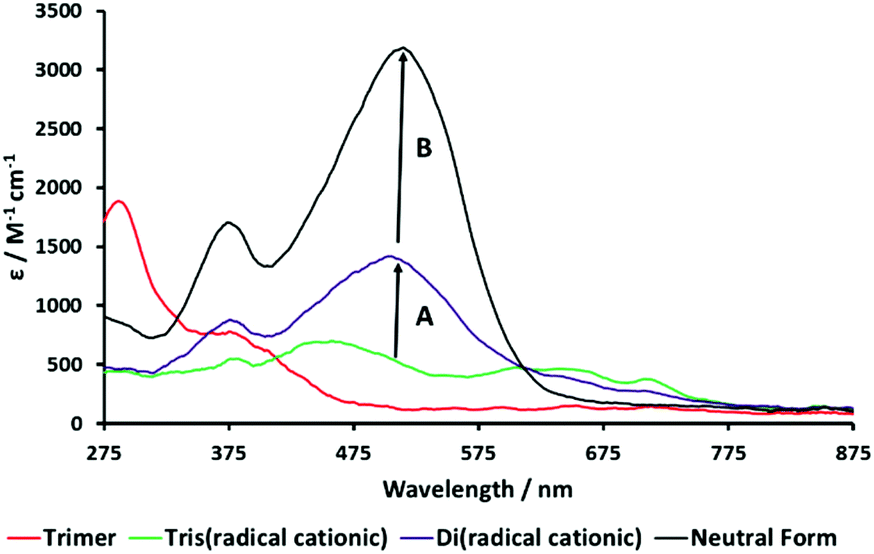 | ||
Fig. 8 UV-vis spectral changes accompanying the stepwise reduction of hexacationic 3 to a neutral species. The change in intensity of the band at 515 nm associated with the formation of quinoidal bipyridinium residues occurs in the ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 (arrows A and B respectively) during two-step reduction of the tris(radical cationic) form of 3 to the neutral species. :2 (arrows A and B respectively) during two-step reduction of the tris(radical cationic) form of 3 to the neutral species. | ||
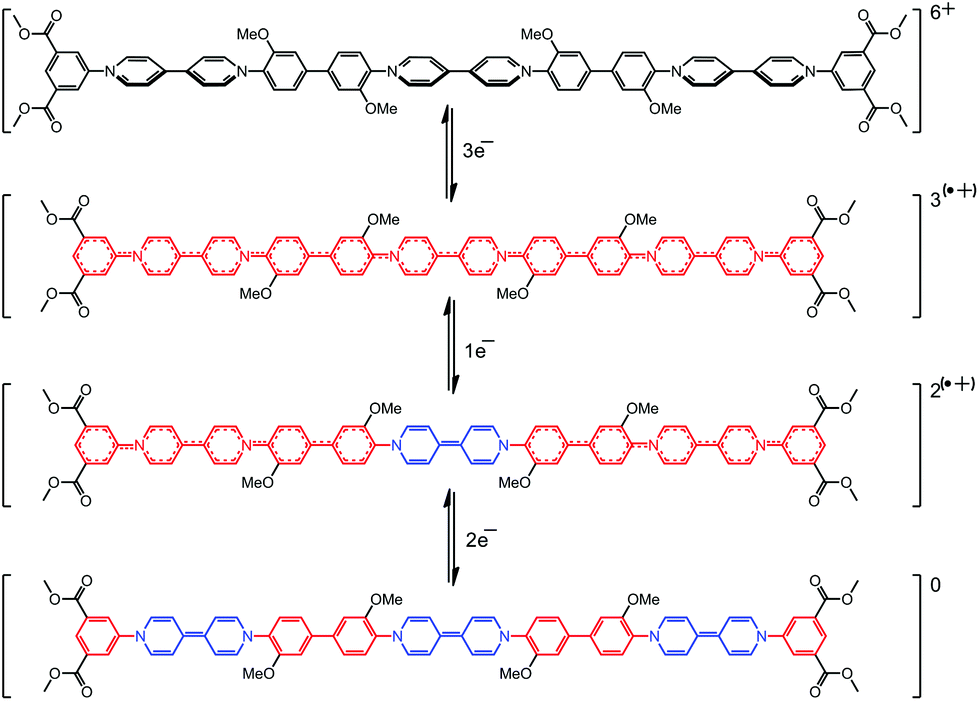 | ||
| Scheme 3 The structures of the stable intermediates observed during the electrochemical interconversions of trimer 3: tris(dicationic), tris(radical cationic), di(radical cationic) and neutral, quinoidal forms (top to bottom respectively). | ||
Conclusion
In this study we report an efficient synthesis of 4,4′-bipyridinium-based aromatic oligomers (1–3) and their spectro-electrochemical properties. Specifically, we use a controlled stepwise synthesis to produce single molecules with three 4,4′-bipyridinium residues containing, for the first time, twelve conjugated aromatic/heterocylic rings. All the compounds undergo reversible stepwise two-electron reduction of each viologen moiety. Addition of the first electron to each viologen to form a radical cation produces a more planar structure with significantly greater electronic communication between the aromatic/heterocyclic groups along the length of each oligomer. Consequently, the tris(radical cation) of trimer 3 is reduced to the neutral, quinoidal form in two well-resolved cathodic steps.Combined results from CV and UV-vis SEC demonstrate that, in these three oligomers, their ground states are not planar, which results in a twisted configuration between the phenyls and viologens. When going to the radical cationic state, because of the greater extent of π conjugation induced by their planar structure, the second E1/2 tends to a slightly more negative value. Spectroscopically, the UV-vis SEC result of 3 demonstrated that the apparently more electron rich, central radical cationic bipyridinium moiety is reduced to the quinoidal, neutral form before the two terminal bipyridinium units which appear comparatively electron poor. In support, computational studies of related conjugated oligomers and polymers have also shown that that maxima for their HOMO/LUMO coefficients are found at the centre of the molecule.68,69 If then the highest occupied spin orbital (HOSO) of the tris(radical cation) form of 3 is also localised in the centre of the molecule then the central 4,4′-bipyridinium would be reduced prior to the higher energy terminal radical cationic units. In addition, the UV-vis SEC measurements have highlighted the potential of these new molecules to find application in display technologies where dramatic and reversible colour changes are required. Work in our laboratory is continued towards integrating conjugated 4,4′-bipyridinium residues into more extended materials systems such as MOFs, mechanically interlocked molecules and high-MW polymers.
Experimental
General methods
Starting materials were purchased from Sigma Aldrich and Alfa Aesar and used without further purification unless otherwise stated. Anhydrous N,N-dimethylformamide (DMF) (Alfa Aesar, 99.8%, packaged under argon) was used as received. Tetra-n-butylammonium hexafluorophosphate (TBAPF6) was recrystallized twice from absolute ethanol and dried at 80 °C under vacuum overnight.Compounds 661 and 962 were synthesized as described previously. All reactions were carried out in oven-dried glassware under dry nitrogen. 1H and 13C NMR were acquired on a Bruker DPX-400 spectrometer operating at 400 MHz and 100 MHz respectively or on a Bruker AV-700 spectrometer operating as 175 MHz for 13C nuclei. Residual 1H signals from the solvent were used as internal calibrants. Infrared spectra were recorded on a Perkin Elmer 17 20-X spectrometer from thin films cast from acetone, and major absorption bands are reported in wavenumbers (cm−1). For spectroelectrochemical (SEC) measurements, IR spectra were recorded on a Bruker Vertex 70v FTIR spectrometer. UV-Vis spectra were acquired on a Scinco S-3100 diode-array spectrophotometer.
Cyclic voltammetry (CV) measurements were performed on 0.2 mM solution of the compound 1, 2 and 3 in anhydrous DMF containing 0.1 M TBAPF6 as supporting electrolyte with a single-compartment three-electrode cell equipped with glassy carbon disc (d = 2 mm) working, coiled platinum wire auxiliary and coiled Ag wire pseudoreference electrodes. Spectroelectrochemical measurements were carried out at 293 K, using an optically transparent thin-layer electrochemical (OTTLE) cell equipped with Pt minigrid working and auxiliary electrodes, a silver microwire pseudoreference electrode, and CaF2 windows. The course of the spectroelectrochemical experiment was monitored by thin-layer cyclic voltammetry conducted with an EmStat 3 (PalmSens BV) potentiostat. The applied potentials stated in the UV-vis SEC are approximated from the CV of each compound. All the melting points were determined on a Gallenkamp melting point apparatus. Mass spectra were recorded Thermo Scientific LQT Orbitrap XL under conditions of electrospray ionisation. X-band EPR spectra were acquired on a Bruker EMX spectrometer using a TM110 cylindrical mode resonator (ER 4103TM). Due to the high dielectric loss of the solvent all samples were contained in 1 mm internal diameter quartz tubes (Wilmad 712-SQ). To allow resolution of hyperfine structure, samples were deoxygenated by a minimum of three freeze–pump–thaw cycles, and spectra were recorded with a 20 μT modulation amplitude. A DPPH standard [g = 2.0036(3)] was used to obtain accurate g-values.70
Synthesis of dicationic unimer 1
1,1′-Bis(2,4-dinitrophenyl)-4,4′-bipyridinium dichloride (2.22 g, 3.96 mmol) was dissolved in a mixture of ethanol (50 mL) and water (50 mL) with excess dimethyl 5-aminoisophthalate (3.52 g, 16.83 mmol). The reaction mixture was heated to reflux and stirred vigorously for 3 days. After cooling to room temperature, the solvent was evaporated, the yellow solid was washed with THF (200 mL) and EtOAc (200 mL). Anion exchange to PF6− was achieved by dissolving the solid (4.60 g) in a mixture of water/methanol (8:2, v/v, 50 mL) and adding 10 g of NH4PF6. The resulting precipitate was filtered off, washed with 300 mL of water and 100 mL of ethyl acetate, and then dried under high vacuum overnight to yield 1 as a white solid (3.06 g, 93%). 1H NMR (acetone-d6, 400 MHz) δ 9.82 (d, J = 6.8 Hz, 4H), 9.14 (d, J = 6.8 Hz, 4H), 8.88 (t, J = 1.3 Hz, 2H), 8.83 (d, J = 1.2 Hz, 4H), 3.99 (s, 12H). 13C NMR (acetone-d6, 100 MHz) δ 164.9, 152.0, 147.7, 144.1, 133.9, 133.4, 131.0, 128.4, 53.5. IR (cm−1) ν = 1730 (α,β-unsaturated ester CO). MS (m/z) calc. for (C30H25N2O8)+: 541.1605, found 541.1601; calc. for (C30H26N2O8)2+: 271.0839, found 271.0840.
Synthesis of 1-(3,5-bis(methoxycarbonyl)phenyl)-[4,4′-bipyridin]-1-ium dichloride 12
1-(2,4-Dinitrophenyl)-[4,4′-bipyridin]-1-ium chloride (9) (3.59 g, 10 mmol) was dissolved in ethanol (50 mL) with a large excess of dimethyl 5-aminoisophthalate (5.04 g, 24.9 mmol). The mixture was heated to reflux and stirred vigorously for 2 days. After cooling to room temperature, the solvent was evaporated and the product reprecipitated twice with ethyl acetate from methanol. The precipitate was collected by filtration, washed with THF (200 mL), and dried in vacuo to yield 12 as a light green solid (3.61 g, 94%) M.p. 243–246 °C (dec). 1H NMR (D2O, 400 MHz) δ 9.29 (d, J = 6.8 Hz, 2H), 8.94 (t, J = 1.2 Hz, 1H), 8.81 (dd, J = 4.8 and 1.6 Hz, 2H), 8.66 (d, J = 1.2 Hz, 2H), 8.64 (d, J = 6.8 Hz, 2H), 8.01 (dd, J = 1.6 and 4.8 Hz, 2H), 4.01 (s, 6H). 13C NMR (D2O, 100 MHz) δ 166.2, 155.5, 149.9, 144.9, 142.5, 142.3, 132.8, 132.81, 132.78, 129.4, 126.2, 53.3. IR (cm−1) ν = 1736 (ester CO). MS (m/z) calc. for (C20H17N2O4)+: 349.1183, found 349.1181.
Synthesis of 8
1-(3,5-Bis(methoxycarbonyl)phenyl)-[4,4′-bipyridin]-1-ium dichloride (6) (1.35 g, 3.51 mmol) was dissolved in 8 mL of EtOH with a large excess of 1-chloro-2,4-dinitrobenzene (10.0 g, 49.4 mmol). The solvent was heated to reflux and stirred strongly for 3 days. After cooling to room temperature, the crystals were filtered off and washed with THF (2 × 100 mL), further washed with EtOAc (2 × 100 mL), and dried in vacuo to yield a light brown solid (1.41 g, 59%). M.p. 216–218 °C. 1H NMR (D2O, 400 MHz) δ 9.53 (d, J = 6.8 Hz, 2H), 9.47 (d, J = 7.2 Hz, 2H), 9.42 (d, J = 2.4 Hz, 2H), 8.99 (t, J = 1.2 Hz, 1H), 8.96 (dd, J = 2.4 and 8.8 Hz, 1H), 8.919 (d, J = 9.6 Hz, 2H), 8.912 (d, J = 9.6 Hz, 2H), 8.72 (d, J = 1.2 Hz, 2H), 8.31 (d, J = 8.8 Hz, 2H), 4.03 (s, 6H). 13C NMR (D2O, 100 MHz) δ 168.7, 155.7, 153.6, 152.4, 149.3, 148.4, 145.3, 145.0, 140.8, 135.7, 135.5, 133.6, 133.2, 132.0. 130.1, 129.9, 125.3, 55.9. IR (cm−1) ν = 1715 (ester, CO), 1541 (NO2). MS (m/z) calc. for (C26H20N4O8)+: 561.1276, found 561.1266.
Synthesis of tetracationic dimer 2
1-(3,5-Bis(methoxycarbonyl)phenyl)-1′-(2,4-dinitrophenyl)-[4,4′bipyridin]-1,1′-diium chloride (8) (984 mg, 1.68 mmol) was dissolved in methanol (50 mL) with 3,3′-dimethoxybenzidine (204 mg, 0.84 mmol). The mixture was heated to reflux and stirred for 4 d. After cooling to room temperature, the solvent was evaporated, and the crude product was precipitated twice from MeOH with ethyl acetate to afford a brown solid. Anion exchange to PF6− was achieved by dissolving the solid in a mixture of water/methanol (10 mL) and adding NH4PF6 (3 g). The resulting precipitate was filtered off, washed with 500 mL of water and 300 mL of ethyl acetate, and then dried under high vacuum overnight to yield 2 as a yellow solid (960 mg, 77%). M.p. 312 °C. 1H NMR (acetone-d6 with 1% trifluoroacetic acid (TFA); the signal of TFA is not reported; 400 MHz) δ 9.92 (d, J = 6.8 Hz, 4H), 9.64 (d, J = 7.2 Hz, 4H), 9.21 (d, J = 6.8 Hz, 4H), 9.12 (d, J = 6.8 Hz, 4H), 8.90 (t, J = 1.4 Hz, 2H), 8.87 (d, J = 1.2 Hz, 4H), 8.04 (d, J = 8 Hz, 2H), 7.91 (d, J = 1.6 Hz, 2H), 7.69 (dd, J = 1.6 and 8.4 Hz, 2H), 4.13 (s, 6H), 4.02 (s, 12H). 13C NMR (CD3CN with 1% TFA; the signal of TFA is not included; 175 MHz) δ 165.3, 153.5, 152.2, 151.5, 148.4, 147.1, 145.4, 143.6, 134.3, 130.6, 128.5, 128.1, 121.6, 113.5, 27.9, 53.9. IR (cm−1) ν = 1722 (α,β-unsaturated ester CO). MS (m/z) calc. for (C54H46N4O10)4+: 227.5798, found 227.5798, calc. for (C54H45N4O10)3+: 303.1040, found 303.1040, calc. for (C54H44N4O10)2+: 3454.1523, found 454.1519.
Synthesis of 1,1′-bis(4′-amino-3,3′-dimethoxy-[1,1′-biphenyl]-4-yl)-[4,4′-bipyridine]-1,1′-diium chloride 11
Zincke salt (6) (570 mg, 1.00 mmol) was dissolved in 50 mL of methanol/water (5:1, v/v) with 3,3′-dimethoxybenzidine (732 mg, 3 mmol). The reaction mixture was heated with stirring at reflux for 3 days, and then concentrated to 3 mL and treated with THF (100 mL). The crude product was precipitated twice from MeOH/EtOAc (1:8, v/v). The precipitate was filtered off and dried under high vacuum to afford 11 as a deep purple solid (653 mg, 95%). M.p. 280 °C (dec.). 1H NMR (CD3OD, 400 MHz) δ 9.48 (d, J = 6.8 Hz, 2H), 9.19 (d, J = 6.8 Hz, 2H), 8.97 (t, J = 1.2 Hz, 1H), 8.82 (d, J = 6.8 Hz, 2H), 8.70 (d, J = 1.2 Hz, 2H), 8.68 (d, J = 7.5 Hz, 2H), 7.63 (d, J = 8.0 Hz, 1H), 7.44 (s, 1H), 7.42 (d, J = 8.0 Hz, 1H), 7.23 (d, J = 8.0 Hz, 1H), 7.20 (s, 1H), 6.92 (d, J = 8.0 Hz, 1H), 4.03 (s, 3H), 4.01 (s, 3H), 3.93 (s, 3H). 13C NMR (CD3OD, 100 MHz) δ 165.7, 153.4, 152.5, 151.4, 149.9, 148.8, 148.5, 147.5, 145.3, 144.4, 137.0, 136.7, 134.6, 133.9, 131.6, 130.8, 130.5, 128.5, 128.1, 127.8, 124.8, 121.5, 120.5, 117.5, 111.9, 110.6, 57.3, 56.4, 53.6. IR (cm−1) ν = 3350 (N–H, primary amine). MS (m/z) calc. for (C38H36N4O4)2+: 612.2731, found 612.2725.
Synthesis of hexacationic trimer 3
Aromatic diamine 11 (403 mg, 0.59 mmol) was dissolved in 100 mL of ethanol/water (1:1, v/v) with precursor (8) (704 mg, 1.20 mmol) and the solution was refluxed for 3 d. The solvent was concentrated to leave approximately 5 mL, and this was added to a rapidly stirred solution of NH4PF6 in a mixture of methanol and water (2:8, v/v, 30 mL). After stirring at room temperature for 1 h, the resulting precipitate was filtered off, washed with water (300 mL) and ethyl acetate (300 mL) and finally purified by column chromatography (elution gradient: acetonitrile to 1% NH4PF6 in acetonitrile w/v). Excess salts were removed by precipitating the crude product twice from acetonitrile/water and the product was then dried under high vacuum to give a black solid, 3, (920 mg, 72.5%). M.p. 325 °C (dec.). 1H NMR (acetone-d6 with 1% TFA; the signal of TFA is not reported; 400 MHz) δ 9.92 (d, J = 6.8 Hz, 4H), 9.64 (d, J = 7.2 Hz, 8H), 9.22 (d, J = 7.2 Hz, 4H), 9.13 (d, J = 6.8 Hz, 4H), 9.10 (d, J = 6.8 Hz, 4H), 8.89 (t, J = 1.2 Hz, 2H), 8.86 (d, J = 1.6 Hz, 4H), 8.05 (t, J = 7.2 Hz, 4H), 7.91 (s, 4H), 7.82 (dd, J = 1.6 and 8.0 Hz, 4H), 4.13 (s, 12H), 4.02 (s, 12H). 13C NMR (acetone-d6 with 1% TFA; the signal of TFA is not included; 175 MHz) δ 165.0, 158.7, 158.3, 153.6, 152.2, 151.9, 148.7, 148.7, 147.7, 145.4, 144.2, 133.9, 132.2, 131.0, 128.5, 128.4, 128.2, 128.2, 121.5, 120.2, 117.4, 114.5, 113.3, 57.5, 53.5. IR (cm−1) ν = 1723 (α,β-unsaturated ester, CO). MS (m/z) calc. for (C78H64N6O12)4+: 319.1140, found 319.1140; calc. for (C78H62N6O12)2+: 637.2207, found 637.2210.
Conflict of interest
The authors declare no competing financial interests.Acknowledgements
We thank the University of Reading for support of the Reading Spectroelectrochemistry Laboratory (Project D14-015).References
- D. B. Amabilino and J. F. Stoddart, Chem. Rev., 1995, 95, 2725–2828 CrossRef CAS.
- F. M. Raymo and J. F. Stoddart, Chem. Rev., 1999, 99, 1643–1663 CrossRef CAS PubMed.
- E. E. Havinga, W. Hoeve and H. Wynberg, Polym. Bull., 1992, 29, 119–126 CrossRef CAS.
- V. Jain, M. Khiterer, R. Montazami, H. M. Yochum, K. J. Shea and J. R. Heflin, ACS Appl. Mater. Interfaces, 2009, 1, 83–89 CAS.
- D. S. Guo, S. Chen, H. Qian, H. Q. Zhang and Y. Liu, Chem. Commun., 2010, 46, 2620–2622 RSC.
- A. Trabolsi, N. Khashab, A. C. Fahrenbach, D. C. Friedman, M. T. Colvin, K. K. Cotí, D. Benítez, E. Tkatchouk, J.-C. Olsen, M. E. Belowich, R. Carmielli, H. A. Khatib, W. A. Goddard, M. R. Wasielewski and J. F. Stoddart, Nat. Chem., 2010, 2, 42–49 CrossRef CAS PubMed.
- W. Zhang, E. DeIonno, W. R. Dichtel, L. Fang, A. Trabolsi, J.-C. Olsen, D. Benitez, J. R. Heath and J. F. Stoddart, J. Mater. Chem., 2011, 21, 1487–1495 RSC.
- C. Kahlfuss, E. Métay, M.-C. Duclos, M. Lemaire, A. Milet, E. Saint-Aman and C. Bucher, Chem. – Eur. J., 2014, 2090–2106 Search PubMed.
- O. Schiemann, N. J. Turro and J. K. Barton, J. Phys. Chem. B, 2000, 104, 7214–7220 CrossRef CAS.
- T. Ha and P. Tinnefeld, Annu. Rev. Phys. Chem., 2012, 63, 595–617 CrossRef CAS PubMed.
- H.-B. Cheng, Y.-M. Zhang, C. Xu and Y. Liu, Sci. Rep., 2014, 4 Search PubMed.
- T. Nakahira and M. Graetzel, J. Phys. Chem., 1984, 88, 4006–4010 CrossRef CAS.
- J. C. Barnes, A. C. Fahrenbach, D. Cao, S. M. Dyar, M. Frasconi, M. A. Giesener, D. Benítez, E. Tkatchouk, O. Chernyashevskyy, W. H. Shin, H. Li, S. Sampath, C. L. Stern, A. A. Sarjeant, K. J. Hartlieb, Z. Liu, R. Carmieli, Y. Y. Botros, J. W. Choi, A. M. Z. Slawin, J. B. Ketterson, M. R. Wasielewski, W. A. Goddard and J. F. Stoddart, Science, 2013, 339, 429–433 CrossRef CAS PubMed.
- B. L. Feringa, W. F. Jager and B. de Lange, Tetrahedron, 1993, 49, 8267–8310 CrossRef CAS.
- J. E. Green, J. Wook Choi, A. Boukai, Y. Bunimovich, E. Johnston-Halperin, E. DeIonno, Y. Luo, B. A. Sheriff, K. Xu, Y. Shik Shin, H.-R. Tseng, J. F. Stoddart and J. R. Heath, Nature, 2007, 445, 414–417 CrossRef CAS PubMed.
- J. Iehl, M. Frasconi, H. P. Jacquot de Rouville, N. Renaud, S. M. Dyar, N. L. Strutt, R. Carmieli, M. R. Wasielewski, M. A. Ratner, J. F. Nierengarten and J. F. Stoddart, Chem. Sci., 2013, 4, 1462–1469 RSC.
- C. L. Bird and A. T. Kuhn, Chem. Soc. Rev., 1981, 10, 49–82 RSC.
- R. J. Mortimer and T. S. Varley, Chem. Mater., 2011, 23, 4077–4082 CrossRef CAS.
- J. Palenzuela, A. Viñuales, I. Odriozola, G. Cabañero, H. J. Grande and V. Ruiz, ACS Appl. Mater. Interfaces, 2014, 6, 14562–14567 CAS.
- E. M. Kosower and J. L. Cotter, J. Am. Chem. Soc., 1964, 86, 5524–5527 CrossRef CAS.
- A. G. Evans, N. K. Dodson and N. H. Rees, J. Chem. Soc., Trans. 2, 1976, 859–863 CAS.
- I. P. Krainov, O. M. Tsyguleva and S. F. Kramarenko, Theor. Exp. Chem., 1988, 23, 699–704 CrossRef.
- P. M. S. Monk, The viologens: physicochemical properties, synthesis and applications of the salts of 4,4′-bipyridine, Wiley, 1998 Search PubMed.
- D. Barrios, R. Vergaz, J. C. Torres-Zafra, C. Vega, J. M. Sanchez-Pena and A. Vinuales, IEEE Photonics J., 2012, 4, 2105–2115 CrossRef.
- K. Wadhwa, S. Nuryyeva, A. C. Fahrenbach, M. Elhabiri, C. Platas-Iglesias and A. Trabolsi, J. Mater. Chem. C, 2013, 1, 2302–2307 RSC.
- Y.-C. Zhang, D.-W. Zhang, H. Wang, Y. Zhou and Z.-T. Li, Polym. Chem., 2015, 6, 4404–4408 RSC.
- K. Murugavel, Polym. Chem., 2014, 5, 5873–5884 RSC.
- M. Kathiresan, L. Walder, F. Ye and H. Reuter, Tetrahedron Lett., 2010, 51, 2188–2192 CrossRef CAS.
- I. Yamaguchi, H. Higashi, S. Kimura and M. Sato, Helv. Chim. Acta, 2010, 93, 819–828 CrossRef CAS.
- M. Hromadová, M. Valášek, N. Fanelli, H. N. Randriamahazaka and L. Pospíšil, J. Phys. Chem. C, 2014, 118, 9066–9072 Search PubMed.
- J. Romanova, V. Liegeois and B. Champagne, Phys. Chem. Chem. Phys., 2014, 16, 21721–21731 RSC.
- J. Romanova, V. Liégeois and B. Champagne, J. Phys. Chem. C, 2014, 118, 12469–12484 CAS.
- A. Beneduci, S. Cospito, A. Crispini, B. Gabriele, F. P. Nicoletta, L. Veltri and G. Chidichimo, J. Mater. Chem., 2013, 1, 2233–2240 CAS.
- J. Fortage, C. Peltier, C. Perruchot, Y. Takemoto, Y. Teki, F. Bedioui, V. Marvaud, G. Dupeyre, L. Pospíšil, C. Adamo, M. Hromadová, I. Ciofini and P. P. Lainé, J. Am. Chem. Soc., 2012, 134, 2691–2705 CrossRef CAS PubMed.
- M. E. Alberto, B. C. De Simone, S. Cospito, D. Imbardelli, L. Veltri, G. Chidichimo and N. Russo, Chem. Phys. Lett., 2012, 552, 141–145 CrossRef CAS.
- P. P. Lainé, F. Bedioui, F. Loiseau, C. Chiorboli and S. Campagna, J. Am. Chem. Soc., 2006, 128, 7510–7521 CrossRef PubMed.
- A. Funston, J. P. Kirby, J. R. Miller, L. Pospíšil, J. Fiedler, M. Hromadová, M. Gál, J. Pecka, M. Valášek, Z. Zawada, P. Rempala and J. Michl, J. Phys. Chem. A, 2005, 109, 10862–10869 CrossRef CAS PubMed.
- M. Pumera, J. Jindřich, M. Valášek and J. Pecka, Electrophoresis, 2005, 26, 4465–4467 CrossRef CAS PubMed.
- M. Valášek, J. Pecka, J. Jindřich, G. Calleja, P. R. Craig and J. Michl, J. Org. Chem., 2005, 70, 405–412 CrossRef PubMed.
- W. W. Porter, T. P. Vaid and A. L. Rheingold, J. Am. Chem. Soc., 2005, 127, 16559–16566 CrossRef CAS PubMed.
- S.-H. Chiu, A. M. Elizarov, P. T. Glink and J. F. Stoddart, Org. Lett., 2002, 4, 3561–3564 CrossRef CAS PubMed.
- P. R. Ashton, V. Baldoni, V. Balzani, A. Credi, H. D. A. Hoffmann, M. V. Martínez Díaz, F. M. Raymo, J. F. Stoddart and M. Venturi, Chem. – Eur. J., 2001, 7, 3482–3493 CrossRef CAS.
- A. H. Flood, S. Nygaard, B. W. Laursen, J. O. Jeppesen and J. F. Stoddart, Org. Lett., 2006, 8, 2205–2208 CrossRef CAS PubMed.
- J. M. Belitsky, A. Nelson, J. D. Hernandez, L. G. Baum and J. F. Stoddart, Chem. Biol., 2007, 14, 1140–1151 CrossRef CAS PubMed.
- C. M. Gothard, C. J. Bruns, N. A. Gothard, B. A. Grzybowski and J. F. Stoddart, Org. Lett., 2012, 14, 5066–5069 CrossRef CAS PubMed.
- T. Ikeda, I. Aprahamian and J. F. Stoddart, Org. Lett., 2007, 9, 1481–1484 CrossRef CAS PubMed.
- K. Da Zhang, J. Tian, D. Hanifi, Y. Zhang, A. C. H. Sue, T.-Y. Zhou, L. Zhang, X. Zhao, Y. Liu and Z. T. Li, J. Am. Chem. Soc., 2013, 135, 17913–17918 CrossRef PubMed.
- V. Kolivoška, M. Valášek, M. Gál, R. Sokolová, J. Bulíčková, L. Pospíšil, G. Mészáros and M. Hromadová, J. Phys. Chem. Lett., 2013, 4, 589–595 CrossRef PubMed.
- V. A. Constantin, D. Bongard and L. Walder, Eur. J. Org. Chem., 2012, 913–921 CrossRef CAS.
- L. Pospíšil, M. Hromadová, N. Fanelli, M. Valášek, V. Kolivoška and M. Gal, Phys. Chem. Chem. Phys., 2011, 13, 4365–4371 RSC.
- V. Kolivoška, M. Gál, Š. Lachmanová, M. Valášek, M. Hromadová and L. Pospíšil, Anal. Chim. Acta, 2011, 697, 23–26 CrossRef PubMed.
- M. Valášek, J. Štursa, R. Pohl and J. Michl, Inorg. Chem., 2010, 49, 10247–10254 CrossRef PubMed.
- V. Kolivoška, M. Gál, L. Pospíšil, M. Valášek and M. Hromadová, Phys. Chem. Chem. Phys., 2011, 13, 11422–11429 RSC.
- W. W. Porter and T. P. Vaid, J. Org. Chem., 2005, 70, 5028–5035 CrossRef CAS PubMed.
- S. Goswami, S. Jana and H.-K. Fun, CrystEngComm, 2008, 10, 507–517 RSC.
- D. Izuhara and T. M. Swager, J. Am. Chem. Soc., 2009, 131, 17724–17725 CrossRef CAS PubMed.
- D. Izuhara and T. M. Swager, Macromolecules, 2011, 44, 2678–2684 CrossRef CAS.
- R. L. LaDuca, M. P. Desciak, R. S. Rarig and J. A. Zubieta, Z. Anorg. Allg. Chem., 2006, 632, 449–453 CrossRef CAS.
- T. Zincke, G. Heuser and W. Möller, Justus Liebigs Ann. Chem., 1904, 333, 296–345 CrossRef.
- H. M. Colquhoun, B. W. Greenland, Z. Zhu, J. S. Shaw, C. J. Cardin, S. Burattini, J. M. Elliott, S. Basu, T. B. Gasa and J. F. Stoddart, Org. Lett., 2009, 11, 5238–5241 CrossRef CAS PubMed.
- E. N. Marvell, G. Caple and I. Shahidi, J. Am. Chem. Soc., 1970, 92, 5641–5645 CrossRef CAS.
- S. K. Nimkar, A. H. Anderson, J. M. Rimoldi, M. Stanton, K. P. Castagnoli, S. Mabic, Y. X. Wang and N. Castagnoli, Chem. Res. Toxicol., 1996, 9, 1013–1022 CrossRef CAS PubMed.
- R. J. Mortimer, Electrochim. Acta, 1999, 44, 2971–2981 CrossRef CAS.
- C. S. Johnson and H. S. Gutowsky, J. Chem. Phys., 1963, 39, 58–62 CrossRef CAS.
- W. R. Dunham, J. A. Fee, L. J. Harding and H. J. Grande, J. Magn. Reson., 1980, 40, 351–359 CAS.
- G. Grampp, B. Y. Mladenova, D. R. Kattnig and S. Landgraf, Appl. Magn. Reson., 2006, 30, 145–164 CrossRef CAS.
- M. Krejčík, M. Daněk and F. Hartl, J. Electroanal. Chem. Interfacial Electrochem., 1991, 317, 179–187 CrossRef.
- S. Tretiak, A. Saxena, R. L. Martin and A. R. Bishop, Phys. Rev. Lett., 2002, 89, 97402 CrossRef CAS PubMed.
- I. Franco and S. Tretiak, J. Am. Chem. Soc., 2004, 126, 12130–12140 CrossRef CAS PubMed.
- J. Krzystek, A. Sienkiewicz, L. Pardi and L. Brunel, J. Magn. Reson., 1997, 125, 207–211 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ob02211h |
| This journal is © The Royal Society of Chemistry 2016 |