
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemical constituents of the soft corals Sinularia vanderlandi and Sinularia gravis from the coast of Madagascar†
Marie Pascaline
Rahelivao
a,
Margit
Gruner
a,
Tilo
Lübken
a,
Daut
Islamov
b,
Olga
Kataeva
ac,
Hanta
Andriamanantoanina
d,
Ingmar
Bauer
a and
Hans-Joachim
Knölker
*a
aDepartment Chemie, Technische Universität Dresden, Bergstrasse 66, 01069 Dresden, Germany. E-mail: hans-joachim.knoelker@tu-dresden.de; Fax: +49 351 463-37030
bA. M. Butlerov Chemistry Institute, Kazan Federal University, Kremlevskaya Str. 18, Kazan 420008, Russia
cA. E. Arbuzov Institute of Organic and Physical Chemistry, Russian Academy of Sciences, Arbuzov Str. 8, Kazan 420088, Russia
dCentre National de Recherche sur l'Environnement, BP 1739, Antananarivo 101, Madagascar
First published on 24th November 2015
Abstract
The crude extracts of the Madagascan soft corals Sinularia vanderlandi and Sinularia gravis (Alcyoniidae) showed activity against Plasmodium falciparum which led us to study their chemical constituents. The new cadinane-type sesquiterpenoid vanderlandin (1) has been obtained from S. vanderlandi along with 24-methylenecholesterol (2). Four new compounds, the spatane-type diterpenoid gravilin (3), the monoalkylmonoacylglycerol 4, the dihomoditerpenoid ketone 5, and isodecaryiol (9), along with the three known compounds (+)-(S)-geranyllinalool (6), (−)-(R)-nephthenol (7), and 11,12-epoxysarcophytol A (8) have been isolated from the methanol extract of S. gravis. The structures were elucidated based on extensive spectroscopic methods, in particular various 2D NMR techniques. The structure of isodecaryiol (9) including its absolute configuration could be confirmed by X-ray diffraction.
Introduction
Soft corals of the genus Sinularia have been shown to contain intriguing chemical components with various biological activities. Sheu et al. isolated the cembrane-type diterpenoids manaarenolide A–I from Sinularia manaarensis with some representatives showing moderate cytotoxic activity against Hepa59T/VGH, KB, Hela, and Med cancer cell lines.1 A series of oxygenated cembranoids, gibberosenes A–G, has been obtained from the Formosan soft coral Sinularia gibberosa.2 The soft corals Sinularia querciformis3,4 and Sinularia granosa3 have also been identified as source of oxygenated cembranoids, some of them with anti-inflammatory activity. A sulfur-containing biscembranoid was obtained from the Formosan soft coral Sinularia flexibilis.5 The cadinane-type sesquiterpenes scabralin A and B from Sinularia scabra have been described by Sheu and coworkers.6 Lin et al. isolated asteriscane-type sesquiterpenoids from the soft coral Sinularia capillosa.7 Soft corals from the coast of Madagascar have only rarely been investigated concerning their secondary metabolites.8–10 This fact and the only very few reports on the chemical constituents of Sinularia vanderlandi and Sinularia gravis (Alcyoniidae) (see ref. 11–13) prompted us to initiate the present study.Results and discussion
Sinularia vanderlandi
Samples of S. vanderlandi were minced, homogenized, and extracted with methanol. In an anti-malaria assay, the crude methanol extract of S. vanderlandi showed an IC50 value of 26.88 ± 4.83 μg mL−1 for the inhibition of the FCM29 strain of Plasmodium falciparum which corresponds to a low activity against this pathogen. After concentration of the crude methanol extract in vacuo, the residue was repeatedly extracted with diethyl ether. The diethyl ether extract was then subjected to column chromatography on silica gel using pentane–diethyl ether (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as eluent. Further purification by a second column chromatographic step under the same conditions led to the isolation of the new cadinane-type sesquiterpenoid14–16 vanderlandin (1) along with 24-methylenecholesterol (2)17–21 (Fig. 1).
:1) as eluent. Further purification by a second column chromatographic step under the same conditions led to the isolation of the new cadinane-type sesquiterpenoid14–16 vanderlandin (1) along with 24-methylenecholesterol (2)17–21 (Fig. 1).
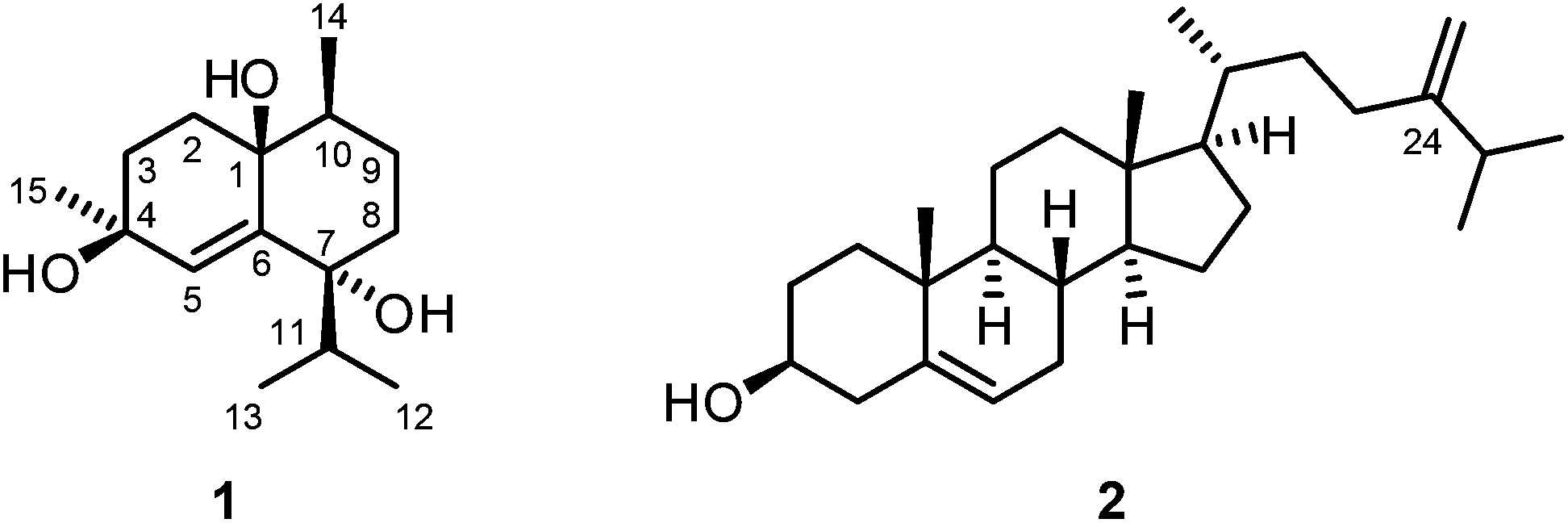 | ||
| Fig. 1 Structures of vanderlandin (1) and 24-methylenecholesterol (2). | ||
Vanderlandin (1) [biogenetic (germacrane) numbering is used according to ref. 16] was isolated as a colorless solid with a melting point of 57–58 °C and a specific optical rotation of [α]20D = −3.8 (c 0.1, MeOH). The EI mass spectrum (m/z = 237 [M − OH]+) in combination with the number and intensities of 1H and 13C NMR signals allowed for the determination of the molecular formula of C15H26O3 which is indicative for three double bond equivalents. A broad IR absorption at ν = 3407 cm−1 revealed the presence of hydroxyl groups. The 1H and 13C NMR spectra in combination with the DEPT spectrum displayed signals for four methyl groups, four methylene groups, and three methine groups (Table S1†). In total, resonances for 15 carbon atoms could be detected in the 13C NMR spectrum including those for three oxygenated quaternary carbon atoms at δC = 83.46, 89.10, and 90.27 ppm. One double bond was identified by 13C NMR signals at δC = 124.33 and 148.05 ppm and a singlet for a vinylic proton at δH = 6.12 ppm. Complete assignment of the proton signals to the corresponding 13C NMR signals was achieved by HSQC and HMBC measurements. A series of HMBC long-range correlations led to the identification of the octahydronaphthalene backbone which is also in agreement with the three double bond equivalents according to the molecular formula. Characteristic HMBC correlations to establish the bicyclic skeleton include the interactions of C-1 with H-2α, H-2β, H-3α, H-3β, H-5, H-9α, and H-9β, of C-4 with H-2α, H-2β, H-3β, and H-5, of C-6 with H-2α, H-2β, H-3α, H-3β, and H-5, and of C-10 with H-2β, H-5, H-8α, H-8β, H-9α, and H-9β (Table S1†). In addition, C-7 correlated with H-5, H-8α, H-8β, H-9α, and H-9β and C-8 exhibited HMBC cross-peaks with H-5, H-9α, H-9β, and H-10. The primary carbon atoms in 1 displayed signals at δC = 17.03 ppm for C-12 and C-13, 13.40 ppm for C-14, and 19.84 ppm for C-15, respectively. According to 1H–1H COSY measurements, H3-12 and H3-13 correlated with H-11 suggesting the presence of an isopropyl group. The location of the isopropyl group at C-7 was determined by HMBC correlations of H-8α, H-8β, H-9α, H-9β, H-11, H3-12, and H3-13 with the quaternary carbon C-7 and, in addition, of H-11 with C-5 and C-8. The attachments of the Me groups at C-10 and at C-4 were confirmed by HMBC correlations of H3-14 with C-9, C-10 and C-1, and of H3-15 with C-4 and C-3, respectively.
Analysis of proton coupling constants and NOE correlations in combination with MM2 molecular modeling studies led to the assignment of the relative configuration of 1 (Table S1,†Fig. 2). The pseudoaxial β position of the Me group at C-10 in a twist-boat-like B-ring was indicated by a strong NOE interaction of H3-14 with H-9β but not with H-9α. NOE correlations of similar intensity of H-10 with both H-9α and H-9β suggesting a pseudoequatorial position of H-10 are in support of this assignment. The absence of any NOE correlation of protons of the isopropyl group with H-9α(ax) most likely accounts for a pseudoequatorial position of this group in cis-relationship to the Me group at C-10. The cis-arrangement of the 1-OH group and the Me group at C-10 was derived from the absence of NOE correlations of H3-14 with H-2α, H-2β, H-3α, and H-3β. In case of a presumed trans-arrangement of the 1-OH group and the Me group at C-10, a much closer proximity of H3-14 to the protons at C-2 and C-3 would be expected which should give rise to pronounced NOE interactions. The configuration of the Me group at C-4 was derived from a strong NOE correlation of H3-15 with H-3α(eq) and only a very weak interaction with H-3β(ax). This is in agreement with a pseudoaxial α position of the Me group at C-4 in the preferred half-chair conformation of the cyclohexene ring (Fig. 2). A twist-boat-like conformation with a pseudoaxial Me group at C-4 in β-position, which would equally explain the observed NOESY results, is energetically disfavored according to molecular modeling studies (MM2 force field). Thus, most likely, the Me group at C-4 adopts a trans-configuration with respect to the alkyl substituents at C-7 and C-10. Based on these arguments, compound 1 was assigned as (−)-(1S/R,4S/R,7R/S,10S/R)-cadin-5-ene-1,4,7-triol, a new representative of the cadinane-type bicyclic sesquiterpenes which we have named vanderlandin. Similar di- and trihydroxylated cadinane sesquiterpenes have been isolated for example from Taiwania cryptomerioides Hayata,22,23 the brown alga Dictyopteris divaricata,24 and more recently, from the rhizome of Acorus calamus L.25
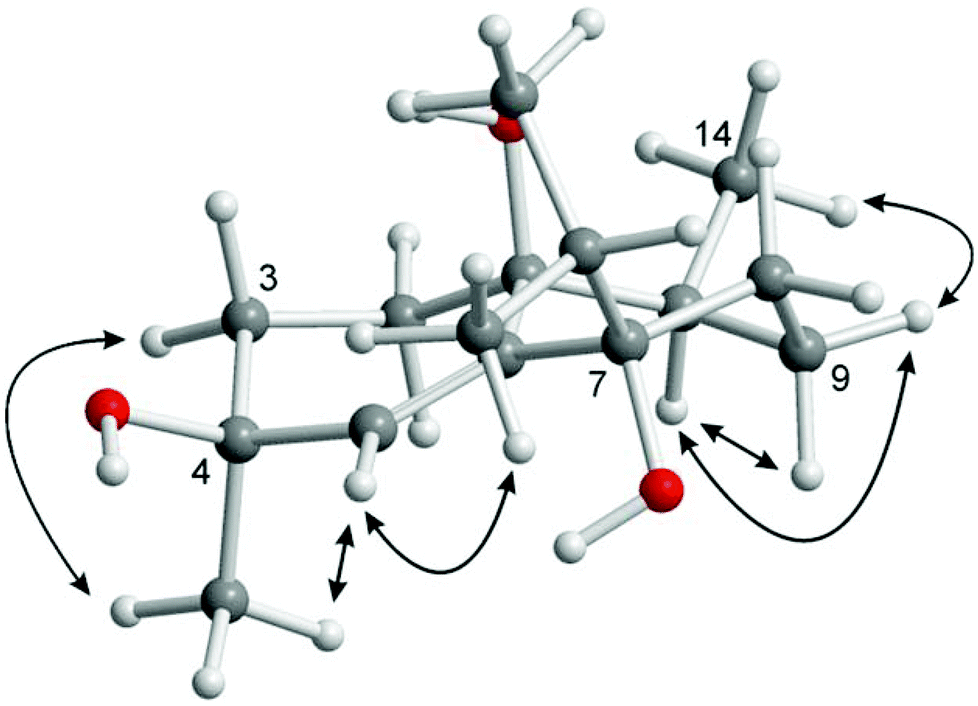 | ||
| Fig. 2 Characteristic NOE correlations of vanderlandin (1) shown at a computer-generated model (MM2 force field). | ||
Sinularia gravis
A specimen of the soft coral S. gravis collected in the inner reef of Mahambo, Madagascar, was minced and extracted with methanol. The anti-malaria assay of the crude methanol extract of S. gravis exhibited an IC50 value of 30.81 ± 9.09 μg mL−1 for the inhibition of the FCM29 strain of Plasmodium falciparum. The combined methanol extracts were partitioned between water and diethyl ether. After removal of the solvent in vacuo, the diethyl ether extract was subjected to column chromatography on silica gel with pentane–diethylether (10:1 and 4:1) as mobile phase to provide four new compounds, gravilin (3), the monoalkylmonoacylglycerol 4, the ketone 5 (Fig. 3), and isodecaryiol (9) (Fig. 5). In addition, the four known compounds 24-methylenecholesterol (2) (Fig. 1),17–21 (+)-(S)-geranyllinalool (6) (Fig. 3),26–36 and the cembranoids (−)-(R)-nephthenol (7)37–44 and 11,12-epoxysarcophytol A (8) (Fig. 5)2,45–50 were isolated.
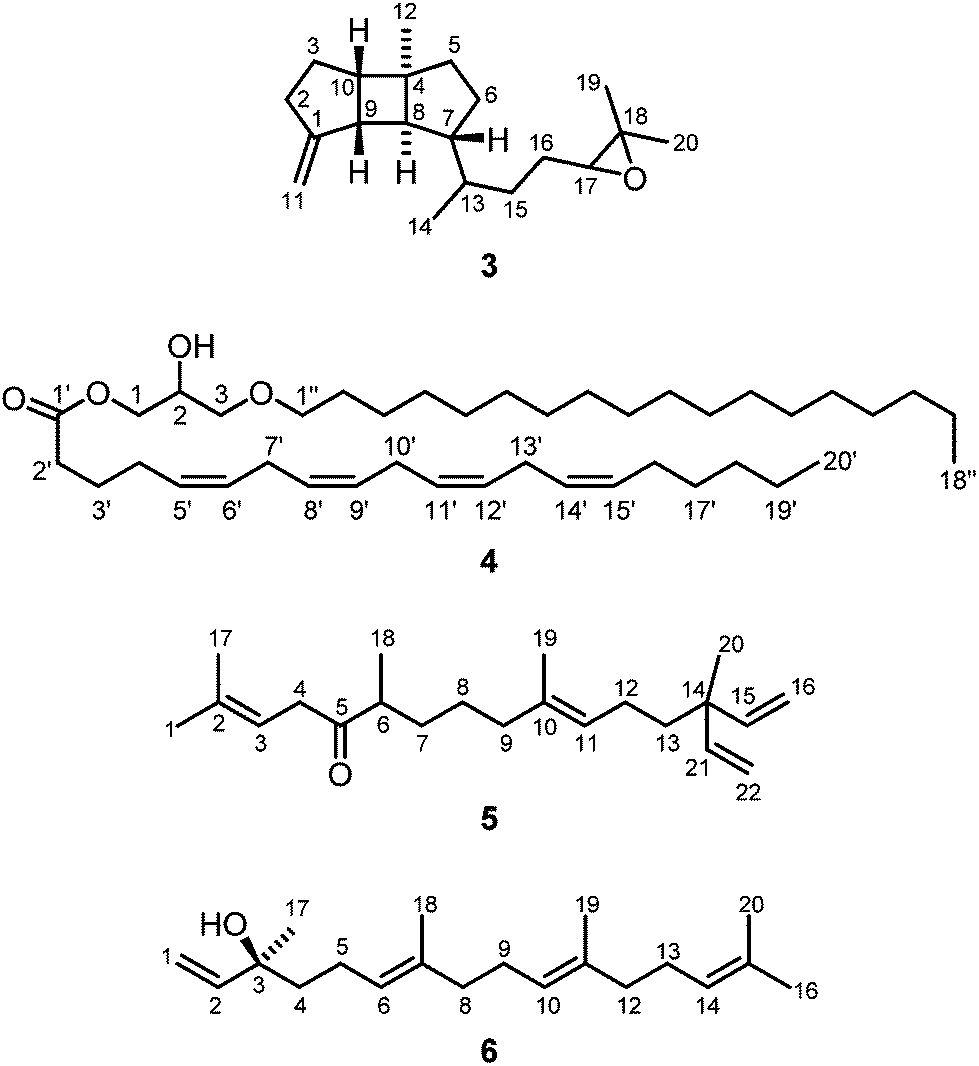 | ||
| Fig. 3 Structures of gravilin (3), monoalkylmonoacylglycerol 4, ketone 5 and (+)-(S)-geranyllinalool (6) isolated from S. gravis. | ||
The novel spatane diterpenoid gravilin (3), the least polar component of the S. gravis extract, was isolated as a colorless solid with a specific optical rotation of [α]20D = +46.0 (c 0.8, MeOH) and a melting point of 100–101 °C. The ESI-MS showed peaks at 289 and 306 mass units for [M + H]+ and [M + NH4]+ ions, respectively. The derived monoisotopic mass of 288 in combination with the number and intensities of 1H and 13C NMR signals led to the molecular formula C20H32O. Signals for four methyl groups, seven methylene groups, and six methine groups were identified by 1H NMR, 13C NMR, and DEPT measurements. Complete proton to carbon assignment was achieved by HSQC and HMBC measurements (Table S2†). On interpretation of the NMR data, it soon became clear that the structure resembles that of spatol, a tricyclic diterpene isolated from the brown seaweed Spatoglossum schmittii.51 The spatane backbone was established on the basis of long range HMBC correlations. Characteristic interactions are those of C-4 with H-3α, H-3β, H2-5, H-6β, H-8, and H-10, of C-8 with H-9 and H-10, of C-9 with H-2α, H-3α, H-3β, and H-10, and of C-10 with H-2β, H-3α, H-3β, H2-5, H-8, and H-9. In place of the methyl group at C-1 in spatol, a methylene group was identified in 3 by vinylic signals at δH = 4.70 (s, H-11a), 4.73 ppm (s, H-11b), and δC = 103.76 ppm (C-11). The attachment of the methylene moiety at C-1 was established based on HMBC correlations of H-11a and H-11b with C-9 and C-2. On the other hand, the methylene group at C-13 in spatol was replaced by a methyl group resonating at δH = 0.85 ppm (d, J = 6.8 Hz, H-14) and δC = 17.87 ppm (C-14). Other than in spatol, the position C-5 is not oxygenated exhibiting a multiplet at δH = 1.52 ppm. This was assigned based on the HSQC correlation with the 13C NMR signal at δC = 42.19 ppm (C-5). The 17,18-epoxide function was identified by the low-field shift of the 13C signals at δC = 64.70 and 58.31 ppm, respectively. HMBC correlations of H-15a, H-15b, H-16a, H-16b, H3-19, and H3-20 with C-17 and H-16a, H-16b, H-17, H3-19, and H3-20 with C-18 have been used to assign the position of the epoxy oxygen atom. In contrast to spatol, no second epoxide function is present. The positions 15 and 16 represent methylene groups with 1H NMR signals at δH = 1.49 (m, H-15a), 1.18–1.22 (m, H-15b), 1.63 (m, H-16a), and 1.31–1.37 ppm (m, H-16b) and with 13C NMR signals at δC = 32.10 (C-15) and 26.43 ppm (C-16). In agreement with other spatane derivatives,51–55 a cis,anti,cis-configuration of the tricyclic 5-4-5 ring system was assigned based on NOESY experiments (Table S2,†Fig. 4). The cis-fusion of the C-ring was supported by NOESY cross-peaks between H-8 and H3-12. The cis-arrangement of the Me group at C-4 relative to the cyclopentane A-ring was proven by NOE correlations of H3-12 with H-2α and H-3α. Therefore, the two cyclopentane moieties must adopt an anti-arrangement relative to each other at the central cyclobutane ring. In contrast to the common trans-orientation of the side chain at C-7 with respect to H-8 and the Me group at C-4, we found a cis-orientation for this substituent. This stereochemical arrangement of the side chain was unambiguously supported by the NOE correlation between H-7 and H-9 and of H-8 with H-13. Only recently, Lin and coworkers isolated the spatane diterpenoid leptoclalin A from Sinularia leptoclados with the same spatane backbone and the same relative configuration at C-7.55 The NMR data of gravilin (3) and leptoclalin A are in good agreement with respect to the chemical shifts for the skeletal carbon and proton signals (Table S3†). This is also true for the signals of H-13 and C-13, thus confirming the uncommon α-configuration of the side chain. The related compounds lobophytumin E and F with the same spatane backbone but an opposite relative configuration at C-7 have been isolated from Lobophytum cristatum.54 Their 1H and 13C NMR data are generally also in good agreement with those of 3 but show a significant deviation for the signals of H-13 and C-13 due to the different configuration at C-7 (see NMR data of lobophytumin F in Table S3†). The configurations at C-13 and C-17 of compound 3 remain to be established as well as the absolute configuration. We assume that the spatane skeleton exhibits the same absolute stereochemistry (see Fig. 3) as determined for many other spatane diterpenoids.51,52,56–59 It is worth noting that gravilin (3), leptoclalin A, and lobophytumins E and F are rare examples of spatane diterpenoids isolated from soft corals. Most of the spatanes have been found in brown algae.
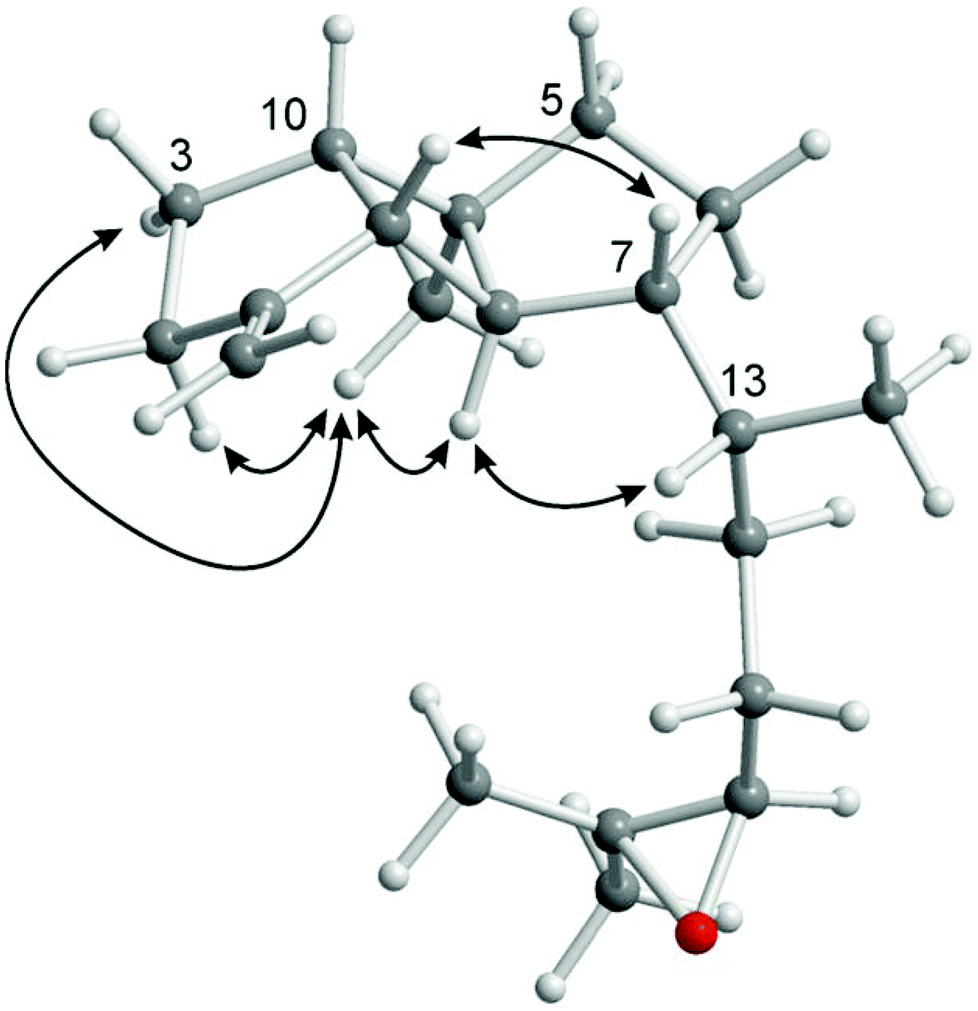 | ||
| Fig. 4 Characteristic NOE correlations of gravilin (3) shown at a computer-generated model (MM2 force field). | ||
Compound 4 (Fig. 3) was isolated as colorless oil. ESI-mass spectrometry performed in positive and negative mode provided peaks for the [M + H]+ ion at m/z = 631.7, [M + NH4]+ at m/z = 648.7, [M + Na]+ at m/z = 653.8 and [M + OAc]− at m/z = 689.4. The resulting monoisotopic mass of 630.7 in combination with the number and intensities of 1H and 13C NMR signals suggested a molecular formula of C41H74O4. The 1H NMR, 13C NMR and DEPT spectra displayed signals for two methyl and nine methine groups (Table S4†). The number of 29 methylene groups was deduced from the 1H NMR spectrum taking the molecular mass into account. The low-field shifted 13C NMR signals at δC = 65.50, 68.87, 71.37, and 71.78 could be assigned to the oxygenated carbon atoms C-1, C-2, C-3 and C-1′′, respectively. In addition, one 13C NMR signal for a carbonyl group was detected at δC = 173.68 ppm. These signals were in agreement with a monoalkylmonoacylglycerol structure bearing a free hydroxyl group. The proton signal of the hydroxyl group was identified at δH = 2.45 ppm (d, J = 4.5 Hz). In this regard, HMBC correlations allowed for the assignment of the hydroxyl group to position C-2 by strong correlations of the OH proton signal with C-1, C-2, and C-3. Further support was obtained from NOE correlations of OH with H-1a, H-1b, H-2, H-3a, and H-3b. 1H and 13C NMR signals for eight methine groups could be attributed to four double bond systems in a skipped arrangement (C-5′, C-6′, C-8′, C-9′, C-11′, C-12′, C-14′, and C-15′). HMBC correlations (C-1′/H2-2′, C-2′/H2-3′, C-3′/H2-4′, C-4′/H-5′) connected the skipped double bond systems with the carbonyl group at C-1′. Towards the methyl terminus, 1H and 13C NMR signals for four remaining methylene groups could be clearly assigned by sequential HMBC correlations starting from the vinylic position C-15′. The configurations of the double bonds in 4 were determined by NOESY measurements. NOE correlations of H2-4′ with H2-7′ and of H2-13′ with H2-16′ indicated a Z-configuration of the Δ5′- and Δ14′-double bonds. Assuming also a Z-configuration of the Δ8′- and Δ11′-double bonds, an arachidonic side chain was suggested as acyl component of the monoalkylmonoacylglycerol which was confirmed by comparison of the 1H and 13C NMR data with literature values.60 Thus, the fully saturated second side chain must be attached to the glycerol core by an ether linkage. The length of this side chain (C18) was determined taking the intensity of the proton NMR signals and the molecular mass into account. The location of the acyl chain at C-1 of the glycerol moiety was assigned based on HMBC correlations of C-1′ with H-1a and H-1b. Analogously, HMBC correlations of C-1′′ with H-3a and H-3b indicated the attachment of the alkyl chain at C-3 of the glycerol unit.
In view of the spectroscopic data, compound 4 was assigned as 2-hydroxy-3-(octadecyloxy)propyl (5Z,8Z,11Z,14Z)-icosa-5,8,11,14-tetraenoate or 1-O-arachidonoyl-3-O-stearylglycerol with an unknown configuration at the stereogenic center C-2 (Fig. 3). This monoalkylmonoacylglycerol isolated from S. gravis from Madagascar has been reported for the first time. The respective 1-alkyl-2-acyl-sn-glycerol isomer with the arachidonoyl chain attached to the central position of the glycerol moiety, thus having a primary hydroxyl group, has been identified by Myher and Kuksis during their GC analysis of mixtures of diradylglycerols obtained from the corresponding glycerophospholipids.61,62 The monoalkylmonoacylglycerol 4 may be derived from monoalkyldiacylglycerol precursors by hydrolytic processes. The latter are well-documented components of soft coral lipids, in particular also in Sinularia species.63,64
Compound 5 (Fig. 3) was isolated from the pentane–diethyl ether (4:1) fraction of S. gravis as a colorless solid. The 1H NMR, 13C NMR, and DEPT spectra displayed signals for five methyl groups, eight methylene groups, five methine groups, and four quaternary carbon atoms (Table S5†). Assignment of all 13C NMR signals to the respective 1H NMR signals could be achieved by an HSQC measurement. The 13C NMR spectrum displayed one signal for a carbonyl group at δC = 213.13 ppm (C-5). Signals for eight vinylic protons and eight vinylic carbon atoms could be identified in the 1H and 13C NMR spectra and assigned to two trisubstituted double bonds and two equivalent monosubstituted terminal double bonds. The connectivity of all groups, and thus the constitution of compound 5, could be unambiguously established by analysis of the HMBC spectrum. For example, the attachment of the methyl groups at C-2, C-6, C-10, and C-14 was suggested by HMBC correlations of C-2 with H3-1 and H3-17, of C-6 with H3-18, of C-10 with H3-19, and of C-14 with H3-20. This assignment was supported by the corresponding HMBC cross-peaks of the 13C signals for the methyl groups with adjacent protons, namely of C-1 with H-3 and H3-17, C-17 with H3-1 and H-3, C-18 with H-6, H-7a, and H-7b, C-19 with H2-9 and H-11, and C-20 with H-15/H-21. The location of the carbonyl group at C-5 was assigned based on the HMBC correlations of H2-4, H-6, and H-7b with C-5. A strong NOE correlation of H3-19 with H2-12 confirmed the E-configuration of the Δ10-double bond. The analytical data allowed to identify compound 5 as (E)-2,6,10,14-tetramethyl-14-vinylhexadeca-2,10,15-trien-5-one (Fig. 3) which has not been described before. It exhibits a geranylgeranyl backbone with two additional carbon atoms in form of a vinyl group attached to the branching position of the terminal isoprene unit. A similar branching motif has been reported for a series of botryococcenes.65–68 The compound is also reminiscent of geranylgeranylacetone (GGA) which is applied as drug for the treatment of gastric ulcers.69 GGA and its derivatives have also been investigated for the treatment of neurodegenerative diseases including paralysis.70
Compound 6 was isolated from the pentane–diethyl ether (10:1) fraction of S. gravis as colorless oil with a specific optical rotation of [α]20D = +23.8 (c 0.07, MeOH). The CD spectrum (MeOH) of 6 showed molar circular dichroism values of Δε = +1.94 (202 nm) and +0.12 (247 nm). Based on the data obtained from 1H NMR, COSY, HSQC, HMBC, and NOESY experiments and by comparison of the 13C NMR chemical shifts with those reported in the literature71 (Table S6†) the structure of 6 was unambiguously elucidated as (+)-(3S,6E,10E)-3,7,11,15-tetramethylhexadeca-1,6,10,14-tetraen-3-ol [(+)-(S)-geranyllinalool] (Fig. 3). The absolute configuration had been determined earlier by Svatoš and coworkers via total synthesis of both enantiomers.32 Geranyllinalool has been isolated before from various plants such as from the oleoresin of the Norwegian spruce Picea abies27,31 and from South African Helichrysum species.28,29 It has been identified as component of various essential oils, for example from pequi fruits collected from the Brazilian Cerrado ecosystem,36 and as ingredient of many fragrances.35 Thus, it plays a decisive role in the fragrance of jasmine.26,30,34 Whilst the laevorotatory (R)-enantiomer has been described frequently, the dextrorotatory (S)-enantiomer is rare. It has been found in jasmine oil26 and in the freshwater aquatic plant Potamogeton pectinatus.33 In the course of the present study, (+)-(S)-geranyllinalool (6) was isolated for the first time from a soft coral (S. gravis).
Compound 7 was also isolated from the pentane–diethyl ether (10:1) fraction of S. gravis as colorless oil with a specific optical rotation of [α]20D = −35.9 (c 1.0, MeOH) and molar circular dichroism values of Δε = −7.78 (207 nm) and +0.94 (240 nm). Analysis of the 1D and 2D NMR data and the mass spectra led to the structure of (−)-(R)-nephthenol (7) (Fig. 5). Comparison of the analytical data including 1H and 13C NMR and EI-MS with those reported for a synthetic sample by Pfander et al.39 proved the identity of the compound also with respect to the absolute configuration which had been determined before by chemical degradation.72 In addition, there is a full agreement of the 13C NMR data of (−)-(R)-nephthenol (7) from S. gravis with those from a sample isolated from the soft coral Litophyton arboreum42 (Table S7†). (−)-(R)-Nephthenol (7) has been isolated first from Nephthea sp.37 and later also from Lithophyton arboreum,42,43Lobophytum pauciflorum,43Sarcophyton decaryi,38,44 and Sinularia erecta.40 It is noteworthy that (+)-(S)-nephthenol has also been isolated from natural sources, namely a Caribbean gorgonian octocoral Eunicea species.41 Herein we describe the first isolation of (−)-(R)-nephthenol (7) from S. gravis.
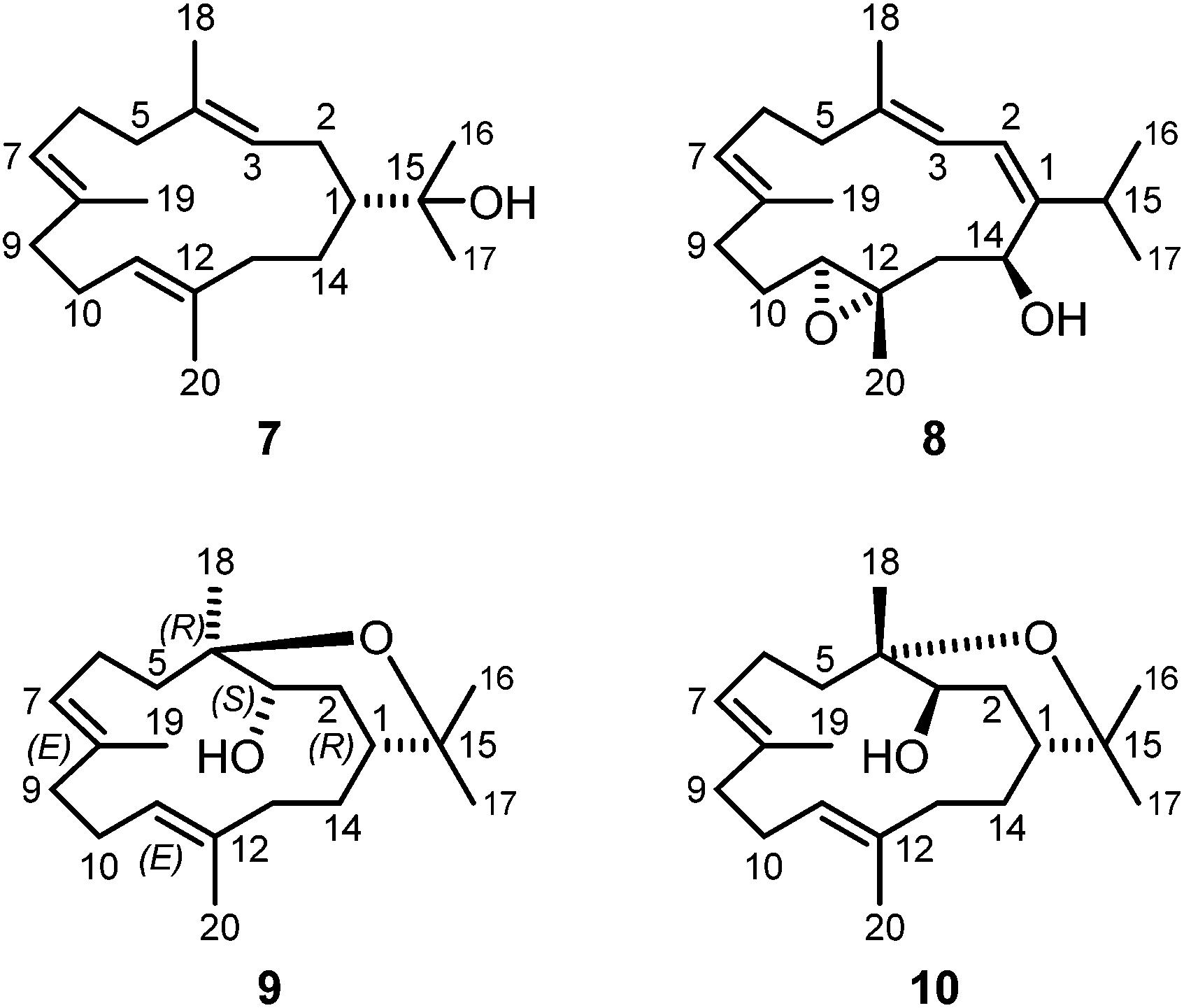 | ||
| Fig. 5 Structures of (−)-(R)-nephthenol (7), 11,12-epoxysarcophytol A (8) and isodecaryiol (9) isolated from S. gravis and decaryiol (10). | ||
Compound 8 was obtained from the pentane–diethyl ether (4:1) fraction of S. gravis as yellow solid with a melting point of 68–70 °C and a specific optical rotation of [α]20D = +220 (c 0.1, MeOH). Based on the spectroscopic data (IR, EI-MS, 1H NMR, 13C NMR, DEPT, COSY, NOESY, HSQC, and HMBC), compound 8 could be unambiguously identified as 11,12-epoxysarcophytol A (Fig. 5). The 1H NMR and 13C NMR data of compound 8 isolated from S. gravis were in full agreement with those of 11,12-epoxysarcophytol A obtained from Sinularia gibberosa (Table S8†).2 The optical rotation value of 8 with [α]20D = +220 (c 0.1, MeOH) is consistent with that reported by Bowden et al.45 with [α]D = +229 (c 0.95) and also with that of the synthetic product described by Li and coworkers50 with [α]20D = +218 (c 0.35, CHCl3). The total synthesis of 11,12-epoxysarcophytol A (8) by Li et al. allowed to elucidate its absolute configuration as depicted in Fig. 5.50 11,12-Epoxysarcophytol A (8) has been isolated from several soft coral species, first from Lobophytum sp.45 and subsequently also from Sarcophyton trocheliophorum,46Cladiella kashmani,47Sinularia gibberosa,2Sarcophyton sp.,48 and Sarcophyton ehrenbergi.49 This is the first report on the isolation of 11,12-epoxysarcophytol A (8) from the soft coral S. gravis.
In addition to 8, the chromatographic separation of the pentane–diethyl ether (4:1) fraction of S. gravis afforded also compound 9 as colorless crystals with a melting point of 94.0–94.5 °C and a specific optical rotation of [α]20D = +14.0 (c 0.1, MeOH). EI- and ESI-MS displayed peaks at m/z = 306 [M]+ and m/z = 307 [M + H]+, respectively. In combination with the number and intensities of the 1H and 13C NMR signals a molecular formula of C20H34O2 was concluded. Extensive 1D and 2D NMR experiments, in particular COSY, HSQC, and HMBC measurements (Fig. 6), led to a structure with the same framework as decaryiol (10),38,40,73–78 a cembranoid which has been isolated first from Sarcophyton decaryi by Kashman and coworkers.38 In particular, the 13C NMR shifts were almost identical (Table S9†). The 1H NMR data were also similar but differed in certain aspects (Table S10†). For example, H-1 in compound 9 gives a triplet of triplets (J = 11.6, 1.9 Hz; theoretically, the triplets are doublets of doublets with identical coupling constants) with one large coupling constant to vicinal protons instead of a doublet of doublets of triplets (J = 11.5, 3.0, 2.0 Hz) with one large and two small coupling constants in compound 10. Slightly different coupling constants were also observed for H-5b and H-6b. In addition, the chemical shifts of H-6b, H-9a, H-9b, and H-10b differ significantly. Most striking, however, was the fact that the value for the optical rotation of +14.0 (c 0.1, MeOH) and the melting point of 94.0–94.5 °C of 9 were not in agreement with those reported for decaryiol (10) (m.p. 126–128.5 °C,38 123–125 °C;74 [α]24D = +69 (c 1.3, CHCl3),38 [α]D = +65 (c 1.1),73 [α]25D = +72 (CHCl3),74 [α]23D = +67.4 (c 1.0, CHCl3)75). A detailed analysis of the 1H NMR coupling constants and NOE correlations revealed a different relative stereochemistry of 9 as compared to 10. The proton at C-3 of compound 9 [δH = 4.20 (dt, J = 11.7, 5.9 Hz)] (just as the corresponding proton of 10) adopts an axial position in the pyran ring according to the large coupling constant of 11.7 Hz with the vicinal axial proton H-2α and a small coupling constant of J = 5.9 Hz with the equatorial proton H-2β. In contrast to 10, H-1 in 9 [δH = 1.58 (tt, J = 11.6, 1.9 Hz)] also occupies an axial position as indicated by two large axial–axial coupling constants with the adjacent protons H-2α and H-14b. This accounts for a cis-relationship between H-1 and H-3 in the pyran ring and consequently also between the hydroxyl group at C-3 and the macrocyclic substituent at C-1. Strong NOE correlations between H-1 and H-3 confirm this assignment (Fig. 7 and 8). The relative configuration at C-4 was assigned based on the NOESY spectrum. NOE correlations from H-3 to H-6b and H-5a suggest a cis-arrangement of H-3 and the macrocyclic side chain at C-4. In support of this, the NOE interaction between H-3 and H3-18 is only weak. Consequently, compound 9 has the opposite configuration at the stereogenic centers C-3 and C-4 relative to C-1 as compared to decaryiol (10). Therefore, we propose for compound 9 the name isodecaryiol.
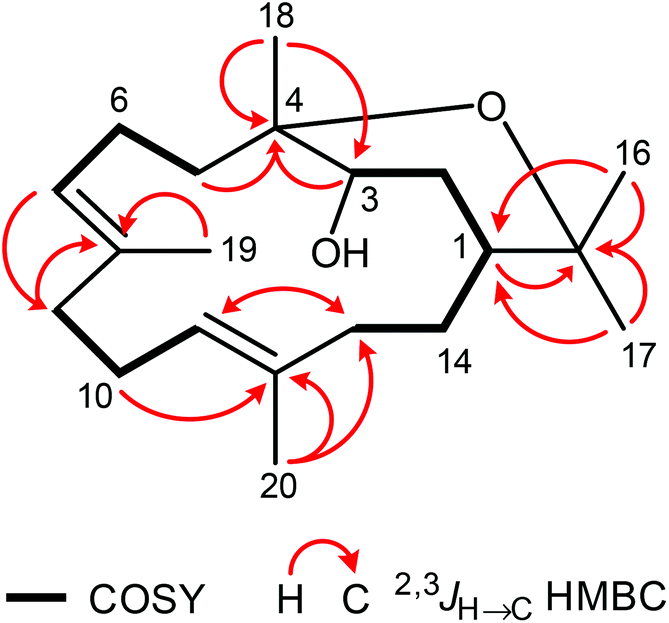 | ||
| Fig. 6 Characteristic COSY and HMBC correlations of 9. | ||
 | ||
| Fig. 7 Characteristic NOE correlations of 9 (structure from X-ray analysis). | ||
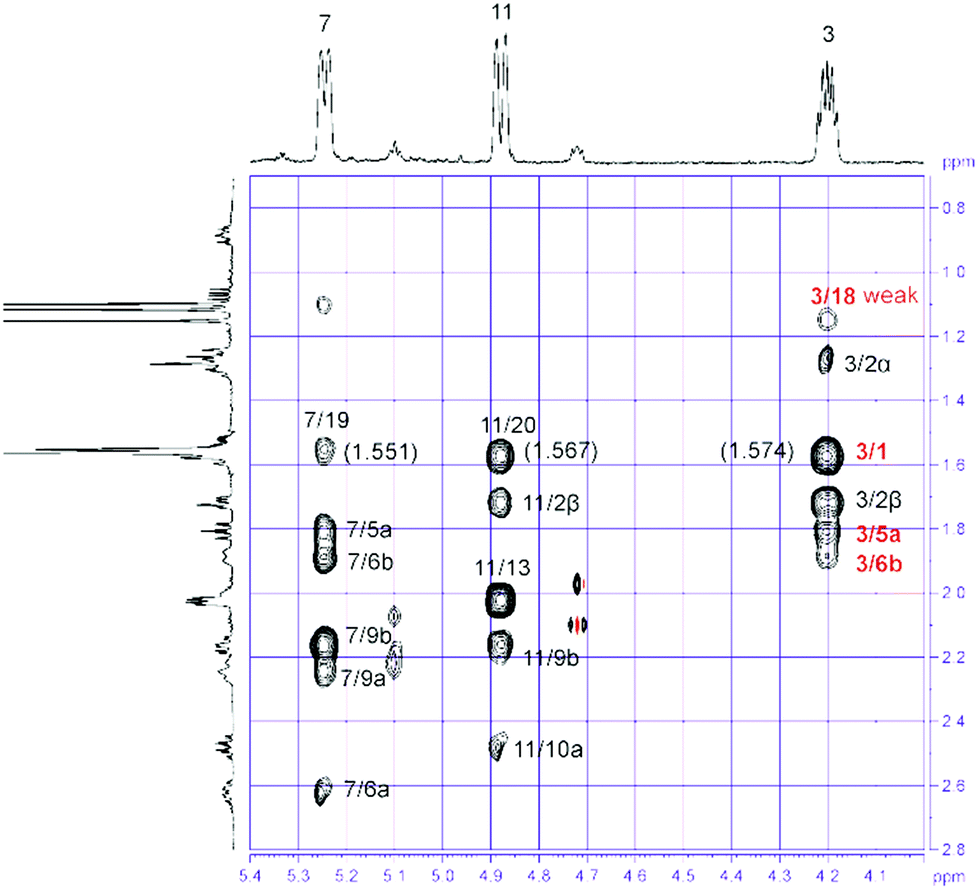 | ||
| Fig. 8 Expansion of the NOESY spectrum of isodecaryiol (9). | ||
The stereochemistry of isodecaryiol (9) has been unequivocally confirmed by an X-ray crystal structure determination (Fig. 9). The unit cell of the crystal has an extremely high number of 12 symmetry-independent molecules (Z′ = 12; see Fig. S71†) which represents a rare case.79 The chirality of the natural product contributes to this high Z′ value.79 The individual molecules have minor conformational differences in the 14-membered ring system and via hydrogen bonds they form tetramers with a cyclic arrangement (Fig. 10). The packing of the molecules in the unit cell is shown in Fig. 11. The absolute configuration of isodecaryiol (9) was determined by the anomal dispersion (Flack parameter:80χ = 0.08(4)). The symmetry-independent molecules have an identical relative and absolute stereochemistry (1R,3S,4R). The R-configuration at C-1 is in agreement with the original assignment for decaryiol (10) by the group of Kashman which was based on chemical correlation with (−)-(R)-nephthenol.38 The S-configuration at C-3 and the R-configuration at C-4 of 9 confirm our assignment based on 1H coupling constants and NOE correlations (see above). 3,4-Epoxynephthenol has been suggested as biogenetic precursor of decaryiol (10).38,73 Price et al. observed that 3,4-epoxynephthenol, isolated from the eggs of Lobophytum microbulatum, is readily transformed to decaryiol (10) under acidic conditions or during prolonged chromatography on silica gel.73 Considering the absolute configuration, (1R,3R,4R)-3,4-epoxynephthenol should be the biogenetic precursor which is transformed into decaryiol (10) by nucleophilic opening of the epoxide ring. Accordingly, isodecaryiol (9) would derive from (1R,3S,4S)-3,4-epoxynephthenol having the opposite orientation of the oxirane ring. The isolation of decaryiol (10) has been reported several times from various soft corals (Sarcophyton decaryi,38Lobophytum microlobulatum,73Sclerophytum sp.,74Sinularia erecta,40,76Nephthea sp.,75,78 and Lobophytum sp.77). Decaryiol (10) inhibits the growth of human cancer cell lines HM02 selectively during the G2/M phase.75 Isodecaryiol (9) constitutes a new representative of cembranoid diterpenes. Most likely, the compound isolated from Nephthea sp., which Wright and coworkers described as decaryiol (10),78 was in fact isodecaryiol (9). The optical rotation value of [α]20D = +27.2 (c 0.01) reported by Wright et al.78 is more close to the value of [α]20D = +14.0 (c 0.1, MeOH) we found for isodecaryiol (9) rather than to the values commonly reported in the literature for decaryiol (10), which are in the range of [α]D = +65 to +72 (see above). In addition, the chemical shifts for H-6b, H-9a, and H-9b in the 1H NMR spectrum (Table S10†) as well as the NOE interactions described by Wright et al.78 fit better to the structure of isodecaryiol (9). However, these authors mentioned an agreement of the 1H NMR data of their natural product with those data reported by Kashman et al. for decaryiol (10),38 but they gave a configuration at C-3 (orientation of the hydroxyl group) which is different from decaryiol (10).78
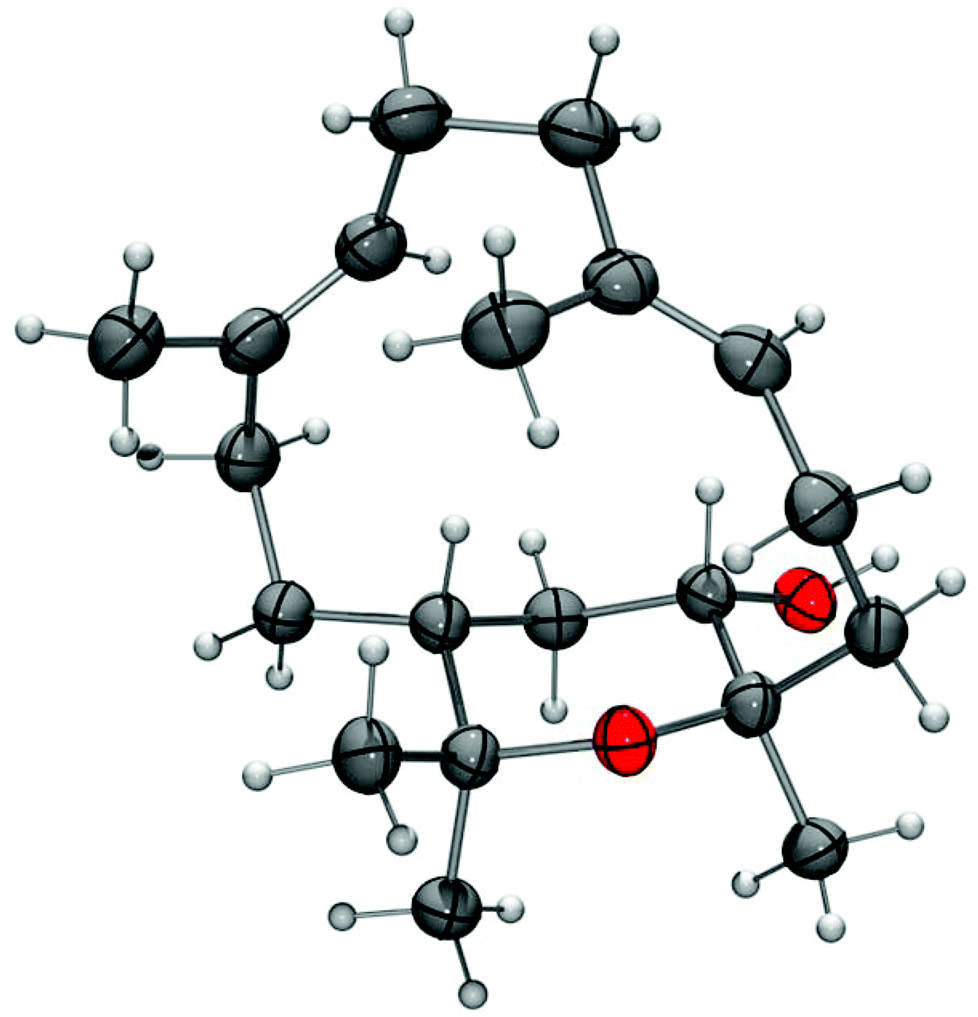 | ||
| Fig. 9 Molecular structure of isodecaryiol (9) in the crystal (thermal ellipsoids at 50% probability level). | ||
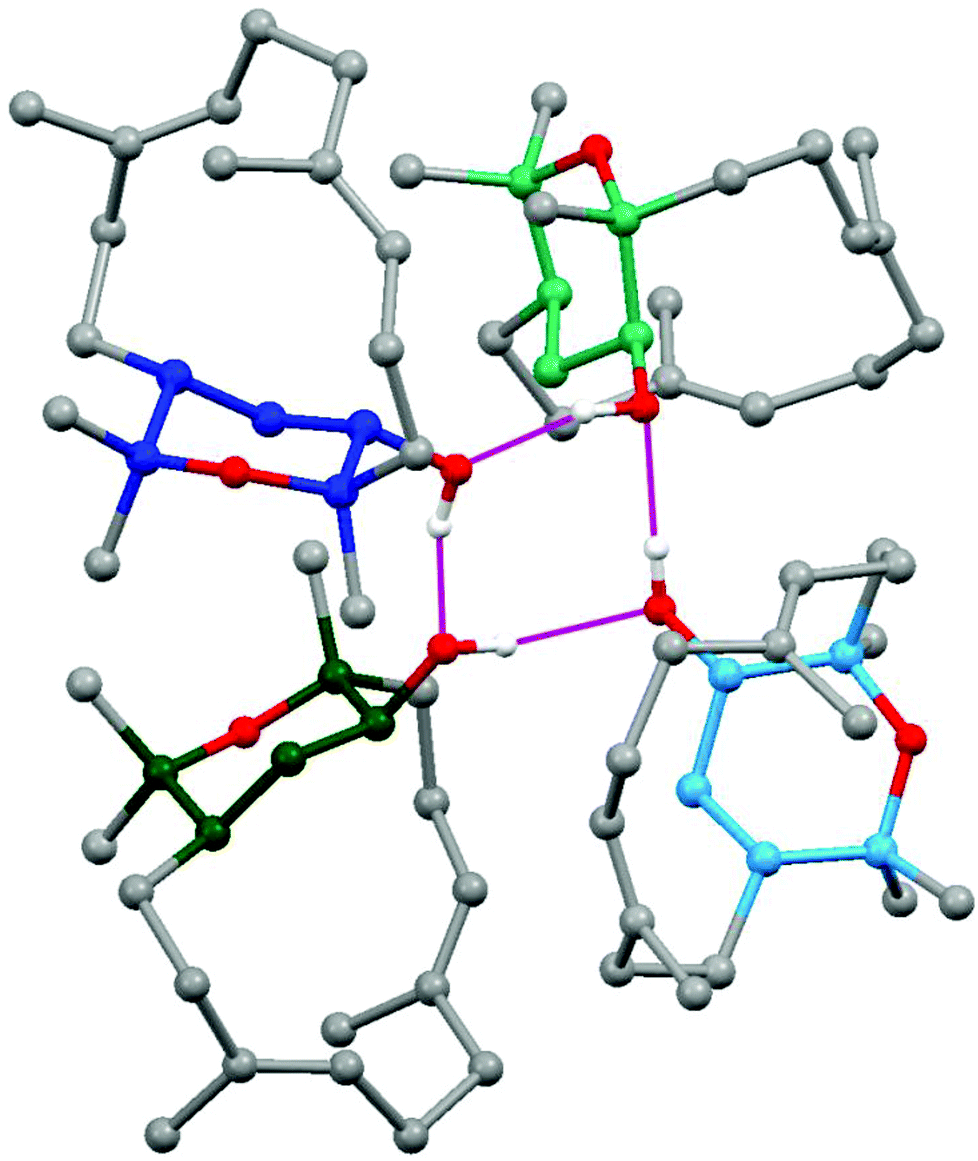 | ||
| Fig. 10 View of the cyclic tetrameric arrangement of isodecaryiol (9) in the crystal via hydrogen bond assembly (only the protons of the hydroxyl groups are shown; all other hydrogen atoms are omitted for clarity). | ||
 | ||
| Fig. 11 Fragment of the crystal packing of isodecaryiol (9) viewed along the c-axis. | ||
Conclusions
Previously, the Madagascan soft corals Sinularia vanderlandi and Sinularia gravis were investigated only very scarcely. From the crude extracts of these species, we have isolated five new natural products: vanderlandin (1), gravilin (3), the monoalkylmonoacylglycerol 4, the dihomoditerpenoid ketone 5, and isodecaryiol (9). The absolute configuration of isodecaryiol (9) has been unequivocally confirmed by an X-ray crystal structure determination. Moreover, for the first time (+)-(S)-geranyllinalool (6) has been isolated from a soft coral. Vanderlandin (1), gravilin (3), and isodecaryiol (9) represent interesting and challenging targets for total synthesis. A detailed study of the biological activities of the new natural products (1, 3–5, and 9) still remains.Experimental
General methods
Thin layer chromatography was performed on aluminum plates from Merck (60 F254) coated with silica gel. For visualization, the plates were analyzed under UV light or treated with a solution of 0.5 g vanillin dissolved in 100 mL of 80/20 (v/v) sulfuric acid/ethanol and subsequently heated. Column chromatography was performed using silica gel from Acros Organics (0.035–0.070 mm). Melting points were measured on a Gallenkamp MPD 350 melting point apparatus. Optical rotations were determined on a Perkin Elmer 341 polarimeter at a wavelength of 589 nm (sodium D line) using a 1.0-decimeter cell with a total volume of 1.0 mL. CD spectra were measured on a JASCO J-815 CD spectrometer. UV spectra were recorded on a Perkin Elmer Lambda 25 UV-Vis spectrometer. Fluorescence spectra were measured on a Varian Cary Eclipse fluorescence spectrometer. IR spectra were recorded on a Thermo Nicolet Avatar 360 FT-IR spectrometer using the ATR technique (attenuated total reflectance). NMR spectra were recorded on a Bruker AVANCE III 600 spectrometer. The chemical shifts δ are reported in ppm using the solvent signal as internal standard (1H: δH 7.250 ppm CHCl3; 13C: δC 77.00 ppm CDCl3). The following abbreviations have been used: s: singlet, d: doublet, t: triplet, q: quartet, quint: quintet, sept: septet, m: multiplet, and br: broad. Assignment of the 1H NMR and 13C NMR signals was achieved using the 2D NMR methods COSY, HSQC, HMBC, and NOESY. The mass spectra were measured by GC-MS coupling with an Agilent Technologies 6890 N GC system equipped with a 5973 Mass Selective Detector (electron impact, 70 eV). ESI-MS were recorded on a Bruker-Esquire mass spectrometer with an ion trap detector; positive and negative ions were detected. X-ray single crystal structure analysis: X-ray diffraction data were collected on a Bruker AXS Kappa APEX Duo diffractometer with microfocus tube, Cu Kα radiation (λ = 1.54184 Å). Programs used: data collection APEX2,81 data reduction SAINT,82 absorption correction SADABS version 2.10,83 structure solution SIR2002,84 structure refinement by full-matrix least-squares against F2 using SHELXL.85 Hydrogen atoms were placed into calculated positions and refined as riding atoms at carbon atoms. The hydrogen atoms of hydroxyl groups were refined using AFIX 148 constraints. The figures were generated using the programs ORTEP-III,86 POV-Ray 3.7, and Mercury 3.1.87:1) was further purified by column chromatography with pentane–diethyl ether (4:1) to yield 4.8 mg of compound 1 and 77.2 mg of 24-methylenecholesterol (2).
A specimen of the soft coral S. gravis (595 g wet weight) was minced and extracted with methanol. After removal of the solvent under reduced pressure, the residue (25 g) was suspended in water and extracted with diethyl ether. The diethyl ether extract was dried with anhydrous Na2SO4 and, after removal of the solvent in vacuo, the residue (7.8 g) was subjected to column chromatography on silica gel and eluted with pentane–diethyl ether mixtures (10:1 and 4:1) to give two fractions. Fraction 1 (1 g) was further separated by column chromatography on silica gel with pentane–diethyl ether (10:1) to afford three subfractions (1-1, 1-2, and 1-3). Subfraction 1-1 (17 mg) was submitted to repeated column chromatography on silica gel with pentane–dichloromethane (9:1) to give 3 mg of compound 3. Subfraction 1-2 afforded 51 mg of (+)-(S)-geranyllinalool (6). Subfraction 1-3 (65 mg) was further purified by column chromatography on silica gel with pentane–diethyl ether (10:1) to give 43 mg of (−)-(R)-nephthenol (7). Fraction 2 (1.24 g) was separated by column chromatography on silica gel using pentane–diethyl ether (4:1) as mobile phase to give four subfractions (2-1, 2-2, 2-3 and 2-4). Subfraction 2-1 (46 mg) was submitted to repeated column chromatography on silica gel with pentane–diethyl ether (17:3) to give 22 mg of the monoalkylmonoacylglycerol 4. Purification of subfraction 2-2 (58 mg) by silica gel column chromatography eluting with pentane–diethyl ether (17:3) followed by a second column chromatography with dichloromethane afforded 3 mg of (E)-2,6,10,14-tetramethyl-14-vinylhexadeca-2,10,15-trien-5-one (5). Subfraction 2-3 (538 mg) was subsequently subjected to column chromatography on silica gel with pentane–diethyl ether (4:1) to give three subfractions (2-3-1, 2-3-2 and 2-3-3). Subfraction 2-3-1 (194 mg) and fraction 2-4 (84 mg) afforded 278 mg of 24-methylenecholesterol (2). Subfraction 2-3-2 was purified by silica gel column chromatography with dichloromethane to give 2 mg of 11,12-epoxysarcophytol A (8). Finally, 10 mg of isodecaryiol (9) were isolated after purification by column chromatography of subfraction 2-3-3 with pentane–diethyl ether (4:1).
Vanderlandin (1). Colorless solid; m.p. 57–58 °C. [α]20D = −3.8 (c 0.1, MeOH). UV (MeOH): λ = 206, 283 (sh) nm. Fluorescence (MeOH): λex = 206 nm, λem = 303, 363 nm. IR (ATR): ν = 3407 (br), 2925, 2854, 1726, 1712, 1457, 1374, 1173, 1074, 1001, 892, 721 cm−1. 1H NMR (600 MHz, CDCl3): δ = 0.97 (3H, d, J = 7.2 Hz, H3-12/H3-13), 0.99 (3H, d, J = 6.8 Hz, H3-12/H3-13), 1.06 (3H, d, J = 7.2 Hz, H3-14), 1.41 (1H, dddd, J = 14.1, 5.0, 3.7, 2.9 Hz, H-9β), 1.50 (1H, ddd, J = 14.1, 11.9, 5.7 Hz, H-9α), 1.54 (3H, s, H3-15), 1.70 (1H, dddd, J = 14.5, 5.3, 3.8, 0.6 Hz, H-8β), 1.78 (1H, dt, J = 13.4, 8.0 Hz, H-3α), 1.85 (1H, ddd, J = 13.7, 8.8, 3.7 Hz, H-2α), 1.88 (1H, sept, J = 6.9 Hz, H-11), 1.89 (1H, ddd, J = 14.7, 11.9, 4.9 Hz, H-8α), 1.90–1.96 (1H, m, H-10), 1.97 (1H, ddd, J = 13.7, 8.9, 7.6 Hz, H-2β), 2.07 (1H, ddd, J = 13.4, 7.5, 3.7 Hz, H-3β), 6.12 (1H, s, H-5), 7.38 (1H, s, OH). 13C NMR and DEPT (150 MHz, CDCl3): δ = 13.40 (C-14), 17.03 (C-12, C-13), 19.84 (C-15), 27.91 (C-9), 30.61 (C-8), 34.46 (C-3), 34.57 (C-2), 35.52 (C-11), 38.50 (C-10), 83.46 (C-7), 89.10 (C-1), 90.27 (C-4), 124.33 (C-5), 148.05 (C-6). MS (EI, 70 eV): m/z (%) = 237 [M − OH]+ (4), 201 (3), 191 (6), 173 (8), 163 (20), 145 (49), 135 (24), 121 (37), 107 (21), 95 (24), 81 (20), 71 (29), 55 (39), 43 (100).
24-Methylenecholesterol (2). Colorless solid; m.p. 129–130 °C. [α]20D = −28.3 (c 0.1, MeOH). CD (MeOH): Δε (λ) +1.55 (202 nm), −0.43 (250 nm). UV (MeOH): λ = 202 nm. Fluorescence (MeOH): λex = 202 nm, λem = 298 nm. IR (ATR): ν = 3416 (br), 2958, 2936, 2868, 2851, 1641, 1458, 1378, 1051, 958, 886 cm−1. 1H NMR (600 MHz, CDCl3): δ = 0.67 (3H, s, H3-18), 0.90–0.94 (1H, m, H-9), 0.94 (3H, d, J = 6.4 Hz, H3-21), 0.96–1.01 (1H, m, H-14), 1.00 (3H, s, H3-19), 1.01 (3H, d, J = 6.8 Hz, H3-26 or H3-27), 1.02 (3H, d, J = 6.8 Hz, H3-26 or H3-27), 1.03–1.10 (2H, m, H-1b, H-15b), 1.10–1.19 (3H, m, H-12b, H-17, H-22b), 1.25–1.30 (1H, m, H-16b), 1.40–1.47 (3H, m, H-8, H-11b, H-20), 1.47–1.60 (5H, m, H-2b, H-7b, H-11a, H-15a, H-22a), 1.80–1.90 (4H, m, H-1a, H-2a, H-16a, H-23b), 1.94–2.03 (2H, m, H-7a, H-12a), 2.05–2.11 (1H, m, H-23a), 2.19–2.26 (2H, m, H-4b, H-25), 2.29 (1H, ddd, J = 12.8, 4.9, 1.9 Hz, H-4a), 3.48–3.55 (1H, m, H-3), 4.64 (1H, br d, J = 1.5 Hz, H-28a), 4.70 (1H, br s, H-28b), 5.33–5.36 (1H, m, H-6). 13C NMR and DEPT (150 MHz, CDCl3): δ = 11.85 (CH3), 18.70 (CH3), 19.39 (CH3), 21.07 (CH2), 21.86 (CH3), 21.99 (CH3), 24.28 (CH2), 28.21 (CH2), 30.96 (CH2), 31.66 (CH2), 31.89 (CH2), 31.89 (CH), 33.79 (CH), 34.67 (CH2), 35.75 (CH), 36.49 (C), 37.24 (CH2), 39.77 (CH2), 42.30 (CH2), 42.35 (C), 50.11 (CH), 55.97 (CH), 56.75 (CH), 71.80 (CH), 105.91 (CH2), 121.70 (CH), 140.75 (C), 156.90 (C). MS (EI, 70 eV): m/z (%) = 398 [M+] (8), 383 (12), 365 (10), 314 (100), 299 (34), 271 (55), 253 (13), 229 (26), 213 (25), 159 (29), 145 (35), 133 (30), 119 (28), 105 (48), 91 (46), 69 (80), 55 (99), 41 (63). ESI-MS (+10 V): m/z = 416 [M + NH4]+, 819 [2M + Na]+.
Gravilin (3). Colorless solid; m.p. 100–101 °C. [α]20D = +46.0 (c 0.8, MeOH). UV (MeOH): λ = 206, 300 (sh) nm. Fluorescence (MeOH): λex = 206 nm, λem = 286 nm. IR (ATR): ν = 2923, 2855, 1739, 1659, 1457, 1377, 1121, 1085, 961, 879 cm−1. 1H NMR (600 MHz, CDCl3): δ = 0.85 (3H, d, J = 6.8 Hz, H3-14), 0.97 (3H, s, H3-12), 1.10–1.15 (1H, m, H-13), 1.18–1.22 (1H, m, H-15b), 1.24 (3H, s, H3-20), 1.29 (3H, s, H3-19), 1.31–1.37 (1H, m, H-16b), 1.49 (1H, m, H-15a),a 1.51 (1H, m, H-6α),a 1.52 (2H, m, H2-5),a 1.54 (1H, m, H-8),a 1.58 (1H, m, H-3β),a 1.62 (1H, m, H-7),a 1.63 (1H, m, H-16a),a 1.80 (1H, dd, J = 13.3, 7.9 Hz, H-3α), 1.86–1.92 (1H, m, H-6β), 2.28 (1H, dd, J = 15.1, 7.9 Hz, H-2β), 2.32 (1H, t, J = 7.5 Hz, H-10), 2.42 (1H, dd, J = 6.9, 3.9 Hz, H-9), 2.48–2.56 (1H, m, H-2α), 2.66 (1H, t, J = 6.4 Hz, H-17), 4.70 (1H, s, H-11a), 4.73 (1H, s, H-11b) [aChemical shift derived from the HSQC spectrum]. 13C NMR and DEPT (150 MHz, CDCl3): δ = 17.87 (C-14), 18.69 (C-20), 21.55 (C-12), 24.91 (C-19), 26.43 (C-16), 27.22 (C-3), 29.93 (C-6), 32.10 (C-15), 33.75 (C-2), 36.38 (C-13), 42.19 (C-5), 43.14 (C-4), 45.77 (C-10), 48.04 (C-9), 55.22 (C-7), 55.29 (C-8), 58.31 (C-18), 64.70 (C-17), 103.76 (C-11), 157.33 (C-1). MS (EI, 70 eV): m/z (%) = 288 [M+] (0.1), 255 (1), 207 (5), 189 (16), 161 (15), 147 (10), 135 (8), 121 (58), 107 (25), 91 (27), 81 (100), 67 (12), 59 (16), 41 (33). ESI-MS (+10 V): m/z = 289 [M + H]+, 306 [M + NH4]+.
1-O-Arachidonoyl-3-O-stearylglycerol (4). Colorless oil. UV (MeOH): λ = 205, 276 (sh) nm. Fluorescence (MeOH): λex = 205 nm, λem = 285 nm. IR (ATR): ν = 3455 (br), 3011, 2922, 2853, 1740, 1650, 1464, 1392, 1236, 1173, 1119, 720 cm−1. 1H NMR (600 MHz, CDCl3): δ = 0.87 (3H, t, J = 7.2 Hz, H3-18′′), 0.88 (3H, t, J = 6.8 Hz, H3-20′), 1.24 (24H, br s, H2-5′′–H2-16′′), 1.26–1.32 (10H, m, H2-18′, H2-19′, H2-3′′, H2-4′′, H2-17′′), 1.32–1.37 (2H, m, H2-17′), 1.53–1.58 (2H, m, H2-2′′), 1.71 (2H, quint, J = 7.5 Hz, H2-3′), 2.04 (2H, q, J = 7.2 Hz, H2-16′), 2.11 (2H, q, J = 7.3 Hz, H2-4′), 2.35 (2H, t, J = 7.5 Hz, H2-2′), 2.45 (1H, d, J = 4.5 Hz, OH), 2.80 (4H, q, J = 5.6 Hz, H2-7′, H2-13′), 2.83 (2H, t, J = 6.0 Hz, H2-10′), 3.41 (1H, dd, J = 9.8, 6.4 Hz, H-3b), 3.42–3.46 (2H, m, H2-1′′), 3.48 (1H, dd, J = 9.8, 4.5 Hz, H-3a), 3.96–4.01 (1H, m, H-2), 4.11 (1H, dd, J = 11.7, 6.4 Hz, H-1b), 4.16 (1H, dd, J = 11.7, 4.1 Hz, H-1a), 5.29–5.42 (8H, m, H-5′, H-6′, H-8′, H-9′, H-11′, H-12′, H-14′, H-15′). 13C NMR and DEPT (150 MHz, CDCl3): δ = 14.07 (C-20′), 14.11 (C-18′′), 22.57 (C-19′), 22.69 (C-17′′), 24.75 (C-3′), 25.61–25.63 (C-7′, C-10′, C-13′), 26.08 (C-3′′), 26.53 (C-4′), 27.22 (C-16′), 29.32 (C-17′), 29.47 (C-4′′), 29.58–29.66 (C-2′′, C-5′′–C-15′′), 31.52 (C-18′), 31.92 (C-16′′), 33.53 (C-2′), 65.50 (C-1), 68.87 (C-2), 71.37 (C-3), 71.78 (C-1′′), 127.53 (C-14′), 127.86, 128.13, 128.25, 128.86, 128.93 (C-6′, C-8′, C-9′, C-11′, C-12′), 128.59 (C-5′), 130.50 (C-15′), 173.68 (C-1′). ESI-MS (+10 V): m/z = 648.7 [M + NH4]+. ESI-MS (+25 V): m/z = 631.7 [M + H]+, 648.8 [M + NH4]+. ESI-MS (+75 V): m/z = 653.8 [M + Na]+. ESI-MS (−50 V): m/z = 689.4 [M + OAc]−.
(E)-2,6,10,14-Tetramethyl-14-vinylhexadeca-2,10,15-trien-5-one (5). Colorless solid. UV (MeOH): λ = 207, 218, 284 (sh) nm. Fluorescence (MeOH): λex = 218 nm, λem = 308 nm. IR (ATR): ν = 3490 (br), 2967, 2927, 2858, 2056, 1710, 1652, 1454, 1375, 1110, 996, 918 cm−1. 1H NMR (600 MHz, CDCl3): δ = 1.05 (3H, d, J = 6.8 Hz, H3-18), 1.27 (3H, s, H3-20), 1.27–1.30 (1H, m, H-7b), 1.30–1.34 (2H, m, H2-8), 1.56 (3H, s, H3-19), 1.57–1.61 (3H, m, H-7a, H2-13), 1.61 (3H, s, H3-17), 1.74 (3H, s, H3-1), 1.93 (2H, t, J = 7.1 Hz, H2-9), 2.02–2.07 (2H, m, H2-12), 2.48–2.58 (1H, m, H-6), 3.12 (2H, d, J = 6.8 Hz, H2-4), 5.05 (2H, d, J = 10.9 Hz, H-16b, H-22b), 5.07–5.14 (1H, m, H-11), 5.20 (2H, d, J = 17.3 Hz, H-16a, H-22a), 5.30 (1H, br t, J = 7.2 Hz, H-3), 5.90 (2H, dd, J = 17.3, 10.9 Hz, H-15, H-21). 13C NMR and DEPT (150 MHz, CDCl3): δ = 15.84 (C-19), 16.44 (C-18), 18.07 (C-17), 22.65 (C-12), 25.45 (C-8), 25.72 (C-1), 27.88 (C-20), 32.52 (C-7), 39.55 (C-9), 41.00 (C-4), 42.05 (C-13), 45.64 (C-6), 73.45 (C-14), 111.70 (C-16, C-22), 116.09 (C-3), 124.45 (C-11), 135.23, 135.39 (C-2, C-10), 145.00 (C-15, C-21), 213.13 (C-5). MS (EI, 70 eV): m/z (%) = 288 (2), 219 (5), 201 (4), 151 (21), 139 (19), 121 (33), 109 (34), 93 (93), 81 (100), 69 (83), 55 (71), 41 (90).
(+)-(S)-Geranyllinalool (6). Colorless oil. [α]20D = +23.8 (c 0.07, MeOH); lit.: [α]20D = +18.2 (c 0.06, CHCl3),32 [α]D = +14.5.26 CD (MeOH): Δε (λ) +1.94 (202 nm), +0.12 (247 nm). UV (MeOH): λ = 211, 280 (sh) nm. Fluorescence (MeOH): λex = 211 nm, λem = 305 nm. IR (ATR): ν = 3414 (br), 2965, 2924, 2855, 1724, 1447, 1376, 1109, 995, 919 cm−1. 1H NMR (600 MHz, CDCl3): δ = 1.27 (3H, s, H3-17), 1.54–1.58 (2H, m, H2-4), 1.58 (3H, s, H3-19), 1.59 (6H, s, H3-18, H3-20), 1.67 (3H, q, J = 1.3 Hz, H3-16), 1.94–1.99 (4H, m, H2-8, H2-12), 1.99–2.03 (2H, m, H2-5), 2.02–2.08 (4H, m, H2-9, H2-13), 5.05 (1H, dd, J = 10.5, 1.1 Hz, H-1b), 5.07–5.11 (2H, m, H-10, H-14), 5.13 (1H, tq, J = 7.2, 1.4 Hz, H-6), 5.21 (1H, dd, J = 17.3, 1.1 Hz, H-1a), 5.91 (1H, dd, J = 17.3, 10.9 Hz, H-2). 13C NMR and DEPT (150 MHz, CDCl3): δ = 16.00 (C-19), 16.03 (C-18), 17.69 (C-20), 22.71 (C-5), 25.69 (C-16), 26.55 (C-9), 26.75 (C-13), 27.90 (C-17), 39.69 (C-8), 39.71 (C-12), 42.06 (C-4), 73.49 (C-3), 111.67 (C-1), 124.09 (C-10), 124.19 (C-6), 124.38 (C-14), 131.27 (C-15), 135.05 (C-11), 135.61 (C-7), 145.05 (C-2). MS (EI, 70 eV): m/z (%) = 290 [M]+ (0.4), 272 (1), 257 (1), 229 (1), 203 (4), 189 (4), 161 (8), 147 (7), 135 (10), 121 (12), 107 (28), 93 (40), 81 (44), 69 (100), 55 (20), 41 (51). ESI-MS (+10 V): m/z = 291 [M + H]+, 308 [M + NH4]+.
(−)-(R)-Nephthenol (7). Colorless oil. [α]20D = −35.9 (c 1.0, MeOH) (lit.: [α]20D = −39.6 (c 1.11, CHCl3),39 [α]23D = −35 (c 1.0, CHCl3)42). CD (MeOH): Δε (λ) −7.78 (207 nm), +0.94 (240 nm). UV (MeOH): λ = 205, 284 (sh) nm. Fluorescence (MeOH): λex = 205 nm, λem = 288 nm. IR (ATR): ν = 3414 (br), 2924, 2855, 1740, 1725, 1665, 1473, 1440, 1379, 1130, 973, 932, 883, 837 cm−1. 1H NMR (600 MHz, CDCl3): δ = 1.15 (1H, s, OH), 1.19 (6H, s, H3-16, H3-17), 1.25–1.28 (1H, m, H-14b), 1.29–1.35 (1H, m, H-1), 1.55 (3H, s, H3-20), 1.55–1.57 (6H, m, H3-18, H3-19), 1.61–1.67 (1H, m, H-14a), 1.85–1.92 (1H, m, H-2b), 1.97–2.04 (2H, m, H-9b, H-13b), 2.05–2.16 (8H, m, H-2a, H2-5, H-6b, H-9a, H2-10, H-13a), 2.16–2.24 (1H, m, H-6a), 4.93 (1H, br t, J = 6.0 Hz, H-7), 4.99 (1H, br t, J = 6.4 Hz, H-11), 5.08–5.12 (1H, m, H-3). 13C NMR and DEPT (150 MHz, CDCl3): δ = 15.30 (C-19), 15.56 (C-18, C-20), 24.00 (C-10), 24.65 (C-6), 27.50 (C-17), 27.65 (C-16), 28.27 (C-14), 28.44 (C-2), 37.70 (C-13), 38.82 (C-5), 39.40 (C-9), 48.45 (C-1), 73.97 (C-15), 124.98 (C-11), 125.76 (C-7), 125.94 (C-3), 133.06 (C-8), 133.37 (C-4), 134.04 (C-12). MS (EI, 70 eV): m/z (%) = 290 [M]+ (1), 272 (26), 257 (15), 229 (14), 202 (12), 189 (19), 175 (12), 161 (25), 147 (26), 136 (37), 121 (74), 107 (78), 93 (99), 81 (84), 67 (96), 59 (100), 41 (69). ESI-MS (+10 V): m/z = 291 [M + H]+, 308 [M + NH4]+.
11,12-Epoxysarcophytol A (8). Yellow solid; m.p. 68–70 °C (lit.: m.p. 75–76 °C).45 [α]20D = +220 (c 0.1, MeOH) (lit.: [α]D = +229 (c 0.95),45 [α]20D = +218 (c 0.35, CHCl3),50 [α]21D = +203.3 (c 0.33, CHCl3)47). UV (MeOH): λ = 217 nm. Fluorescence (MeOH): λex = 217 nm, λem = 293 nm. IR (ATR): ν = 3425 (br), 2961, 2922, 2869, 1724, 1641, 1473, 1453, 1378, 1112, 995, 917, 871, 683 cm−1. 1H NMR (600 MHz, CDCl3): δ = 1.06 (3H, d, J = 6.8 Hz, H3-16), 1.08 (3H, d, J = 6.8 Hz, H3-17), 1.29 (3H, s, H3-20), 1.45–1.52 (1H, m, H-10b), 1.58 (3H, d, J = 0.8 Hz, H3-19), 1.73 (3H, d, J = 0.8 Hz, H3-18), 1.82–1.89 (1H, m, H-10a), 1.97 (1H, dd, J = 15.4, 4.5 Hz, H-13a), 2.04–2.09 (1H, m, H-9b), 2.10 (1H, dd, J = 15.4, 7.5 Hz, H-13b), 2.13–2.19 (1H, m, H-5b), 2.21 (1H, br s, OH), 2.22–2.28 (4H, m, H-5a, H-6a, H-6b, H-9a), 2.66 (1H, sept, J = 6.8 Hz, H-15), 3.18 (1H, t, J = 6.4 Hz, H-11), 4.72 (1H, br t, J = 6.0 Hz, H-14), 5.10 (1H, br t, J = 6.4 Hz, H-7), 5.75 (1H, br dd, J = 10.5, 1.7 Hz, H-3), 5.98 (1H, d, J = 10.9 Hz, H-2). 13C NMR and DEPT (150 MHz, CDCl3): δ = 15.02 (C-19), 17.26 (C-18), 19.44 (C-20), 23.79 (C-17), 24.12 (C-10), 24.22 (C-16), 25.96 (C-6), 27.63 (C-15), 36.45 (C-9), 38.33 (C-5), 42.15 (C-13), 58.60 (C-11), 59.98 (C-12), 65.76 (C-14), 118.32 (C-2), 119.52 (C-3), 126.90 (C-7), 133.64 (C-8), 136.78 (C-4), 148.45 (C-1).
Isodecaryiol (9). Colorless crystals, m.p. 94.0–94.5 °C. [α]20D = +14.0 (c 0.1, MeOH). UV (MeOH): λ = 206, 250 (sh) nm. Fluorescence (MeOH): λex = 206 nm, λem = 301 nm. IR (ATR): ν = 3336 (br), 2972, 2914, 2856, 1474, 1428, 1378, 1281, 1246, 1195, 1178, 1142, 1119, 1067, 1017, 981, 968, 937, 920, 894, 849, 829, 804, 744 cm−1. 1H NMR (600 MHz, CDCl3): δ = 0.89 (1H, ddt, J = 13.3, 11.6, 3.6 Hz, H-14b), 1.10 (3H, s, H3-16), 1.12 (3H, s, H3-17), 1.15 (3H, s, H3-18), 1.23–1.32 (1H, m, H-2α), 1.27 (1H, OH, br s), 1.28–1.34 (1H, m, H-14a), 1.53 (1H, ddd, J = 14.4, 6.0, 3.3 Hz, H-5b), 1.55 (3H, s, H3-20), 1.56 (3H, s, H3-19), 1.58 (1H, tt, J = 11.6, 1.9 Hz, H-1), 1.72 (1H, ddd, J = 12.5, 5.4, 2.0 Hz, H-2β), 1.81 (1H, ddd, J = 14.5, 11.2, 3.3 Hz, H-5a), 1.88 (1H, dddd, J = 15.4, 7.6, 3.9, 1.9 Hz, H-6b), 1.99–2.03 (1H, m, H-10b), 2.03 (2H, br dd, J = 8.9, 3.7 Hz, H-13a, H-13b), 2.16 (1H, ddd, J = 14.0, 11.7, 3.8 Hz, H-9b), 2.22–2.29 (1H, m, H-9a), 2.48 (1H, dtd, J = 15.0, 11.4, 3.6 Hz, H-10a), 2.62 (1H, dddd, J = 15.2, 11.4, 9.9, 3.6 Hz, H-6a), 4.20 (1H, dt, J = 11.7, 5.9 Hz, H-3), 4.88 (1H, br d, J = 11.6 Hz, H-11), 5.24 (1H, br d, J = 9.4 Hz, H-7). 13C NMR and DEPT (150 MHz, CDCl3): δ = 14.82 (C-19), 15.20 (C-20), 22.25 (C-17), 23.76 (C-6), 24.22 (C-18), 25.19 (C-14), 25.27 (C-10), 28.93 (C-2), 29.61 (C-16), 36.42 (C-13), 38.06 (C-5), 39.30 (C-9), 39.97 (C-1), 70.40 (C-3), 75.26 (C-15), 76.79 (C-4), 127.70 (C-7), 128.08 (C-11), 132.59 (C-8), 133.02 (C-12). MS (EI, 70 eV): m/z (%) = 306 [M]+ (4), 288 (10), 273 (3), 263 (4), 205 (3), 192 (4), 175 (5), 161 (7), 151 (13), 136 (24), 123 (20), 107 (30), 95 (32), 81 (55), 69 (74), 55 (430), 43 (100). ESI-MS (+10 V): m/z = 307 [M + H]+.
Crystal data for 9. C20H34O2, M = 306.47 g mol−1, crystal size: 0.27 × 0.25 × 0.22 mm3, monoclinic, space group C2, a = 24.3170(11) Å, b = 13.7760(6) Å, c = 67.036(3) Å, β = 91.6033(13)°, V = 22
447.5(17) Å3, Z = 48, Z′ = 12, ρcalcd = 1.088 g cm−3, μ = 0.519 mm−1, T = 100(2) K, λ = 1.54178 Å, θ range = 1.98–67.68°, reflections collected 182016 (Rint = 0.0214), 2461 refined parameters, R1 = 0.0347, wR2 = 0.1178 [I > 2 σ(I)]; maximal residual electron density: 0.42 e Å−3; Flack parameter: χ = 0.08 (4). The thermal displacement parameters of the independent molecules A and E (Fig. S71†) are indicative of a disorder, which was impossible to resolve as the whole molecule should be disordered. The disorder resulted in an alert B in the Hirshfeld test. CCDC-1424360 contains the supplementary crystallographic data for this structure.
Acknowledgements
Marie Pascaline Rahelivao thanks the European Commission (Erasmus Mundus Programme), the Gesellschaft von Freunden und Förderern der TU Dresden (GFF TU Dresden), and the TUD graduate academy for their support by providing scholarships. We would like to thank Dr. Shirley Parker-Nance (Nelson Mandela Metropolitan University, Port Elizabeth, and South African Institute for Aquatic Biodiversity, Grahamstown, South Africa) for the identification of the soft corals. We are grateful to Mr. Michel Ratsimbason (Centre National d'Application de Recherche Pharmaceutique, Antananarivo, Madagascar) for the antimalaria assay. We also thank Dr. Arndt W. Schmidt for his support in preparing the manuscript.Notes and references
- J.-H. Su, A. F. Ahmed, P.-J. Sung, C.-H. Chao, Y.-H. Kuo and J.-H. Sheu, J. Nat. Prod., 2006, 69, 1134–1139 CrossRef CAS PubMed.
- A. F. Ahmed, Z.-H. Wen, J.-H. Su, Y.-T. Hsieh, Y.-C. Wu, W.-P. Hu and J.-H. Sheu, J. Nat. Prod., 2008, 71, 179–185 CrossRef CAS PubMed.
- Y. Lu, C.-Y. Huang, Y.-F. Lin, Z.-H. Wen, J.-H. Su, Y.-H. Kuo, M. Y. Chiang and J.-H. Sheu, J. Nat. Prod., 2008, 71, 1754–1759 CrossRef CAS PubMed.
- Y.-Y. Lin, Y.-H. Jean, H.-P. Lee, W.-F. Chen, Y.-M. Sun, J.-H. Su, Y. Lu, S.-Y. Huang, H.-C. Hung, P.-J. Sung, J.-H. Sheu and Z.-H. Wen, PLoS One, 2013, 8, e62926 CAS.
- B.-W. Chen, C.-H. Chao, J.-H. Su, C.-Y. Huang, C.-F. Dai, Z.-H. Wen and J.-H. Sheu, Tetrahedron Lett., 2010, 51, 5764–5766 CrossRef CAS.
- J.-H. Su, C.-Y. Huang, P.-J. Li, Y. Lu, Z.-H. Wen, Y.-H. Kao and J.-H. Sheu, Arch. Pharmacal Res., 2012, 35, 779–784 CrossRef CAS PubMed.
- D. Chen, W. Chen, D. Liu, L. van Ofwegen, P. Proksch and W. Lin, J. Nat. Prod., 2013, 76, 1753–1763 CrossRef CAS PubMed.
- J. J. Poza, R. Fernández, F. Reyes, J. Rodríguez and C. Jiménez, J. Org. Chem., 2008, 73, 7978–7984 CrossRef CAS PubMed.
- A. Longeon, M.-L. Bourguet-Kondracki and M. Guyot, Tetrahedron Lett., 2002, 43, 5937–5939 CrossRef CAS.
- Y. Hou and L. Harinantenaina, Curr. Med. Chem., 2010, 17, 1191–1219 CrossRef CAS PubMed.
- V. Anjaneyulu, P. V. S. Rao and P. Radhika, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 1999, 38, 357–360 Search PubMed.
- V. Anjaneyulu, P. V. S. Rao, P. Radhika, H. Laatsch and L. N. Misra, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 2000, 39, 121–124 Search PubMed.
- P. Radhika, M. Cabeza, E. Bratoeff and G. García, Steroids, 2004, 69, 439–444 CrossRef CAS PubMed.
- G. Rücker, Angew. Chem., Int. Ed. Engl., 1973, 85, 895–907 Search PubMed.
- M. Bordoloi, V. S. Shukla, S. C. Nath and R. P. Sharma, Phytochemistry, 1989, 28, 2007–2037 CrossRef CAS.
- Dictionary of Marine Natural Products with CD-ROM, ed. J. W. Blunt and M. H. G. Munro, Taylor & Francis, Boca Raton, FL, USA, 2008 Search PubMed.
- G. W. Patterson, Lipids, 1971, 6, 120–127 CrossRef CAS.
- G. W. Patterson, Comp. Biochem. Physiol., 1968, 24, 501–505 CAS.
- K. Iguchi, S. Saitoh and Y. Yamada, Chem. Pharm. Bull., 1989, 37, 2553–2554 CrossRef CAS.
- W. Lu, C. Zhang, L. Zeng and J. Su, Steroids, 2004, 69, 803–808 CrossRef CAS PubMed.
- M. P. Rahelivao, M. Gruner, H. Andriamanantoanina, B. Andriamihaja, I. Bauer and H.-J. Knölker, Mar. Drugs, 2015, 13, 4197–4216 CrossRef PubMed.
- Y. T. Lin, Y. S. Cheng and Y. H. Kuo, J. Chin. Chem. Soc., 1970, 17, 111–113 CrossRef CAS.
- Y. H. Kuo, Y. S. Cheng, S. T. Kao and Y. T. Lin, J. Chin. Chem. Soc., 1973, 20, 83–86 CrossRef CAS.
- F. Song, X. Fan, X. Xu, J. Zhao, Y. Yang and J. Shi, J. Nat. Prod., 2004, 67, 1644–1649 CrossRef CAS PubMed.
- W. Dong, D. Yang and R. Lu, Planta Med., 2010, 76, 454–457 CrossRef CAS PubMed.
- E. Demole and E. Lederer, Bull. Soc. Chim. Fr., 1958, 1128–1137 CAS.
- B. Kimland and T. Norin, Acta Chem. Scand., 1967, 21, 825–826 CrossRef CAS.
- F. Bohlmann and A. Suwita, Phytochemistry, 1979, 18, 2046–2049 CrossRef CAS.
- F. Bohlmann, C. Zdero, W.-R. Abraham, A. Suwita and M. Grenz, Phytochemistry, 1980, 19, 873–879 CrossRef CAS.
- M. Verzele, G. Maes, A. Vuye, M. Godefroot, M. van Alboom, J. Vervisch and P. Sandra, J. Chromatogr., A, 1981, 205, 367–386 CrossRef CAS.
- V. I. Roshchin, R. A. Baranova and V. A. Solov'ev, Chem. Nat. Compd., 1986, 22, 156–163 Search PubMed.
- A. Svatoš, K. Urbanová and I. Valterová, Collect. Czech. Chem. Commun., 2002, 67, 83–90 CrossRef.
- P. Waridel, J.-L. Wolfender, J.-B. Lachavanne and K. Hostettmann, Phytochemistry, 2003, 64, 1309–1317 CrossRef CAS PubMed.
- L. Jirovetz, G. Buchbauer, T. Schweiger, Z. Denkova, A. Slavchev, A. Stoyanova, E. Schmidt and M. Geissler, Nat. Prod. Commun., 2007, 2, 407–412 CAS.
- A. Lapczynski, S. P. Bhatia, C. S. Letizia and A. M. Api, Food Chem. Toxicol., 2008, 46, S176–S178 CrossRef CAS PubMed.
- K. C. Geőcze, L. C. A. Barbosa, P. H. Fidêncio, F. O. Silvério, C. F. Lima, M. C. A. Barbosa and F. M. D. Ismail, Food Res. Int., 2013, 54, 1–8 CrossRef.
- F. J. Schmitz, D. J. Vanderah and L. S. Ciereszko, J. Chem. Soc., Chem. Commun., 1974, 407–408 RSC.
- S. Carmely, A. Groweiss and Y. Kashman, J. Org. Chem., 1981, 46, 4279–4284 CrossRef CAS.
- R. Schwabe, I. Farkas and H. Pfander, Helv. Chim. Acta, 1988, 71, 292–297 CrossRef CAS.
- A. Rudi, T. Lev-Ari Dayan, M. Aknin, E. M. Gaydou and Y. Kashman, J. Nat. Prod., 1998, 61, 872–875 CrossRef CAS PubMed.
- Y.-P. Shi, A. D. Rodríguez and O. L. Padilla, J. Nat. Prod., 2001, 64, 1439–1443 CrossRef CAS.
- K. H. Shaker, M. Müller, M. A. Ghani, H.-M. Dahse and K. Seifert, Chem. Biodiversity, 2010, 7, 2007–2015 CAS.
- Y. Kashman, M. Bodner, Y. Loya and Y. Benayahu, Isr. J. Chem., 1977, 16, 1–3 CrossRef CAS.
- A. Groweiss, Y. Kashman, D. J. Vanderah, B. Tursch, P. Cornet, J. C. Braekman and D. Daloze, Bull. Soc. Chim. Belg., 1978, 87, 277–283 CrossRef CAS.
- B. F. Bowden, J. C. Coll and D. M. Tapiolas, Aust. J. Chem., 1983, 36, 2289–2295 CrossRef CAS.
- G. J. Greenland and B. F. Bowden, Aust. J. Chem., 1994, 47, 2013–2021 CrossRef CAS.
- C. A. Gray, M. T. Davies-Coleman and M. H. Schleyer, J. Nat. Prod., 2000, 63, 1551–1553 CrossRef CAS.
- F. Cao, J. Zhou, K.-X. Xu, M.-Q. Zhang and C.-Y. Wang, Nat. Prod. Commun., 2013, 8, 1675–1678 CAS.
- C. Bhujanga Rao, D. C. Babu, T. V. Bharadwaj, D. Srikanth, K. S. Vardhan, T. V. Raju, R. A. Bunce and Y. Venkateswarlu, Nat. Prod. Res., 2014, 29, 70–76 CrossRef PubMed.
- J. Lan, Z. Liu, H. Yuan, L. Peng, W.-D. Z. Li, Y. Li, Y. Li and A. S. C. Chan, Tetrahedron Lett., 2000, 41, 2181–2184 CrossRef CAS.
- W. H. Gerwick, W. Fenical, D. Van Engen and J. Clardy, J. Am. Chem. Soc., 1980, 102, 7991–7993 CrossRef CAS.
- W. H. Gerwick and W. Fenical, J. Org. Chem., 1983, 48, 3325–3329 CrossRef CAS.
- Y. Venkateswarlu and M. A. F. Biabani, Phytochemistry, 1995, 40, 331–333 CrossRef CAS.
- L. Li, L. Sheng, C.-Y. Wang, Y.-B. Zhou, H. Huang, X.-B. Li, J. Li, E. Mollo, M. Gavagnin and Y.-W. Guo, J. Nat. Prod., 2011, 74, 2089–2094 CrossRef CAS PubMed.
- T.-C. Tsai, Y.-J. Wu, J.-H. Su, W.-T. Lin and Y.-S. Lin, Mar. Drugs, 2013, 11, 114–123 CrossRef CAS PubMed.
- W. H. Gerwick, W. Fenical and M. U. S. Sultanbawa, J. Org. Chem., 1981, 46, 2233–2241 CrossRef CAS.
- R. G. Salomon, B. Basu, S. Roy and N. D. Sachinvala, J. Am. Chem. Soc., 1991, 113, 3096–3106 CrossRef CAS.
- S. D. Rosa, C. Iodice, M. Khalaghdoust, S. Oryan and A. Rustaiyan, Phytochemistry, 1999, 51, 1009–1012 CrossRef.
- H. Yamase, K. Umemoto, T. Ooi and T. Kusumi, Chem. Pharm. Bull., 1999, 47, 813–818 CrossRef CAS.
- G. A. Frangulyan, A. V. Komkov, E. P. Prokof'ev, V. M. Belotserkovets, E. É. Lavut and V. I. Panov, Chem. Nat. Compd., 1987, 23, 168–173 CrossRef.
- J. J. Myher and A. Kuksis, Can. J. Biochem. Cell Biol., 1984, 62, 352–362 CAS.
- J. J. Myher and A. Kuksis, J. Chromatogr., A, 1989, 471, 187–204 CrossRef CAS PubMed.
- A. B. Imbs, H. V. Luu and L. Q. Pham, Chem. Nat. Compd., 2007, 43, 610–611 CrossRef CAS.
- A. B. Imbs, Russ. J. Mar. Biol., 2013, 39, 153–168 CrossRef CAS.
- J. R. Maxwell, A. G. Douglas, G. Eglinton and A. McCormick, Phytochemistry, 1968, 7, 2157–2171 CrossRef CAS.
- R. E. Cox, A. L. Burlingame, D. M. Wilson, G. Eglinton and J. R. Maxwell, J. Chem. Soc., Chem. Commun., 1973, 284–285 RSC.
- P. Metzger and E. Casadevall, Tetrahedron Lett., 1983, 24, 4013–4016 CrossRef CAS.
- P. Metzger, E. Casadevall, M. J. Pouet and Y. Pouet, Phytochemistry, 1985, 24, 2995–3002 CrossRef CAS.
- S. Endo, N. Hiramatsu, K. Hayakawa, M. Okamura, A. Kasai, Y. Tagawa, N. Sawada, J. Yao and M. Kitamura, Mol. Pharmacol., 2007, 72, 1337–1348 CrossRef CAS PubMed.
- H. Serizawa, A. B. Argade, A. Datwani, N. Spencer, Y. Pan and F. Ermini, WO2014163643, 2014 Search PubMed.
- M.-C. Blanc, P. Bradesi and J. Casanova, Magn. Reson. Chem., 2005, 43, 176–179 CrossRef CAS PubMed.
- B. Tursch, J. C. Braekman and D. Daloze, Bull. Soc. Chim. Belg., 1975, 84, 767–774 CrossRef CAS.
- J. C. Coll, B. F. Bowden, G. M. König, R. Braslau and I. R. Price, Bull. Soc. Chim. Belg., 1986, 95, 815–834 CrossRef CAS.
- D. V. Rao, T. S. Rao and C. B. Rao, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 1990, 29, 683–684 Search PubMed.
- H. Gross, S. Kehraus, M. Nett, G. M. König, W. Beil and A. D. Wright, Org. Biomol. Chem., 2003, 1, 944–949 CAS.
- A. Rudi, G. Shmul, Y. Benayahu and Y. Kashman, Tetrahedron Lett., 2006, 47, 2937–2939 CrossRef CAS.
- E. Fattorusso, A. Romano, O. Taglialatela-Scafati, C. Irace, C. Maffettone, G. Bavestrello and C. Cerrano, Tetrahedron, 2009, 65, 2898–2904 CrossRef CAS.
- H. I. Januar, E. Chasanah, C. A. Motti, D. M. Tapiolas, C. H. Liptrot and A. D. Wright, Mar. Drugs, 2010, 8, 2142–2152 CrossRef CAS PubMed.
- K. M. Steed and J. W. Steed, Chem. Rev., 2015, 115, 2895–2933 CrossRef CAS PubMed.
- H. D. Flack, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 1983, 39, 876–881 CrossRef.
- Bruker, APEX2 Software Suite for Crystallographic Programs, Bruker AXS Inc., Madison, WI, USA, 2009 Search PubMed.
- Bruker, Area Detector Control and Integration Software. Version 5.1. In: SMART and SAINT, Bruker Analytical X-ray Instruments Inc., Madison, WI, USA, 1996 Search PubMed.
- G. M. Sheldrick, SADABS. Program for Absorption Correction. Version 2.10, University of Göttingen, Germany, 1996 Search PubMed.
- M. C. Burla, M. Camalli, B. Carrozzini, G. L. Cascarano, C. Giacovazzo, G. Polidori and R. Spagna, J. Appl. Crystallogr., 2003, 36, 1103 CrossRef CAS.
- G. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565–565 CrossRef CAS.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van de Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS.
- W. Trager and J. B. Jensen, Science, 1976, 193, 673–675 CAS.
- W. Trager and J. B. Jensen, J. Parasitol., 2005, 91, 484–486 CrossRef PubMed.
- Y. Corbett, L. Herrera, J. Gonzalez, L. Cubilla, T. L. Capson, P. D. Coley, T. A. Kursar, L. I. Romero and E. Ortega-Barria, Am. J. Trop. Med. Hyg., 2004, 70, 119–124 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Tables S1–S10; Fig. S1–S70 (copies of the 1H, 13C, and 2D NMR spectra for the compounds 1, 3–5, and 9); Fig. S71 (symmetry-independent molecules of compound 9); details of the antimalarial assay. CCDC 1424360. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ob02280k |
| This journal is © The Royal Society of Chemistry 2016 |