
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multivalency effects on Pseudomonas aeruginosa biofilm inhibition and dispersal by glycopeptide dendrimers targeting lectin LecA†
Myriam
Bergmann
a,
Gaëlle
Michaud
a,
Ricardo
Visini
a,
Xian
Jin
a,
Emilie
Gillon
b,
Achim
Stocker
a,
Anne
Imberty
b,
Tamis
Darbre
*a and
Jean-Louis
Reymond
*a
aDepartment of Chemistry and Biochemistry, University of Berne, Freiestrasse 3, 3012 Berne, Switzerland. E-mail: jean-louis.reymond@dcb.unibe.ch; Fax: +41 31 631 8057
bCentre de Recherches sur les Macromolécules Végétales, UPR5301, CNRS and Université Grenoble Alpes, 601 rue de la Chimie, F38041 Grenoble, France
First published on 21st September 2015
Abstract
The galactose specific lectin LecA partly mediates the formation of antibiotic resistant biofilms by Pseudomonas aeruginosa, an opportunistic pathogen causing lethal airways infections in immunocompromised and cystic fibrosis patients, suggesting that preventing LecA binding to natural saccharides might provide new opportunities for treatment. Here 8-fold (G3) and 16-fold (G4) galactosylated analogs of GalAG2, a tetravalent G2 glycopeptide dendrimer LecA ligand and P. aeruginosa biofilm inhibitor, were obtained by convergent chloroacetyl thioether (ClAc) ligation between 4-fold or 8-fold chloroacetylated dendrimer cores and digalactosylated dendritic arms. Hemagglutination inhibition, isothermal titration calorimetry and biofilm inhibition assays showed that G3 dendrimers bind LecA slightly better than their parent G2 dendrimers and induce complete biofilm inhibition and dispersal of P. aeruginosa biofilms, while G4 dendrimers show reduced binding and no biofilm inhibition. A binding model accounting for the observed saturation of glycopeptide dendrimer galactosyl groups and LecA binding sites is proposed based on the crystal structure of a G3 dendrimer LecA complex.
Introduction
Pseudomonas aeruginosa is an opportunistic Gram negative human pathogen causing lethal airways infections in immunocompromised and cystic fibrosis patients by forming antibiotic resistant biofilms.1 One of the therapeutic approaches to fight these infections consists of developing biofilm inhibitors to restore antibiotic sensitivity with a reduced effect of the resistance phenomena.2 It has been shown that tissue attachment and biofilm formation in P. aeruginosa are mediated in part by the galactose specific lectin LecA (PA-IL)3 and the fucose specific lectin LecB (PA-IIL),4 as evidenced by the impaired biofilm formation in deletion mutants5 and case reports of treating P. aeruginosa infections using lectin-binding saccharide solutions,6 leading to the hypothesis that targeted inhibitors of these lectins might allow control of biofilms.7Following this hypothesis ligands of LecA,8 LecB,8 or both9 have been reported featuring various monovalent or multivalent glycosides displayed on a multivalent scaffold.10 However only very few examples have been reported to actually interfere with Pseudomonas aeruginosa biofilm formation.11 In particular we recently reported glycopeptide dendrimers displaying four α-L-C-fucoside groups (FD2: (Fuc-α-CH2CO-Lys-Pro-Leu)4(Lys-Phe-Lys-Ile)2Lys-His-Ile-NH2)12 or analogs with four galactoside groups (GalAG2: (Gal-β-OC6H4CO-Lys-Pro-Leu)4(Lys-Phe-Lys-Ile)2Lys-His-Ile-NH2; GalBG2: (Gal-β-S CH2CH2CO-Lys-Pro-Leu)4(Lys-Phe-Lys-Ile)2Lys-His-Ile-NH2)13 at the end of a common second generation (G2) peptide dendrimer scaffold.14 These dendrimers bound tightly to their respective lectin, and inhibited the formation and induced partial dispersion of P. aeruginosa biofilms, representing an interesting example of bioactive synthetic dendrimers.15
Tight lectin binding and biofilm inhibition by these tetravalent G2 dendrimers depended on a multivalency effect since the lower generation analogs (G0 and G1) were essentially inactive.16 In the case of the GalAG2 lectin binding was enhanced by a specific CH–π interaction between the (ε)-CH of His50 on LecA and the aromatic ring of the GalA aglycone leading to further binding interactions between the terminal tripeptide arm of the dendrimer and LecA, however optimization of the amino acid sequence of this tripeptide only resulted in modest activity improvements.17 Considering that many of the reported high affinity multivalent glycosidic ligands for lectins feature an 8-fold or higher multivalency, we asked the question whether the binding affinity and biological activity of dendrimers GalAG2 and GalBG2 might be increased in higher valency analogs as observed in other series of tight binding LecA ligands.8 Various G3 and G4 analogs of GalAG2 and GalBG2 were prepared using the multiple chloroacetyl cysteine (ClAc) thioether ligation as the key step.18 Hemagglutination inhibition, isothermal titration calorimetry and biofilm inhibition assays are reported that show that G3 dendrimers bind LecA slightly better than their parent G2 dendrimers and induce complete biofilm inhibition and dispersal of P. aeruginosa biofilms, while G4 dendrimers have reduced binding and no biofilm inhibition. A binding model accounting for the observed saturation of glycopeptide dendrimer galactosyl groups and LecA binding sites is proposed based on a crystal structure of a G3 dendrimer LecA complex.
Results and discussion
Synthesis
Although we previously reported a single case of a G3 glycopeptide dendrimer by direct SPPS,12c the synthesis was difficult and the isolated yield was very low due to incomplete coupling of the glycosidic end group to the multiple N-termini even with low loading resin (∼0.2 mmol g−1) and large excess of glycosylated reagent. This is probably a result of the steric crowding in the G3 dendrimer with some of the amino termini not exposed to the coupling reagents. This step proved to be a limiting factor across various peptide dendrimers and carbohydrate building blocks even for G2 peptide dendrimers despite the fact that the reaction is a standard amide bond formation step. We therefore turned our attention to a convergent approach using our recently reported multivalent ClAc ligation method as an efficient strategy to access G3 and G4 peptide dendrimers (Fig. 1).18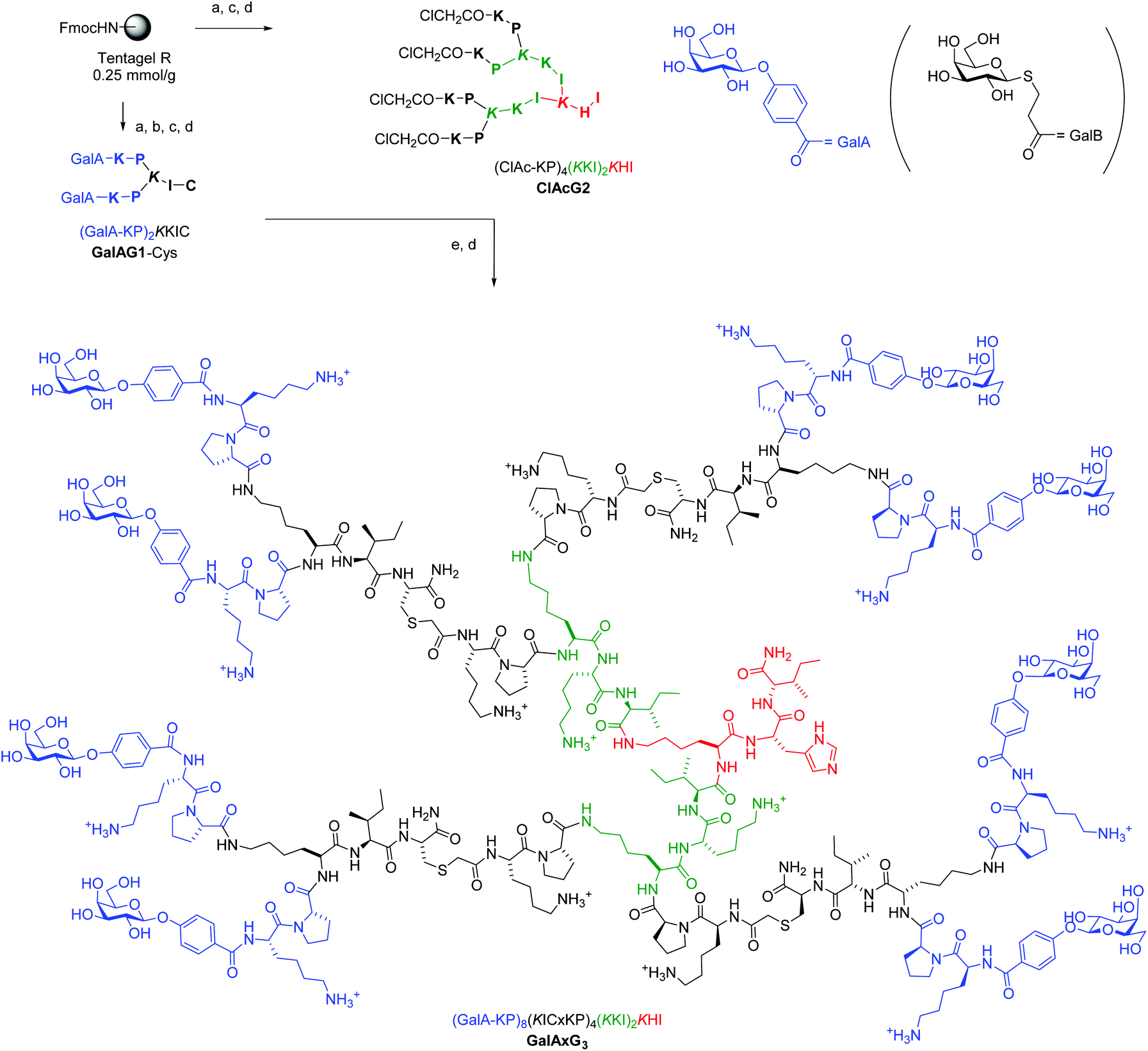 | ||
Fig. 1 Convergent synthesis and structural formula of the octavalent G3 glycopeptide dendrimer GalAxG3 with the corresponding sequence notation. Sequences are written with N-terminus at left and C-terminus at right, the C-terminus of the peptide is carboxamide (CONH2). One letter codes are used for standard amino acids, the branching diamino acid lysine is in italics and extended on both amino groups, x = –S–CH2–CO–. The detailed structure of the terminal galactosides abbreviated GalA and GalB in the abbreviated sequence notation of the dendrimers used in Table 1 and 2 are shown at top right. Conditions: (a) SPPS: Fmoc deprotection with piperidine/DMF 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 (v/v), 2 × 15 min; amino acid coupling (3 eq. Fmoc-aa-OH, 3 eq. PyBOP, 5 eq. DIEA in NMP), 2–4 hours; carbohydrate coupling: 4 eq. Ac4GalA-OH, 3 eq. HCTU, 5 eq. DIPEA in NMP, overnight; (b) deacetylation: MeOH/25% NH3/H2O (8:1:1, v/v/v); (c) cleavage: TFA/TIS/H2O (95:2.5:2.5, v/v/v); (d) RP-HPLC purification; (e) ClAc ligation: ClAcG2 (1 eq.), GalAG1-Cys (6 eq.), KI (20 eq.) DIPEA (55 eq.) in DMF/H2O (1:1, v/v), RT, overnight under an argon atmosphere. :4 (v/v), 2 × 15 min; amino acid coupling (3 eq. Fmoc-aa-OH, 3 eq. PyBOP, 5 eq. DIEA in NMP), 2–4 hours; carbohydrate coupling: 4 eq. Ac4GalA-OH, 3 eq. HCTU, 5 eq. DIPEA in NMP, overnight; (b) deacetylation: MeOH/25% NH3/H2O (8:1:1, v/v/v); (c) cleavage: TFA/TIS/H2O (95:2.5:2.5, v/v/v); (d) RP-HPLC purification; (e) ClAc ligation: ClAcG2 (1 eq.), GalAG1-Cys (6 eq.), KI (20 eq.) DIPEA (55 eq.) in DMF/H2O (1:1, v/v), RT, overnight under an argon atmosphere. | ||
SPPS was first used to prepare a G1 glycopeptide dendrimer featuring the N-terminal dipeptide LysPro of GalAG2 known to engage in direct contact with the lectin, which was acylated at its two N-termini with either 4-carboxyphenyl-β-galactoside to yield GalAG1-Cys or carboxypropyl-β-thiogalactoside to yield GalBG1-Cys. The carbohydrate building block coupling step was not problematic at the G1 level and both products were obtained in good yield after purification by preparative HPLC. The amino acid sequence of GalAG2 was then used to design the 2-fold, 4-fold and 8-fold chloroacetylated core dendrimers ClAcG1, ClAcG2 and ClAcG3 consisting of dipeptide branches. The chloroacetylated dendrimer cores ClAcPSG2 and ClAcPSG3 were also prepared since they were found previously to give high yields in ClAc ligation reactions.18 The isolated yields of these various chloroacetylated core dendrimers after SPPS and HPLC purification were above 25% for G1 and G2, but only 3–5% for G3 cores, reflecting the generally more difficult synthesis of G3 peptide dendrimers (Table 1).
| Name | Sequencea | MS calc./obs.b | Yield mgc (%) |
|---|---|---|---|
| a Sequence notation for peptide dendrimers using single letter amino acid codes for L-amino acids and indicating the branching lysine in italics. See Fig. 1 for the correspondence between sequence notation and complete structural formula, illustrated for dendrimer GalAxG3. Further abbreviations: ClAc = ClCH2CO–, x = –CH2SCH2CO–, GalA = 4-(β-galactosyloxy)benzoyl, GalB = (β-galactosyl)SCH2CH2CO, the peptide C-terminus is carboxamide (CONH2). b ESI+ or MALDI. c Yields are for purified compounds after SPPS (upper part) or ClAc ligation (lower part) and preparative HPLC. | |||
| Components from SPPS | |||
| GalAG1-Cys | (GalA-KP)2KIC | 1376.6/1377 | 63 (41.6%) |
| GalBG1-Cys | (GalB-KP)2KIC | 1312.6/1312.6 | 57 (39.5%) |
| ClAcG1 | (ClAc-KI)2KHI | 1031.1/1030 | 41.4 (36.55%) |
| ClAcG2 | (ClAc-KP)4(KKI)2KHI | 2341.6/2341 | 64 (25%) |
| ClAcG3 | (ClAc-KP)8(KLF)4(KKI)2KHI | 5102.7/5102 | 19.3 (3.4%) |
| ClAcPSG2 | (ClAc-PS)4(KPS)2KPS | 1996.8/1996.9 | 66 (30%) |
| ClAcPSG3 | (ClAc-PS)8(KPS)4(KPS)2KPS | 4289/4290 | 25 (5%) |
| GalAPSG2 | (GalA-KPL)4(KPS)2KPS | 3437.9/3478 | 26.4 (6%) |
| Dendrimers prepared by ClAc ligation | |||
| GalAxG2 | (GalA-KP)4(KICxKI)2KHI | 3711.3/3711 | 6.9 (64%) |
| GalAxG3 | (GalA-KP)8(KICxKP)4(KKI)2KHI | 7702/7701 | 7.3 (70%) |
| GalAxG4 | (GalA-KP)16(KICxKP)8(KLF)4(KKI)2KHI | 15823.6/15825 |
1.1 (12%) |
| GalAxPSG3 | (GalA-KP)8(KICxPS)4(KPS)2KPS | 7357.3/7358 | 9.7 (74.5%) |
| GalAxPSG4 | (GalA-KP)16(KICxPS)8(KPS)4(KPS)2KPS | 15009.9/15010.6 | 9.1 (80.8%) |
| GalBxG2 | (GalB-KP)4(KICxKI)2KHI | 3583.4/3582.7 | 6.7 (64.5%) |
| GalBxG3 | (GalB-KP)8(KICxKP)4(KKI)2KHI | 7446.2/7445 | 6.2 (64%) |
The ClAc ligation was then performed to append to different dendrimer cores multiple copies of the GalAG1-Cys dendrimer bearing a phenyl-β-galactoside LecA ligand. Dendrimers ClAcG1 and ClAcG2 were also ligated to GalBG1-Cys to obtain G2 and G3 analogs with a thiopropyl-β-galactoside endgroup. The ligation reactions generally proceeded cleanly and gave isolated yields in the range 64–80%, except for GalAxG4 which was obtained in only 12% yield due to a difficult purification process.
Hemagglutination inhibition assays
Binding of the various glycopeptide dendrimers to lectin LecA was first investigated in a hemagglutination inhibition assay (Table 2, left part). In this assay compounds are tested in two-fold dilution series for their inhibition of the agglutination of erythrocytes induced by the lectin, providing minimal hemagglutination inhibition concentration (MIC) values. The MIC values are then scaled to the reference MIC value of free galactose in each assay to yield relative potencies (r.p.) of inhibition. Although absolute MIC values of both test compounds and the reference galactose may vary depending on the erythrocyte source and lectin concentration, r.p. values are quite reproducible and a good indicator of multivalent binding potential. Here the primary r.p. values were further converted to a secondary r.p. value by scaling to the r.p. of the parent monovalent glycopeptides GalAG0 and GalBG0 as references. This allowed the quantification of multivalency effects independently of affinity effects on monovalent ligand binding. These effects were quite substantial in the case of GalAG0, which inhibits hemagglutination at a 40-fold lower concentration than free D-galactose. A similar effect also occurs with p-nitrophenyl-β-galactoside and reflects a productive CH–π interaction of the aromatic aglycone with the C(ε)–H of His50 on the lectin.17 The r.p. values were furthermore scaled to the number of galactosyl endgroup (nGal) to correct for multivalency.| Ligand | Sequencea | Hemagglutination assayb | Isothermal titration calorimetry (ITC)c | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| n Gal | MIC μM | r.p./n | N | % bnd Gal | ΔH [kcal mol−1] | −TΔS | ΔG | K D [nM] | r.p./nGal | ||
| a Sequence notation for peptide dendrimers using single letter amino acid codes for L-amino acids and indicating the branching lysine in italics. See Fig. 1 for the correspondence between sequence notation and complete structural formula, illustrated for dendrimer GalAxG3. Further abbreviations: GalA = 4-(β-galactosyloxy)benzoyl, GAlB = (β-galactosyl)SCH2CH2CO, x = –CH2SCH2CO–, the peptide C-terminus is carboxamide (CONH2). b Minimal hemagglutination inhibition concentrations (MIC) were determined in two different series with the MIC for galactose being 42 mM or 3.125 mM, marked with *, nGal is the number of galactosyl groups per compound, r.p./n is the relative potency per galactosyl group relative to free galactose as a reference: MIC(galactose)/(n × MIC(compound)), r.p./nGalA or B is the relative potency per galactosyl group relative to GalAG0 or GalBG0 as a reference: MIC(GalAG0 or GalBG0)/(n × MIC(compound)). c Thermodynamic parameters and dissociation constants KD are reported as an average of two independent runs from ITC in 20 mM Tris, 100 mM NaCl, 100 μM CaCl2, pH = 7.5. Titration concentrations (ligand/LecA) are the following: NPG (3 mM/0.3 mM), GalAG0 (0.5 mM/0.0516 mM), GalAG1 (0.25 mM/0.0486 mM), GalAG2 (0.03 mM/0.018 mM), GalAxG2 (0.03 mM/0.016 mM), GalAxG3 (0.015 mM/0.017 mM), GalAxG3PS (0.015 mM/0.0146 mM), GalAxG4PS (0.01 mM/0.019 mM), GalBG0 (1.0 mM/0.091 mM), GalBG1 (0.25 mM/0.049 mM), GalBG2 (0.03 mM/0.018 mM), GalBxG2 (0.03 mM/0.021 mM), GalBxG3 (0.015 mM/0.013 mM). N = stoichiometry value in the ligand/galactose binding site on LecA. r.p./nGal = relative potency per galactosyl group relative to the parent monovalent ligand GalAG0 or GalBG0. | |||||||||||
| D-Galactose | 1 | 42000 (3125*) |
— | — | — | — | — | — | — | — | |
| NPG | p-Nitrophenyl-β-galactoside | 1 | — | — | 0.91 ± 0.03 | — | −10.6 ± 0.5 | 4.04 | 6.53 | 16000 ± 500 |
— |
| IPTG | Isopropyl-β-thiogalactoside | 1 | 10400 |
— | — | — | — | — | — | — | — |
| GalAG0 | (GalA-KPL) | 1 | 80* | 1 | 0.65 ± 0.02 | 100 | −17.8 ± 0.3 | 10.2 | −7.5 | 2960 ± 50 | 1 |
| GalAG1 | (GalA-KPL)2KHI | 2 | 31* | 1.3 | 0.302 ± 0.003 | 93 | −29 ± 0.5 | 19.3 | −9.7 | 83 ± 12 | 18 |
| GalAG2 | (GalA-KPL)4(KFKI)2KHI | 4 | 0.78* | 26 | 0.136 ± 0.001 | 84 | −69 ± 1.1 | 57 | −11.7 | 2.5 ± 0.1 | 296 |
| GalAxG2 | (GalA-KP)4(KICxKI)2KHI | 4 | 8.3 | 31 | 0.16 ± 0.009 | 98 | −54 ± 1 | 43 | −11.1 | 6.9 ± 1.4 | 107 |
| GalAxG3 | (GalA-KP)8(KICxKP)4(KKI)2KHI | 8 | 1.04 | 125 | 0.064 ± 0.01 | 79 | −115 ± 8 | 103.0 | −11.7 | 2.5 ± 0.2 | 148 |
| GalAxG4 | (GalAKP)16(KICxKP)8(KLF)4(KKI)2KHI | 16 | 1.28 | 50 | — | — | — | — | — | — | — |
| GAlAPSG2 | (GalA-KPL)4(KPS)2KPS | 4 | 0.78 | 40 | — | — | — | — | — | — | — |
| GalAxPSG3 | (GalA-KP)8(KICxPS)4(KPS)2KPS | 8 | 0.35 | 375 | 0.096 ± 0.001 | >100 | −92 ± 0.5 | 81 | −11.4 | 4.2 ± 0.2 | 88 |
| GalAxPSG4 | (GalAKP)16(KICxPS)8(KPS)4(KPS)2KPS | 16 | 0.14 | 475 | 0.033 ± 0.001 | 41 | −217 ± 17 | 206 | −11.5 | 3.4 ± 0.4 | 54 |
| GalBG0 | (GalB-KPL) | 1 | 2500* | 1 | 0.71 ± 0.01 | 100 | −14 ± 0.04 | 7.8 | −6.0 | 37000 ± 800 |
1 |
| GalBG1 | (GalB-KPL)2KHI | 2 | 630* | 1.9 | 0.37 ± 0.02 | >100 | −20 ± 0.1 | 12 | −8.2 | 1060 ± 160 | 18 |
| GalBG2 | (GalB-KPL)4(KFKI)2KHI | 4 | 125* | 4.8 | 0.18 ± 0.02 | >100 | −43 ± 1 | 33 | −10.1 | 40 ± 1 | 230 |
| GalBxG2 | (GalB-KP)4(KICxKI)2KHI | 4 | 125* | 4.8 | 0.14 ± 0.001 | 79 | −47 ± 6 | 37 | −11.2 | 33 ± 25 | 280 |
| GalBxG3 | (GalB-KP)8(KICxKP)4(KKI)2KHI | 8 | 37.5* | 8.3 | 0.08 ± 0.01 | 90 | −90 ± 9 | 79 | −11.4 | 5.9 ± 2 | 785 |
In the phenyl-galactoside (GalA) series there was no significant increase in r.p./nGal in the divalent dendrimer GalAG1, but the tetravalent dendrimer GalAG2 was 26-fold stronger than GalAG0 on a per galactose basis. A comparable multivalency effect was obtained with the tetravalent ClAc analog GalAxG2 (r.p./nGal = 31), showing that the modification of the peptide dendrimer backbone performed to enable the convergent ClAc ligation did not influence hemagglutination inhibition significantly. Increasing the dendrimer size to octavalency resulted in a more modest, 4-fold increase in relative potency per galactosyl endgroup with GalAxG3 (r.p./nGal = 125), respectively a 14-fold increase in its analog GalAxPSG3 (r.p./nGal = 375). However there was no further increase in relative potency at the G4 level, with the 16-valent GalAxG4 (r.p./nGal = 50) showing a 2.5-fold drop and GalAxPSG4 (r.p./nGal = 475) a 1.3-fold increase in potency relative to the corresponding G3 dendrimers. In the thiopropyl-galactoside (GalB) series multivalency effects in hemagglutination inhibition were much weaker, with r.p./nGal values increasing only up to 8-fold in GalBxG3.
Isothermal titration calorimetry (ITC)
Multivalency effects in the dendrimer LecA interactions were also investigated by ITC using p-nitrophenyl-β-galactoside (KD = 16 μM) as a reference. Binding enthalpies ΔH and dissociation constants KD were determined experimentally by titrating the various ligands into a LecA solution, and used to calculate binding entropies ΔS and binding free energies ΔG (Table 2, right part, and ESI Fig. S47†). The binding enthalpy ΔH amounted to approximately 14 kcal mol−1 per galactosyl end group across all the ligands tested. The GalA ligands showed generally stronger binding compared to the corresponding GalB ligands, implying a smaller entropic penalty on binding consistent with their more rigid structures and the burial of a larger hydrophobic surface area upon lectin binding. The percentage of bound galactose per dendrimer was also obtained, a value which was somewhat imprecise due to small errors in concentration determination caused by varying amounts of water in the lyophilized powders of the purified dendrimers. Values >80% were interpreted as complete engagement of all dendrimer galactosyl groups with the lectin, with incomplete binding observed only in the case of the G4 dendrimer GalAPSG4 (40% bound galactose).In each series the multivalency effect on binding was quantified by the relative potency of binding per galactosyl endgroup (r.p./nGal), which was calculated from the ratio between the KD of the reference monovalent glycotripeptide ligand GalAG0 or GalBG0 and the KD of the multivalent dendrimer. In the GalA series the relative potency increase in the G0/G1/G2 series (1/18/296) was much stronger than that observed in the hemagglutination assay (1/1.3/26). A further 1.4-fold increase in relative potency occurred in the homologous ClAc synthesis series GalAxG2/GalAxG3 (107/148), although both compounds were less potent than the parent GalAG2 dendrimer. Further generation increase was detrimental to binding, as evidenced by a relative potency decrease by 1.6-fold in the sequence GalAxPSG3/GalAxPSG4 (88/54). In the GalB series the relative potency increase in the G0/G1/G2 series (1/18/230) was comparable to that observed with the GalA series, in particular with a 13-fold increase in relative potency when moving from the divalent G1 dendrimer to the tetravalent G2 dendrimer, which was comparable to the 16-fold increase observed in the GalA series. As for the GalA series the gain in relative potency was much smaller between the tetravalent G2 and octavalent G3 GalB type dendrimers and amounted to only 2.8-fold (280/785).
Taken together, the ITC data confirmed the hemagglutination inhibition assays in showing that, while a very strong multivalency effect occurred at the level of the tetravalent G2 dendrimers, only a relatively modest increase in relative potency per galactosyl endgroup (1.4 to 4-fold) was possible by moving to higher multivalency dendrimers. It should be noted that ITC might not be able to measure KD values below 1 nM and might therefore obscure an increase in potency in the relatively tight binding GalA series. Nevertheless this uncertainly should not affect the results in the weaker binding GalB series reaching KD = 5.9 nM for GalBxG3 reflecting a 2.8-fold increase in relative potency per galactosyl group compared to its G2 analog.
Biofilm inhibition
P. aeruginosa biofilms were induced by growing the bacteria in 0.25% (w/v) nutrient broth no. 2, Oxoid, medium in 96-well sterile, U-bottomed polystyrene microtiter plates. Biofilm inhibition was determined by quantifying the amount of biofilm formed after 24 h of growth in the presence or absence of glycodendrimers or control compounds at different concentrations. The biofilm was quantified using the WST-8 assay indicating the amount of viable cells,19 rather than with the crystal violet assay indicating the overall biomass.19b,19c,20The results are reported as minimal biofilm inhibition concentration (MBIC), which is the lowest concentration inducing complete biofilm inhibition (Table 3). In addition to biofilm inhibition, the ability of the compound to disperse already established biofilms was tested at a fixed concentration of 50 μM by treating established biofilms with the compounds for 24 h, followed by quantification of the live attached cells as above. The monovalent glycosides D-galactose, isopropyl thiogalactoside (IPTG), GalAG0 and GalBG0 and dendrimers FD2, GalAG2 and GalBG2 were used as controls since their biofilm inhibition has been previously quantified using the steel coupon assay.5b,12b,13 In particular D-galactose was inactive and the monovalent glycosides were only very weakly active and showed partial biofilm inhibition or dispersal only at millimolar to molar concentrations, while FD2, GalAG2 and GalBG2 showed MBIC values of 20 μM (80 μM on a per carbohydrate basis).
| Compound |
n
gala |
MBICa | MBIC × nGalb |
Biofilm dispersalc |
|---|---|---|---|---|
| a MBIC: minimal biofilm inhibition concentration. b MBIC corrected for the number of galactosyl groups. c Biofilm dispersal with 50 μM ligand. | ||||
| D-Galactose | 1 | >450 mM | — | Inactive (100 mM) |
| IPTG | 1 | >360 mM | — | 25% (100 mM) |
| FD2 | 4 | 20 μM | 80 μM | 100% |
| GalAG0 | 1 | >3 mM | — | 25% (0.5 mM) |
| GalAG1 | 2 | 225 μM | 450 μM | n.d. |
| GalAG2 | 4 | 20 μM | 80 μM | 50% |
| GalAxG2 | 4 | 40 μM | 160 μM | Inactive |
| GalAxG3 | 8 | 9 μM | 72 μM | 100% |
| GalAxG4 | 16 | n.d. | n.d. | n.d. |
| GalAPSG2 | 4 | >45 μM | >180 μM | n.d. |
| GalAxPSG3 | 8 | >45 μM | >360 μM | n.d. |
| GalAxPSG4 | 16 | >45 μM | >720 μM | 45% |
| GalBG0 | 1 | >1.35 mM | >2.6 mM | Inactive (0.25 mM) |
| GalBG1 | 2 | n.d. | n.d. | n.d. |
| GalBG2 | 4 | 20 μM | 80 μM | 60% |
| GalBxG2 | 4 | 60 μM | 240 μM | Inactive |
| GalBxG3 | 8 | 13 μM | 104 μM | 100% |
G2 dendrimers GalAxG2 and GalBxG2 obtained by the convergent ClAc ligation approach had 2–3 fold higher MBIC values than their parent dendrimers GalAG2 and GalBG2, showing a slightly detrimental effect of the modified peptide backbone on the antibiofilm activity. Nevertheless the corresponding G3 dendrimers GalAxG3 and GalBxG3 showed good biofilm inhibition properties with MBIC ∼10 μM, which is comparable to the inhibition by GalAG2 and GalBG2 on a per galactose basis. These dendrimers were also very active in the biofilm dispersal assay. Dendrimer GalAxPSG4, which was the only G4 dendrimer obtained in sufficient yields to perform biofilm inhibition studies, did not show any antibiofilm activity, however the same was true for all dendrimers with a Pro-Ser independent of their size, indicating a sequence rather than a dendrimer generation effect.
These data showed that the positive dendritic effect on biofilm inhibition in the series G0 (monovalent, no activity), G1 (divalent, weak activity for GalA only), and G2 (tetravalent, strong activity in both GalA and GalB) did not extend to higher generations. Indeed the expansion to an octavalent, G3 dendrimer, although still beneficial for binding as observed in the hemagglutination and ITC experiments, did not induce a significant increase in biofilm inhibition, and expansion to G4 resulted in a loss of activity. These data clearly marked tetravalency of G2 dendrimers as the optimal multivalency in this system. Although this pattern paralleled that of the relative binding potency to LecA in these series, the lack of biofilm inhibition activity with dendrimers built around a Pro-Ser core despite their strong LecA binding showed that strong, multivalent LecA binding was necessary but not sufficient for biofilm inhibition.
X-ray crystallography and molecular modeling
The fact that the relative binding potency and biofilm inhibition per galactosyl group do not increase strongly beyond G2 dendrimers might reflect a steric crowding effect preventing optimal interactions with LecA at the level of G3 and G4 dendrimers. However the percentage of bound galactosyl groups deduced from ITC data indicated that essentially all galactosyl groups of G2 and G3 dendrimers were bound to LecA (>80% bound galactose), while incomplete binding only occurred at the level of GalAxPSG4 (40% bound galactose). A crystallography and modeling study was therefore undertaken to propose a binding mode for the octavalent G3 dendrimers capable of accounting for the complete saturation of all eight galactosyl groups on the side of the G3 dendrimers and all four galactose binding sites on the side of lectin LecA.After screening several complexes and conditions, good quality crystals of the complex GalAxPSG3·LecA were obtained by soaking crystals of LecA with the dendrimer, providing a structure at 1.9 Å resolution. In this structure LecA tetramers are arranged in a 3D checker-board lattice leaving large cavities available facing the galactose binding sites and which can be occupied by a macromolecular ligand (Fig. 2A). All galactose binding sites are indeed occupied by a phenyl-β-galactosyl ligand, which under the conditions used for crystallization must be part of the terminal arms of GalAxPSG3. The binding interactions at the level of the phenyl-β-galactosyl groups comprise all the interactions observed previously in other aromatic galactoside LecA complexes, including hydrogen-bonding interactions between several hydroxyl groups on galactose and residues H50, D100 and N107 on LecA, as well as the critical CH–π interaction between the C(ε)–H of His50 of LecA and the aromatic group of the phenyl galactoside (Fig. 2B).
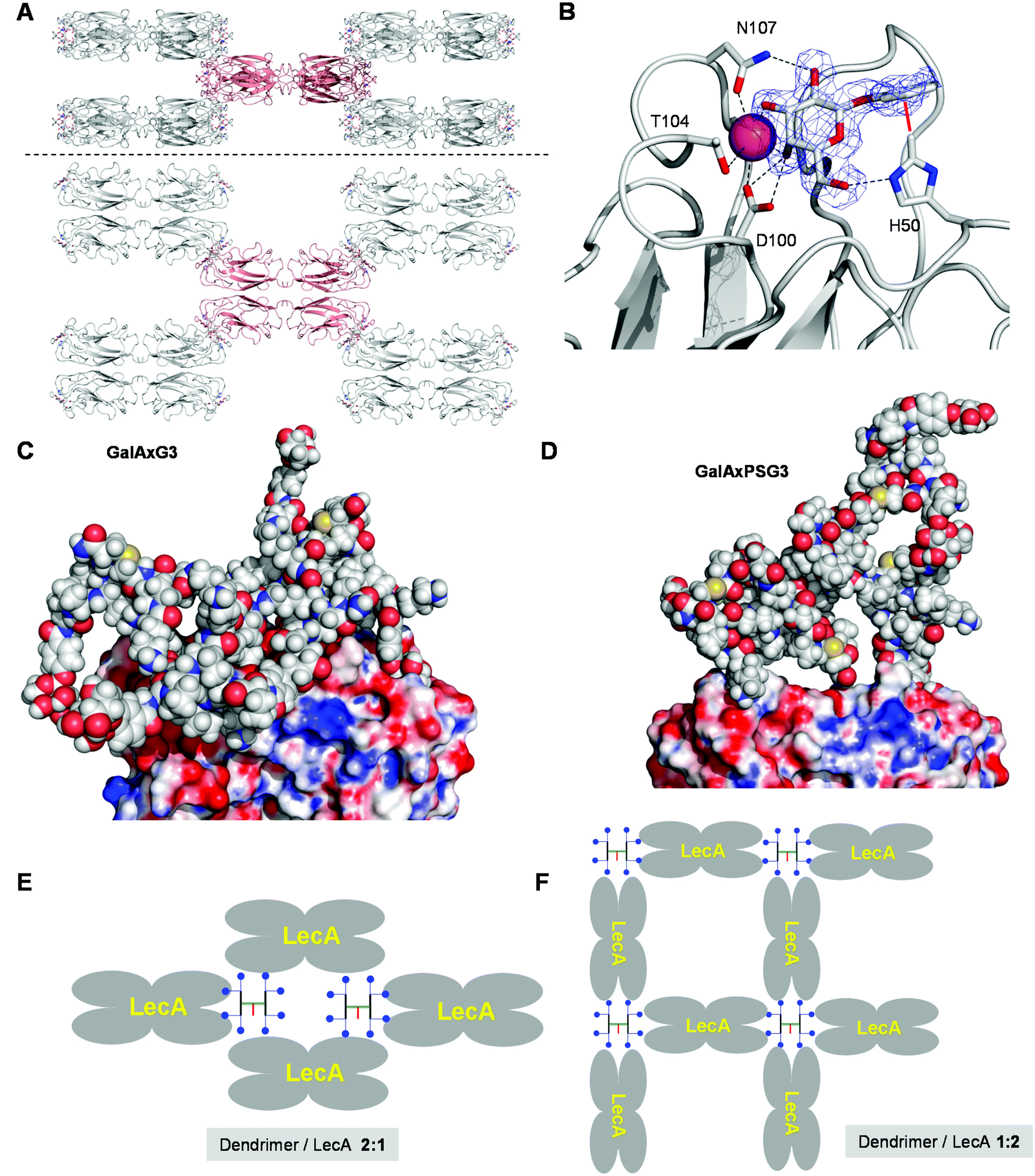 | ||
| Fig. 2 Crystal structure and molecular modeling of G3 dendrimer LecA interactions. A. Side view and top view of the 3D checker-board lattice of the GalAxPSG3. LecA complex in the crystal (PDB 5D21). One LecA tetramer (red) is shown surrounded by its eight neighbours making contact at each of the eight corners. B. Details of the visible electron density of GalAxPSG3 and dendrimer–lectin binding interactions. C. Snapshot of the MD simulation of a chelate bound GalAxG3·LecA complex. D. Snapshot of a chelate bound GalAxPSG3·LecA complex. E. Schematic representation of LecA crystal lattice bound to a G3 dendrimer. F. Schematic representation of an extensive G3 dendrimer·LecA lattice accounting for saturation of all galactosyl groups and galactose binding sites. | ||
No electron density is visible beyond the aromatic groups, indicating a disordered conformation of the dendrimer or a mixture of different bound states. The surface of LecA surrounding the galactose binding sites is almost entirely covered by crystal water molecules, implying that GalAxG3PS binds LecA exclusively via its phenyl-galactosyl groups and the visible binding interactions. The positive dendritic effect on binding affinity observed with GalAxG3PS and other G2 and G3 dendrimers in both hemagglutination and ITC (Table 3) can therefore only be understood in terms of a chelate binding mode in which two arms of the dendrimer bind to a pair of galactose binding sites on the same side of the LecA tetramer. In the context of the crystal structure the space available in the checker-board lattice of the crystal is in any case large enough to accommodate GalAxG3PS.
To test if chelate-bound structures might be possible we ran independent molecular dynamics (MD) simulations of LecA (as the tetramer) in complex with GalAxG3 and GalAxPSG3 in a geometry compatible with the observed crystal structure. These complexes remained stable over more than 5 ns of MD simulations. In particular, the protein tetramer structure retained the initial conformation seen in the X-ray model, with a global Cα RMSD oscillating around 1.4 Å. Sampled structures show how the dendrimer core can arrange into a rather compact molten globule-like state, while single branches stretch out to accommodate their ends into the binding pockets of the two LecA subunits (Fig. 2C). The GalAxPSG3 dendrimer presents a more compact structure than GalAxG3 due to the large number of more rigid proline residues (Fig. 2D).
The above modeling studies suggest that G3 dendrimers bind LecA in a chelate bound mode with a pair of galactosyl groups combining with a pair of galactose binding sites on the same side of LecA. G3 dendrimers are clearly too small to bridge between the two pairs of galactose binding sites on opposite sides of the same tetramer. The crystal structure of the GalAxPSG3·LecA complex most likely corresponds to a 2:1 dendrimer/LecA tetramer stoichiometry engaging only a single pair of galactosyl groups per dendrimer (Fig. 2E). ITC data by contrast show that all galactoside groups engage in a binding interaction with LecA, which can only occur with the opposite 1:2 dendrimer/LecA stoichiometry. Such stoichiometry might occur in a cross-linked network accounting for the formation of precipitates during ITC with all higher generation dendrimers (Fig. 2F).
Conclusion
In summary, convergent synthesis using the ClAc ligation as the key step enabled an efficient synthesis of G3 and G4 glycopeptide dendrimer ligands for the galactose specific P. aeruginosa lectin LecA bearing either 4-carboxyphenyl-β-galactoside (GalA) or carboxypropyl-β-thiogalactoside (GalB) end groups. LecA complexation studies by the hemagglutination assay and by ITC showed that the strong positive effect on the relative binding affinity per galactosyl group observed up to the tetravalent G2 dendrimers only partially extended to the octavalent G3 dendrimers, and that G4 dendrimers had decreased relative binding affinities. Similar effects were observed in P. aeruginosa biofilm inhibition and dispersal assays, with dendrimers GalAxG3 and GalBxG3 showing comparable activities to G2 dendrimers in biofilm inhibition and slightly better activities in biofilm dispersal. A crystal structure and modeling study was used to propose a lattice binding model accounting for the saturation of all dendrimer galactosyl groups and LecA galactose binding sites for these potent G3 dendrimers. Further studies with additional glycopeptide dendrimers diversified using the efficient ClAc ligation approach are underway to decipher the structure–activity relationships in this system in more detail and will be reported in due course.Materials and methods
Synthesis
Amino acids were used as the following derivatives: Fmoc-His(Trt)-OH, Fmoc-Leu-OH, Fmoc-Phe-OH, Fmoc-Ile-OH, Fmoc-Pro-OH, Fmoc-Lys(Boc)-OH, Fmoc-Lys(Fmoc)-OH, Fmoc-Cys(Trt)-OH, Fmoc-Ser(tBu)-OH. Chemicals were used as supplied and solvents were of analytical grade. Analytical RP-UHPLC was performed in the Dionex ULTIMATE 3000 RS chromatography system (ULTIMATE 3000 RS photo diode array detector) using a Dionex Acclaim® RSLC 120 C18, 3.0 × 50 mm, particle size 2.2 μm, 120 Å pore size, flow rate 1.2 ml min−1 column. Eluent A contained water with 0.1% TFA; eluent D contained acetonitrile and water (90:10) with 0.1% TFA. Compounds were detected by UV absorption at 214 nm. Preparative RP-HPLC was performed with HPLC-grade acetonitrile and MilliQ deionized water using a Dr Maisch GmbH Reprospher C18-DE, 100 × 30 mm, particle size 5 μm, 100 Å pore size column installed on a Waters Prep LC Controller system (flow rate 40 ml min−1). Eluent A contained water with 0.1% TFA; eluent B contained acetonitrile and water (60:40) with 0.1% TFA. MS spectra were provided by the Service of Mass Spectrometry of the Department of Chemistry and Biochemistry, University of Bern.
Solid phase peptide synthesis
Dendrimers were synthesized in the solid phase using Fmoc chemistry in plastic syringes (10 mL) mounted on the axis of a rotatory agitator for gentle stirring during reactions.21 Rink amide NovaSyn® TGR resin (loading: 0.23 mmol g−1) (purchased from Novabiochem) was acylated with each Fmoc-protected α-amino acid (3 eq.) in the presence of benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate (PyBOP) (3 eq.) and N,N-diisopropylethylamine (DIEA) (5 eq.) in N-methyl-2-pyrrolidone (NMP). The Fmoc-protecting groups were removed with a solution of 20% piperidine in DMF. When necessary the resin was chloroacetylated twice with a solution of chloroacetic acid anhydride (10 eq.) in NMP for 20 min. At the end of the other sequences the terminal amino acids were coupled with a protected sugar derivative (5 eq.) in the presence of DIEA (5 eq.) and 2-(6-chloro-1H-benzotriazole-1-yl)-1,1,3,3-tetramethylaminium hexafluorophosphate) (HCTU) (3 eq.). The carbohydrate was deprotected with a solution of MeOH/NH3/H2O (v/v 8:1:1). The resin was dried and the cleavage was carried out with TFA/triisopropylsilane/H2O (95:2.5:2.5). Peptide dendrimers containing Cys residues were cleaved with TFA/triisopropylsilane/H2O/1,2-ethanedithiol (94:1:2.5:2.5). Peptide dendrimers were precipitated with methyl tert-butyl ether and purified by preparative HPLC. The compounds GalAG0, GalAG1, GalAG2, GalBG0, GalBG1 and GalBG2 were prepared as previously described.13
Thioether ligation
A solution of the core dendrimer (sequence containing chloroacetyl endgroups, around 3 mg, 1 eq.) and KI (20 eq.) in DMF/H2O (1:1, v/v) (300 μl) was prepared in a 2 mL glass vial. The mixture was degassed with Ar for 10 min. In another 2 mL glass vial arm dendrimer (Cys containing sequence, 1.5 eq. per chloroacetyl endgroup in the core sequence) was weighed and degassed with Ar for 10 min. The core dendrimer/KI solution was transferred to the glass vial containing the arm dendrimer via a gas-tight syringe. DIEA (55 eq.) was added and the solution was stirred overnight at room temperature under an Ar atmosphere. The reaction was followed by analytical RP-UHPLC. After 16 to 23 h, the reaction was quenched by the addition of 3.5 mL of eluent A. After filtration, the solution was directly purified by preparative RP-HPLC.
GalAG1-Cys (GalA-KP) 2 KIC was obtained from 500 mg resin (loading 0.22 mmol g−1) as a foamy white solid after preparative RP-HPLC (63 mg, 45.76 μmol, 42%). Analytical RP-UHPLC: tR = 1.257 min (A/D 100/0 to 0/100 in 2.2 min, λ = 214 nm). MS (ESI+) calc. for C63H97N11O21S [M + H]+: 1376.6, found 1376.6, [M + H]+/2688.6
GalBG1-Cys (GalB-KP) 2 KIC was obtained from 500 mg resin (loading 0.22 mmol g−1) as a foamy white solid after preparative RP-HPLC (57 mg, 43.43 μmol, 40%). Analytical RP-UHPLC: tR = 1.217 min (A/D 100/0 to 0/100 in 2.2 min, λ = 214 nm). MS (ESI+) calc. for C55H97N11O19S3 [M + H]+: 1312.6, found 1312, [M + H]+/2656.8
ClAcG1 (ClAc-KI) 2 KHI was obtained from 500 mg resin (loading 0.22 mmol g−1) as a foamy white solid after preparative RP-HPLC (41.4 mg, 40.18 μmol, 33%). Analytical RP-UHPLC: tR = 0.990 min (A/D 80/20 to 0/100 in 2.2 min, λ = 214 nm). MS (ESI+) calc. for C46H81Cl2N13O9 [M + H]+: 1031.1, found 1030.4, [M + H]+/2515.8; [M + H]+/3344.6
ClAcG2 (ClAc-KP) 4 (KKI) 2 KHI was obtained from 500 mg resin (loading 0.22 mmol g−1) as a foamy white solid after preparative RP-HPLC (64 mg, 27.34 μmol, 25%). Analytical RP-UHPLC: tR = 1.345 min (A/D 100/0 to 0/100 in 2.2 min, λ = 214 nm). MS (ESI+) calc. for C106H183Cl4N29O21 [M + H]+: 2341.6, found 2341.0.
ClAcG3 (ClAc-KP) 8 (KLF) 4 (KKI) 2 KHI was obtained from 500 mg resin (loading 0.25 mmol g−1) as a foamy white solid after preparative RP-HPLC (19.3 mg, 3.78 μmol, 3%). Analytical RP-UHPLC: tR = 1.647 min (A/D 100/0 to 0/100 in 2.2 min, λ = 214 nm). MS (ESI+) calc. for C242H391Cl8N57O45 [M + H]+: 5102.7, found 5102.
ClAcPSG2 (ClAc-PS) 4 (KPS) 2 KPS was obtained from 500 mg resin (loading 0.22 mmol g−1) as a foamy white solid after preparative RP-HPLC (66 mg, 33.07 μmol, 30%). Analytical RP-UHPLC: tR = 1.285 min (A/D 90/10 to 0/100 in 2.2 min, λ = 214 nm). MS (ESI+) calc. for C82H127Cl4N21O28 [M + H]+: 1996.8, found 1995.9, 2033.9 [M + K]+, 2056.4 [M + K + Na]+.
ClAcPSG3 (ClAc-PS) 8 (KPS) 4 (KPS) 2 KPS was obtained from 500 mg resin (loading 0.22 mmol g−1) as a foamy white solid after preparative RP-HPLC (25 mg, 5.83 μmol, 5%). Analytical RP-UHPLC: tR = 1.38 min (A/D 90/10 to 0/100 in 2.2 min, λ = 214 nm). MS (ESI+) calc. for C178H275Cl8N45O60 [M + H]+: 4289, found 4290 and several sodium and potassium adducts.
GalAPSG2 (GalA-KPL) 4 (KPS) 2 KPS was obtained from 500 mg resin (loading 0.25 mmol g−1) as a foamy white solid after preparative RP-HPLC (26.4 mg, 7.68 μmol, 6%). Analytical RP-UHPLC: tR = 2.635 min (A/D 100/0 to 0/100 in 7.5 min, λ = 214 nm). MS (ESI+) calc. for C162H251N29O52 [M + H]+: 3437.89, found 3437.8. Calc. for [M + K]+: 3475.89, found 3975.7.
GalAxG2 (GalA-KP) 4 (KICxKI) 2 KHI was obtained as a foamy white solid after preparative RP-HPLC (6.9 mg, 1.86 μmol, 68%). Analytical RP-UHPLC: tR = 1.944 min (A/D 100/0 to 0/100 in 4.5 min, λ = 214 nm). MS (ESI+) calc. for C172H273N35O51S2 [M + H]+: 3711.3, found 3711.0.
GalAxG3 (GalA-KP) 8 (KICxKP) 4 (KKI) 2 KHI was obtained as a foamy white solid after preparative RP-HPLC (6.6 mg, 0.86 μmol, 67%). Analytical RP-UHPLC: tR = 1.799 min (A/D 100/0 to 0/100 in 4.5 min, λ = 214 nm). MS (ESI+) calc. for C358H567N73O105S4 [M + H]+: 7702, found 7701.0; [M + H] + adducts with trifluoroacetic acid (TFA).
GalAxG4 (GalA-KP)
16
(KICxKP)
8
(KLF)
4
(KKI)
2
KHI was obtained as a foamy white solid after preparative RP-HPLC (1.1 mg, 0.07 μmol, 12%). Analytical RP-UHPLC: tR = 2.094 min (A/D 100/0 to 0/100 in 4.5 min, λ = 214 nm). MS (ESI+) calc. for C746H1159N145O213S8 [M + H]+: 15824, found 15825.
GalAxPSG3 (GalA-KP) 8 (KICxPS) 4 (KPS) 2 KPS was obtained as a foamy white solid after preparative RP-HPLC (8.2 mg, 1.11 μmol, 74%). Analytical RP-UHPLC: tR = 2.519 min (A/D 100/0 to 0/100 in 7.5 min, λ = 214 nm). MS (ESI+) calc. for C334H511N65O112S4 [M + H]+: 7357.3, found 7358.0, [M + K]+ 7395.0
GalAxPSG4 (GalA-KP)
16
(KICxPS)
8
(KPS)
4
(KPS)
2
KPS was obtained as a foamy white solid after preparative RP-HPLC (9.1 mg, 0.61 μmol, 81%). Analytical RP-UHPLC: tR = 1.878 min (A/D 100/0 to 0/100 in 4.5 min, λ = 214 nm). MS (ESI+) calc. for C682H1043N133O228S8 [M + H]+: 15010, found 15010.3.
GalBxG2 (GalB-KP) 4 (KICxKI) 2 KHI was obtained as a foamy white solid after preparative RP-HPLC (5.2 mg, 1.45 μmol, 50%). Analytical RP-UHPLC: tR = 1.839 min (A/D 100/0 to 0/100 in 4.5 min, λ = 214 nm). HRMS (ESI+) calc. for C156H273N35O47S6 [M + H]+: 3583.4, found 3582.84.
GalBxG3 (GalB-KP) 8 (KICxKP) 4 (KKI) 2 KHI was obtained as a foamy white solid after preparative RP-HPLC (4.9 mg, 0.66 μmol, 51%). Analytical RP-UHPLC: tR = 1.784 min (A/D 100/0 to 0/100 in 4.5 min, λ = 214 nm). MS (ESI+) calc. for C326H567N73O97S12 [M + H]+: 7446, found 7445.0.
Hemagglutination assays
Isothermal titration calorimetry (ITC)
Lyophilized LecA was dissolved in buffer (Tris 20 mM, NaCl 100 mM, CaCl2 100 μM, pH = 7.5). Protein concentration was checked by the measurement of absorbance at 280 nm using 1 Abs = 2.116 mg ml−1 (Mw = 12893 g mol−1) using a NanoDrop instrument. The ligands were dissolved directly into the same buffer. ITC was performed with an iTC200 calorimeter (MicroCal Inc.). Titration was performed on 0.0146–0.3 mM LecA in the 200 μl sample cell using 2 μl injections of 0.01–01 mM ligand every 120 s at 25 °C. The data were fitted with MicroCal Origin 8 software, according to standard procedures using a single-site model. The change in free energy ΔG was calculated from the equation: ΔG = ΔH − TΔS where T is the absolute temperature, ΔH and ΔS are the change in enthalpy and entropy respectively. Two independent titrations were performed for each ligand tested.
Biofilm inhibition
A modified version of the method described by Diggle et al. was employed.5b 96-well sterile, U-bottomed polystyrene microtitre plates (TPP Switzerland) were prepared by adding 200 μl of sterile deionized water to the peripheral wells to decrease evaporation from test wells. Aliquots of 180 μl of culture medium (10% (w/v) nutrient broth no. 2, Oxoid) containing desired concentrations of the test compound were added to the internal wells. The inoculum of P. aeruginosa strain PAO1 was prepared from 5 ml of an overnight culture grown in LB broth. Aliquots of 20 μl of overnight cultures, pre-washed in 10% (w/v) nutrient broth and normalized to an OD600 of 1, were inoculated into the test wells. The plates were incubated in a humid environment for 25 hours at 37 °C. The wells were washed with 200 μl sterile deionized water before staining with 200 μl 10% (w/v) nutrient broth containing 0.5 mM WST-8 and 20 μM phenazine ethosulfate for 3 hours at 37 °C. Afterwards, the supernatants from the well were transferred to a polystyrene flat bottomed 96-well plate (TPP Switzerland) and the absorbance was measured at 450 nm with a plate reader (SpectraMax250 from Molecular Devices).Crystallization of GalAxPSG3·LecA
LecA was expressed and purified by affinity chromatography along an optimized protocol and in accordance with a previous report.17 The crystals of the GalAxPSG3·LecA complex were obtained by soaking. For this, LecA crystals were grown under a condition containing 1.5 M ammonium sulfate at a pH of 4.6 which is the SaltRx II 13 condition. The crystals grew within 3–4 days. Drops of 4 μL containing the crystals were supplemented with GalAPSG3 at 15 binding equivalents of the compound, respectively. The soaked crystals were incubated at 18 °C for 3–4 days, transferred into a solution of 1.5 M ammonium sulfate supplemented with 30% v/v glycerol at pH 4.6 and immediately flash frozen in liquid nitrogen for storage. Further details on data collection statistics are given in Table S1†. Crystals were cryocooled at 100 K after soaking them for as short a time as possible in glycerol 30% v/v in a precipitant solution. All data were collected at the SLS synchrotron (Villigen, Switzerland) at beamline PX-II/III. The structures were solved with CCP4 and Phenix.22,23Molecular modeling
The crystal structure of GalAxPSG3·LecA obtained above was used to model the dendrimer·LecA complexes. Two systems were considered: (i) LecA in complex with GalAxG3 and (ii) LecA in complex with GalAxPSG3. The initial configurations were built using Corina,24 imposing two binding units of GalA in two binding pockets of the respective LecA subunits. The missing hydrogen atoms were added to the protein structure using the default protonation states at pH = 7. The amber force field (FF99SB)25 was used to describe intra- and inter-molecular interactions. The electrostatic point-charges for the GalA unit, and for the branching lysine and modified cysteine amino acids present in the dendrimer structures were obtained using the standard RESP procedure.26 The GalAxPSG3·LecA and GalAxG3·LecA systems were solvated by 60531 and 71608 water molecules and placed in a truncated octahedral water box. The TIP3P water model27 was used to add the solvent. Particle mesh Ewald routines were used to treat long-range electrostatic interactions.28 A cutoff of 11 Å was used for the van der Waals interactions and the real part of the electrostatic potential. The simulations were performed using Langevin dynamics for temperature regulation.29 Bonds comprising hydrogen atoms were constrained with the SHAKE constraint algorithm.30 All simulations were performed using the AMBER1231 program and the trajectories were analyzed using VMD.32 First, energy minimization on the entire system was carried out using the steepest descent algorithm for the first 1000 steps followed by 2500 steps of conjugate gradient algorithm. The solvent was then heated up from 0 to 300 K in 200 ps MD, using a time step of 2 fs. This step assigns weak positional restraints on the solute. After this procedure the whole system was energy minimized. Finally production runs of 5 ns were performed at 300 K.
Acknowledgements
This work was supported financially by the University of Bern, the Swiss National Science Foundation, and the COST Actions D34 and CM1102 MultiGlycoNano. We thank the staff at the Swiss Light Source, Beamline X06DA (PXIII), Villigen, Switzerland, for support during data collection.References
- V. E. Wagner and B. H. Iglewski, Clin. Rev. Allergy Immunol., 2008, 35, 124–134 CrossRef CAS PubMed.
- (a) M. N. Hurley, M. Camara and A. R. Smyth, Eur. Respir. J., 2012, 40, 1014–1023 CrossRef CAS PubMed; (b) J. Valle, S. Da Re, N. Henry, T. Fontaine, D. Balestrino, P. Latour-Lambert and J. M. Ghigo, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12558–12563 CrossRef CAS PubMed.
- (a) N. Garber, U. Guempel, A. Belz, N. Gilboa-Garber and R. J. Doyle, Biochim. Biophys. Acta, 1992, 1116, 331–333 CrossRef CAS PubMed; (b) G. Cioci, E. P. Mitchell, C. Gautier, M. Wimmerova, D. Sudakevitz, S. Perez, N. Gilboa-Garber and A. Imberty, FEBS Lett., 2003, 555, 297–301 CrossRef CAS PubMed; (c) A. Imberty, M. Wimmerova, E. P. Mitchell and N. Gilboa-Garber, Microbes Infect., 2004, 6, 221–228 CrossRef CAS PubMed.
- (a) E. Mitchell, C. Houles, D. Sudakevitz, M. Wimmerova, C. Gautier, S. Perez, A. M. Wu, N. Gilboa-Garber and A. Imberty, Nat. Struct. Biol., 2002, 9, 918–921 CrossRef CAS PubMed; (b) R. Loris, D. Tielker, K. E. Jaeger and L. Wyns, J. Mol. Biol., 2003, 331, 861–870 CrossRef CAS PubMed.
- (a) M. Mewe, D. Tielker, R. Schonberg, M. Schachner, K. E. Jaeger and U. Schumacher, J. Laryngol. Otol., 2005, 119, 595–599 Search PubMed; (b) S. P. Diggle, R. E. Stacey, C. Dodd, M. Camara, P. Williams and K. Winzer, Environ. Microbiol., 2006, 8, 1095–1104 CrossRef CAS PubMed.
- (a) H. P. Hauber, M. Schulz, A. Pforte, D. Mack, P. Zabel and U. Schumacher, Int. J. Med. Sci., 2008, 5, 371–376 CrossRef CAS PubMed; (b) C. Chemani, A. Imberty, S. de Bentzmann, M. Pierre, M. Wimmerova, B. P. Guery and K. Faure, Infect. Immun., 2009, 77, 2065–2075 CrossRef CAS PubMed.
- A. Bernardi, J. Jimenez-Barbero, A. Casnati, C. De Castro, T. Darbre, F. Fieschi, J. Finne, H. Funken, K. E. Jaeger, M. Lahmann, T. K. Lindhorst, M. Marradi, P. Messner, A. Molinaro, P. V. Murphy, C. Nativi, S. Oscarson, S. Penades, F. Peri, R. J. Pieters, O. Renaudet, J. L. Reymond, B. Richichi, J. Rojo, F. Sansone, C. Schaffer, W. B. Turnbull, T. Velasco-Torrijos, S. Vidal, S. Vincent, T. Wennekes, H. Zuilhof and A. Imberty, Chem. Soc. Rev., 2013, 42, 4709–4727 RSC.
- S. Cecioni, A. Imberty and S. Vidal, Chem. Rev., 2015, 115, 525–561 CrossRef CAS PubMed.
- B. Gerland, A. Goudot, G. Pourceau, A. Meyer, S. Vidal, E. Souteyrand, J. J. Vasseur, Y. Chevolot and F. Morvan, J. Org. Chem., 2012, 77, 7620–7626 CrossRef CAS PubMed.
- (a) A. Imberty, Y. M. Chabre and R. Roy, Chem. – Eur. J., 2008, 14, 7490–7499 CrossRef CAS PubMed; (b) R. J. Pieters, Org. Biomol. Chem., 2009, 7, 2013–2025 RSC; (c) V. Wittmann and R. J. Pieters, Chem. Soc. Rev., 2013, 42, 4492–4503 RSC.
- G. M. L. Consoli, G. Granata, V. Cafiso, S. Stefani and C. Geraci, Tetrahedron Lett., 2011, 52, 5831–5834 CrossRef CAS.
- (a) E. M. V. Johansson, E. Kolomiets, F. Rosenau, K.-E. Jaeger, T. Darbre and J.-L. Reymond, New J. Chem., 2007, 31, 1291–1299 RSC; (b) E. M. Johansson, S. A. Crusz, E. Kolomiets, L. Buts, R. U. Kadam, M. Cacciarini, K. M. Bartels, S. P. Diggle, M. Camara, P. Williams, R. Loris, C. Nativi, F. Rosenau, K. E. Jaeger, T. Darbre and J. L. Reymond, Chem. Biol., 2008, 15, 1249–1257 CrossRef CAS PubMed; (c) E. Kolomiets, M. A. Swiderska, R. U. Kadam, E. M. Johansson, K. E. Jaeger, T. Darbre and J. L. Reymond, ChemMedChem, 2009, 4, 562–569 CrossRef CAS PubMed; (d) E. M. V. Johansson, R. U. Kadam, G. Rispoli, S. A. Crusz, K.-M. Bartels, S. P. Diggle, M. Camara, P. Williams, K.-E. Jaeger, T. Darbre and J.-L. Reymond, MedChemComm, 2011, 2, 418–420 RSC.
- R. U. Kadam, M. Bergmann, M. Hurley, D. Garg, M. Cacciarini, M. A. Swiderska, C. Nativi, M. Sattler, A. R. Smyth, P. Williams, M. Camara, A. Stocker, T. Darbre and J. L. Reymond, Angew. Chem., Int. Ed., 2011, 50, 10631–10635 CrossRef CAS PubMed.
- (a) T. Darbre and J. L. Reymond, Curr. Top. Med. Chem., 2008, 8, 1286–1293 CrossRef CAS PubMed; (b) J.-L. Reymond and T. Darbre, Org. Biomol. Chem., 2012, 10, 1483–1492 RSC; (c) J. L. Reymond, M. Bergmann and T. Darbre, Chem. Soc. Rev., 2013, 42, 4814–4822 RSC.
- C. C. Lee, J. A. MacKay, J. M. J. Frechet and F. C. Szoka, Nat. Biotechnol., 2005, 23, 1517–1526 CrossRef CAS PubMed.
- R. Visini, X. Jin, M. Bergmann, G. Michaud, F. Pertici, O. Fu, A. Pukin, T. R. Branson, D. M. Thies-Weesie, J. Kemmink, E. Gillon, A. Imberty, A. Stocker, T. Darbre, R. J. Pieters and J. L. Reymond, ACS Chem. Biol., 2015 DOI:10.1021/acschembio.5b00302.
- R. U. Kadam, M. Bergmann, D. Garg, G. Gabrieli, A. Stocker, T. Darbre and J.-L. Reymond, Chem. – Eur. J., 2013, 18, 17054–17063 CrossRef PubMed.
- N. A. Uhlich, T. Darbre and J. L. Reymond, Org. Biomol. Chem., 2011, 9, 7071–7084 CAS.
- (a) N. W. Roehm, G. H. Rodgers, S. M. Hatfield and A. L. Glasebrook, J. Immunol. Methods, 1991, 142, 257–265 CrossRef CAS PubMed; (b) E. Peeters, H. J. Nelis and T. Coenye, J. Microbiol. Methods, 2008, 72, 157–165 CrossRef CAS PubMed; (c) S. Stepanovic, D. Vukovic, V. Hola, G. Di Bonaventura, S. Djukic, I. Cirkovic and F. Ruzicka, APMIS, 2007, 115, 891–899 CrossRef PubMed; (d) M. Ishiyama, Y. Miyazono, K. Sasamoto, Y. Ohkura and K. Ueno, Talanta, 1997, 44, 1299–1305 CrossRef CAS PubMed.
- B. Adam, G. S. Baillie and L. J. Douglas, J. Med. Microbiol., 2002, 51, 344–349 CrossRef PubMed.
- N. Maillard, A. Clouet, T. Darbre and J. L. Reymond, Nat. Protoc., 2009, 4, 132–142 CrossRef CAS PubMed.
- M. D. Winn, C. C. Ballard, K. D. Cowtan, E. J. Dodson, P. Emsley, P. R. Evans, R. M. Keegan, E. B. Krissinel, A. G. W. Leslie, A. McCoy, S. J. McNicholas, G. N. Murshudov, N. S. Pannu, E. A. Potterton, H. R. Powell, R. J. Read, A. Vagin and K. S. Wilson, Acta Crystallogr., 2011, 67, 235–242 CrossRef CAS PubMed.
- P. D. Adams, P. V. Afonine, G. Bunkoczi, V. B. Chen, I. W. Davis, N. Echols, J. J. Headd, L.-W. Hung, G. J. Kapral, R. W. Grosse-Kunstleve, A. J. McCoy, N. W. Moriarty, R. Oeffner, R. J. Read, D. C. Richardson, J. S. Richardson, T. C. Terwilliger and P. H. Zwart, Acta Crystallogr., 2010, 66, 213–221 CrossRef CAS PubMed.
- J. Sadowski, J. Gasteiger and G. Klebe, J. Chem. Inf. Comput. Sci., 1994, 34, 1000–1008 CrossRef CAS.
- V. Hornak, R. Abel, A. Okur, B. Strockbine, A. Roitberg and C. Simmerling, Proteins, 2006, 65, 712–725 CrossRef CAS PubMed.
- C. I. Bayly, P. Cieplak, W. Cornell and P. A. Kollman, J. Phys. Chem., 1993, 97, 10269–10280 CrossRef CAS.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- (a) T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS; (b) U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef CAS.
- D. S. Cerutti, R. Duke, P. L. Freddolino, H. Fan and T. P. Lybrand, J. Chem. Theory Comput., 2008, 4, 1669–1680 CrossRef CAS PubMed.
- J. P. Ryckaert, G. Ciccotti and H. J. C. Berendsen, J. Comput. Phys., 1977, 23, 327–341 CrossRef CAS.
- J. C. Phillips, R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kalé and K. Schulten, J. Comput. Chem., 2005, 26, 1781–1802 CrossRef CAS PubMed.
- B. Hess, C. Kutzner, D. van der Spoel and E. Lindahl, J. Chem. Theory Comput., 2008, 4, 435–447 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ob01682g |
| This journal is © The Royal Society of Chemistry 2016 |