
Stereoselective total synthesis of (−)-nupharamine utilizing an α-chlorosulfide and a sulfinimine for C–C bond formation†
Sadagopan
Raghavan
* and
Sheelamanthula
Rajendar
Division of Natural Product Chemistry, Indian Institute of Chemical Technology, Hyderabad 500007, India. E-mail: sraghavan@iict.res.in
First published on 14th October 2015
Abstract
An efficient stereoselective synthesis of the nuphar alkaloid, (−)-nupharamine, is reported. The key features include the Lewis acid catalyzed reaction of an α-chlorosulfide with a silyl ketene acetal for C–C bond formation, creation of the stereocenter at C2 by a diastereoselective reaction of allyl indium with a sulfinimine and reductive amination for the introduction of the C6 stereocenter of the piperidine ring.
Introduction
Nuphar alkaloids comprise the family of sesquiterpenoid and triterpenoid alkaloids possessing piperidine, indolizidine and quinolizidine ring systems that are isolated from aquatic plants of the genus Nuphar (Nymphaeaceae).1 Nupharamine (1), was isolated from the Japanese water lily, Nuphar japonica,2 found in Japan and Korea. It has been used as a diuretic and for the treatment of stomach ache.3 Other members of the family have been shown to possess antibiotic,4a antifungal,4b potent immunosuppressive,4c central paralytic effects,5a,b,c and antitumor activities.5d,e The structural motif common to nuphar alkaloids is the trisubstituted piperidine ring with a methyl group at C-3 and a 3-furyl substituent at the C-6 position. Nuphar alkaloids have been targets for asymmetric synthesis due to their significant biological properties. The total synthesis of nupharamine 1 (Fig. 1) has been reported by several groups, utilizing a Diels–Alder strategy,6 an intramolecular aza-Wittig reaction,7 a cross-metathesis/reductive amination reaction,8 an intramolecular Mannich reaction9 and allenic hydroxylamine cyclisation10 as key steps. | ||
| Fig. 1 Nuphar alkaloid, nupharamine. | ||
Results and discussion
Herein, we describe the stereoselective synthesis of (−)-nupharamine 1 by employing a Lewis acid catalyzed reaction of an α-chlorosulfide with a silyl ketene acetal, diastereoselective allylation of a t-butyl sulfinimine for C–N bond construction and reductive amination as key steps. The retrosynthetic analysis is depicted in Scheme 1. Nupharamine was visualized to be obtained by reductive amination of an amino ketone obtained from sulfinamide 2 which can be obtained by an allylation followed by cross-metathesis from sulfinimine 3. Compound 3 was envisaged to be obtained from β-keto sulfide 4 which in turn can be traced to sulfide 5 and silyl enol ether 6. | ||
| Scheme 1 Retrosynthetic analysis of nupharamine. | ||
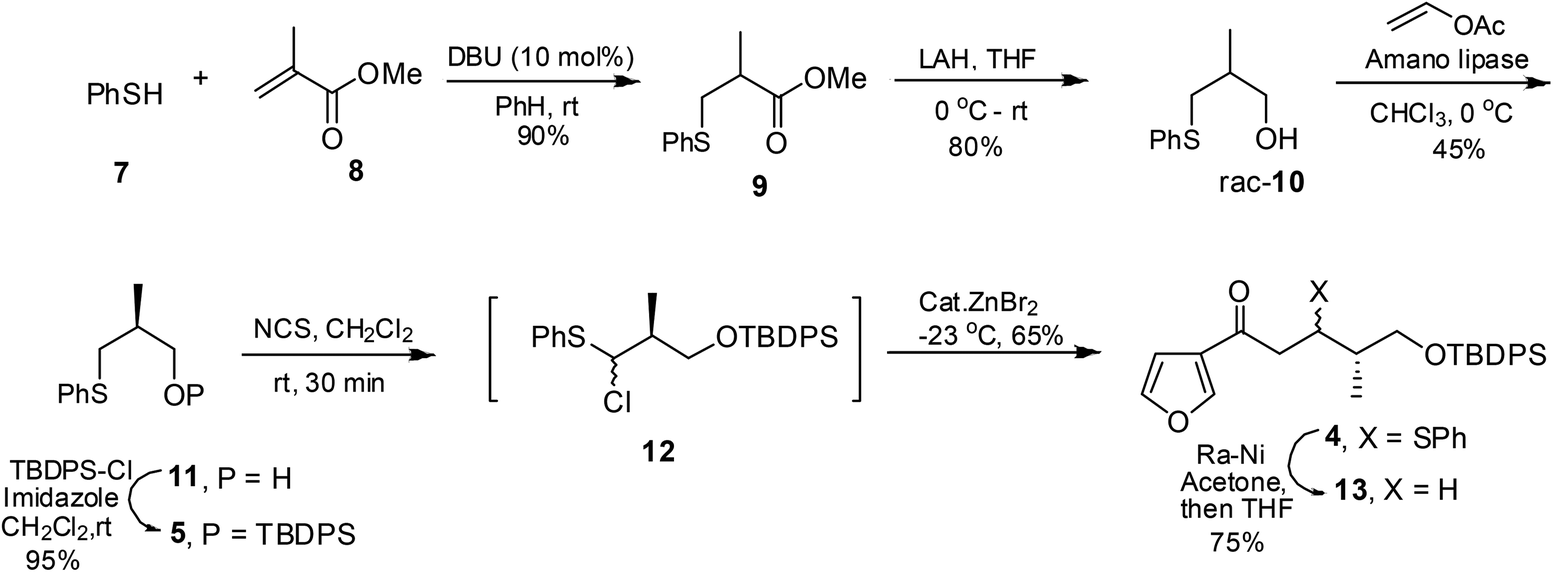
The synthesis began with the Michael addition of thiophenol 7 to methyl methacrylate 8 to furnish racemic ester 9. LAH reduction furnished alcohol 10, enzymatic resolution of which using vinyl acetate and Amano lipase furnished optically pure sulfide 11 (45% yield, 99% ee).11 The protection of 11 under standard conditions using TBDPS-Cl afforded the silyl ether 5. The treatment of sulfide 5 with N-chlorosuccinimide furnished α-chlorosulfide 1212,13 which without isolation was reacted with silyl ketene acetal 6, prepared from the corresponding ketone,14 in the presence of catalytic amounts of ZnBr2 to yield keto sulfide 4,15 as an inseparable mixture of diastereomers in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. The thiophenyl residue in 4 was chemoselectively hydrogenolyzed using RANEY®-Ni pre-treated with acetone16 to furnish compound 13, Scheme 2.
:1 ratio. The thiophenyl residue in 4 was chemoselectively hydrogenolyzed using RANEY®-Ni pre-treated with acetone16 to furnish compound 13, Scheme 2.
 | ||
| Scheme 2 Synthesis of compound 13. | ||
The keto group in 13 was protected as its ketal using ethylene glycol17 in the presence of catalytic amounts of p-toluenesulfonic acid to afford 14. Deprotection of the silyl ether in 14 using TBAF furnished the alcohol 15, which on oxidation using DMP18 yielded the aldehyde 16. Treatment of 16 with (R)-N-tert-butanesulfinamide19 in the presence of Ti(OEt)4 furnished sulfinimine 3. Attempted reactions of 3 with prenyl lithium20 expecting to obtain homoallylic amine derivative 2 did not bear fruit and the terminal alkene 18 was obtained exclusively.21
Exploring an alternative route, the three component allylation was attempted using aldehyde 16, sulfinamide 17 and allyl indium, generated in situ, as the reacting partners.22 The reaction proceeded cleanly to yield homoallyl amine derivative 19 selectively. The reaction outcome can be rationalized by invoking transition state I, wherein the allyl indium attacks the sulfinimine from the less hindered face by chelating to the oxygen atom. The trisubstituted alkene 2 was prepared via intermolecular olefin cross-metathesis23 between olefin 19 and 2-methyl-2-butene using the second generation Hoveyda–Grubbs’ catalyst (HG-II, 1.5 mol%) under solvent-free reaction conditions at 40 °C. Compound 2 on treatment with 4 N HCl24 in dioxane resulted in the concurrent hydration of the trisubstituted olefin, deprotection of the sulfinamide and 1,3-dioxolane groups to yield the amine·HCl, which on neutralization, followed by reduction using NaBH4 in MeOH produced nupharamine 1 as the sole product7 in 70% yield. The hydride delivery to the imine from the face opposite to the bulky C-2 substituent as depicted in transition state II would explain the formation of 1, Scheme 3. The spectroscopic data and optical rotation of nupharamine 1 were in good agreement with those reported in the literature,8 {[α]25D = −37.5 (c 0.7, CHCl3), lit.8 [α]22D = −38.7 (c 0.75, CHCl3)}.
 | ||
| Scheme 3 Synthesis of nupharamine. | ||
Conclusions
The α-chlorosulfide prepared from sulfide 5 has been employed in the reaction with silyl ketene acetal for C–C bond formation. The t-butyl sulfinamide auxiliary has been employed for the stereoselective creation of the C-2 stereogenic center by reaction with allyl indium. A reductive amination reaction has been utilized to create the C-6 stereocenter. The synthetic route disclosed can be readily adapted to prepare other members of nuphar alkaloids.Experimental
General information
All materials were used as received from a commercial supplier without further purification. All anhydrous reactions were performed using oven-dried or flame dried glassware. Tetrahydrofuran (THF) was distilled over Na/Ph2CO under a nitrogen atmosphere. Dichloromethane (CH2Cl2), and triethylamine (TEA) were dried over CaH2 and distilled prior to use. All reactions were monitored by using E. Merck analytical thin layer chromatography (TLC) plates and analyzed with 254 nm UV light and/or anisaldehyde–sulfuric acid or potassium permanganate or PMA treatment. Silica gel for column chromatography was purchased from Acme (Silica Gel 60–120, 100–200 mesh). All 1H and 13C NMR spectra were recorded in CDCl3 using Gemini 200, Avance 300, Inova 400, and Inova 500 spectrometers. Chemical shifts (δ) are reported in parts per million (ppm) relative to residual CHCl3 as an internal reference (1H: δ 7.26 ppm, 13C: δ 77.00 ppm). Coupling constants (J) are reported in Hertz (Hz). Peak multiplicity is indicated as follows: s (singlet), d (doublet), t (triplet), q (quartet) and m (multiplet). Mass spectra were recorded using a Waters mass spectrometer. HPLC spectra were recorded using a Waters 2998 spectrometer. High resolution mass spectra (HRMS) were recorded using an Applied Bio-Sciences HRMS spectrometer and a Thermo LTQ-Orbitrap mass spectrometer. All IR-spectra were recorded using a Nexus 870-FT-IR Thermo Nicolet spectrometer.:2, v/v) as the eluent to afford compound 9 (37.8 g, 180 mmol) in 90% yield as an oil. TLC Rf = 0.4 (5% EtOAc–hexanes). IR (neat): 2948, 1736, 1581, 1437, 1210, 1165, 742, 692 cm−1. 1H NMR (300 MHz, CDCl3): δ 7.32–7.30 (d, J = 6.8 Hz, 2H), 7.25 (t, J = 6.8 Hz, 2H), 7.16 (t, J = 6.8 Hz, 1H), 3.64 (s, 3H), 3.23 (dd, J = 12.8, 6.7 Hz, 1H), 2.87 (dd, J = 12.8, 7.5 Hz, 1H), 2.70–2.59 (m, 1H), 1.26 (d, J = 6.7 Hz, 3H). 13C NMR (75 MHz, CDCl3): δ 174.8, 135.6, 129.8, 128.7, 126.2, 51.5, 39.4, 37.2, 16.5. MS (ESI): 233 [M + Na]+. HRMS (ESI): m/z calcd for C11H14O2NaS 233.0612, found 233.0602.
:2, v/v) as the eluent afforded racemic alcohol 10 (22.9 g, 126 mmol) in 80% yield as a colorless liquid. TLC Rf = 0.12 (20% EtOAc–hexanes). IR (neat): 3390, 2959, 2876, 1583, 1478, 1030, 739, 691 cm−1. 1H NMR (300 MHz, CDCl3): δ 7.35 (t J = 6.7 Hz, 2H), 7.28 (t, J = 6.7 Hz, 2H), 7.17 (t, J = 6.7 Hz, 1H), 3.64 (dd, J = 10.5, 5.2 Hz, 1H), 3.59 (dd, J = 10.5, 6.0 Hz, 1H), 3.07 (dd, J = 12.8, 6.7 Hz, 1H), 2.84 (dd, J = 12.8, 6.7 Hz, 1H), 2.00–1.91 (m, 1H), 1.63 (bs, 1H), 1.04 (d, J = 6.7 Hz, 3H). 13C NMR (75 MHz, CDCl3): δ 136.6, 128.7, 125.6, 66.4, 37.1, 35.3, 16.3. MS (ESI) 205 [M + Na]+. HRMS (ESI): m/z calcd for C10H14ONaS 205.0663, found 205.0669.
:2, v/v) as the eluent afforded alcohol 11 (8.19 g, 45 mmol) in 45% yield as a colorless liquid. [α]25D = +11.5 (c 1.0, CH2Cl2).
:0.5, v/v) as the eluent to afford compound 5 (16.7 g, 40 mmol) in 95% yield as a yellow oil. TLC Rf = 0.42 (5% EtOAc–hexanes). [α]25D = +18.5 (c 1.0, CHCl3). IR (neat): 2957, 2930, 2858, 1472, 1109, 1084, 738, 700, 503 cm−1. 1H NMR (300 MHz, CDCl3): δ 7.65–7.59 (m, 4H), 7.38–7.24 (m, 8H), 7.20 (t, J = 7.5 Hz, 2H), 7.09 (t, J = 7.5 Hz, 1H), 3.63 (dd, J = 9.8, 4.5 Hz, 1H), 3.52 (dd, J = 9.8, 6.7 Hz, 1H), 3.21 (dd, J = 12.8, 6.0 Hz, 1H), 2.66 (dd, J = 12.8, 7.5 Hz, 1H), 2.01–1.86 (m, 1H), 1.06 (s, 9H), 1.09 (d, J = 6.7 Hz, 3H). 13C NMR (75 MHz, CDCl3): δ 137.2, 135.5, 133.5, 129.5, 128.7, 128.5, 127.6, 125.3, 67.4, 36.9, 35.7, 26.8, 19.2, 16.2. MS (ESI): 443 [M + Na]+. HRMS (ESI): m/z calcd for C26H32ONaSSi 443.1835, found 443.1846.
:0.5, v/v) as the eluent to afford sulfide 4 (3.43 g, 6.5 mmol) as an inseparable mixture of diastereomers in 65% yield as a gummy liquid. TLC Rf = 0.3 (5% EtOAc–hexanes). IR (neat): 3448, 2959, 2930, 2857, 1725, 1467, 1375, 1259, 1111, 1038, 803, 703, 506 cm−1. 1H NMR (300 MHz, CDCl3): δ 7.9 (s, 1H), 7.8 (s, 1H), 7.75 (s, 1H), 7.65 (s, 1H), 7.63–7.44 (m, 8H), 7.39–7.19 (m, 12H), 7.19–7.0 (m, 10H), 6.66 (s, 1H), 6.59 (s, 1H), 3.72–3.65 (m, 4H), 3.55–3.47 (m, 2H), 3.04 (dd, J = 15.8, 6.9 Hz, 1H), 2.91 (dd, J = 15.8, 7.9 Hz, 1H), 2.88 (dd, J = 12.8, 6.9 Hz, 1H), 2.82 (dd, J = 12.8, 5.8 Hz, 1H), 2.10–2.04 (m, 1H), 2.03–1.95 (m, 1H), 0.97 (d, J = 8.8 Hz, 3H), 0.93 (s, 9H), 0.86 (d, J = 6.9 Hz, 3H), 0.83 (s, 9H).13C NMR (75 MHz, CDCl3): δ 192.6, 192.2, 147.4, 147.3, 143.8, 143.7, 135.5, 135.3, 134.7, 133.5, 130.0, 129.7, 129.5, 129.4, 129.3, 129.1, 128.7, 128.5, 127.5, 127.4, 125.7, 125.3, 108.5, 108.3, 67.3, 66.5, 46.0, 44.4, 37.2, 37.0, 35.7, 35.4, 26.8, 26.6, 19.2, 19.0, 16.3, 16.2. MS (ESI): 551 [M + Na]+. HRMS (ESI): m/z calcd for C32H36O3NaSSi 551.2047, found 551.2048.
:0.5, v/v) as the eluent to afford compound 13 (6.3 g, 15 mmol) in 75% yield as a yellow liquid. TLC Rf = 0.2 (5% EtOAc–hexanes). [α]25D = −3.5 (c 1.0, CHCl3). IR (neat): 2932, 2860, 1677, 1468, 1389, 1154, 1107, 749, 701, 503 cm−1. 1H NMR (300 MHz, CDCl3): δ 7.93 (s, 1H), 7.68–7.63 (m, 4H), 7.46–7.33 (m, 7H), 6.74 (d, J = 1.5 Hz, 1H), 3.53 (dd, J = 10.5, 6.1 Hz, 1H), 3.49 (dd, J = 10.5, 5.2 Hz, 1H), 2.70 (t, J = 6.7 Hz, 2H), 1.92–1.79 (m, 1H), 1.78–1.66 (m, 1H), 1.65–1.51 (m, 1H), 1.05 (s, 9H), 0.95 (d, J = 6.7 Hz, 3H). 13C NMR (75 MHz, CDCl3): δ 194.7, 146.8, 143.9, 135.6, 133.8, 129.6, 127.6, 108.8, 68.4, 38.1, 35.3, 27.7, 27.0, 19.4, 17.0. MS (ESI): 443 [M + Na]+. HRMS (ESI): m/z calcd for C26H32O3NaSi 443.2013, found 443.2018.
:1, v/v) as the eluent to afford compound 14 (5.42 g, 11.7 mmol) in 78% yield as an oil. TLC Rf = 0.32 (5% EtOAc–hexanes). [α]25D = +4.1 (c 1.0, CHCl3). IR (neat): 2930, 1459, 1395, 1215, 1163, 1104, 767, 702, 606 cm−1. 1H NMR (200 MHz, CDCl3): δ 7.68–7.61 (m, 5H), 7.44–7.33 (m, 7H), 6.30 (s, 1H), 4.02–3.94 (m, 2H), 3.90–3.83 (m, 2H), 3.48 (dd, J = 10.1, 5.8 Hz, 1H), 3.41 (dd, J = 10.1, 6.7 Hz, 1H), 1.98–1.78 (m, 2H), 1.70–1.48 (m, 2H), 1.26–1.13 (m, 1H), 1.02 (s, 9H), 0.91 (d, J = 6.7 Hz, 3H). 13C NMR (75 MHz, CDCl3): δ 143.1, 139.9, 135.5, 134.0, 129.6, 129.4, 127.5, 108.7, 107.8, 68.7, 64.6, 37.0, 35.7, 26.9, 26.8, 19.2, 16.7. MS (ESI): 465 [M + H]+. HRMS (ESI): m/z calcd for C28H37O4Si 465.2456, found 465.2465.
:2, v/v) as the eluent to afford the alcohol 15 (2.37 g, 10.5 mmol) in 90% yield as an oil. TLC Rf = 0.1 (20% EtOAc–hexanes). [α]25D = −11.7 (c 2.0, CHCl3). IR (neat): 3421, 2954, 2883, 1663, 1465, 1187, 1044, 874, 800, 602 cm−1. 1H NMR (300 MHz, CDCl3): δ 7.38 (s, 1H), 7.36 (s, 1H), 6.32 (s, 1H), 4.01–3.96 (m, 2H), 3.92–3.87 (m, 2H), 3.48 (dd, J = 10.5, 5.6 Hz, 1H), 3.42 (dd, J = 10.5, 6.1 Hz, 1H), 2.01–1.95 (m, 1H), 1.91–1.85 (m, 1H), 1.66–1.59 (m, 1H), 1.54–1.47 (m, 1H), 1.26–1.19 (m, 1H), 0.90 (d, J = 6.7 Hz, 3H). 13C NMR (75 MHz, CDCl3): δ 143.1, 139.8, 127.6, 108.6, 107.7, 67.6, 64.5 36.8, 35.5, 26.6, 16.5. MS (ESI): 249 [M + Na]+. HRMS (ESI): m/z calcd for C12H18NaO4 249.1097, found 249.1113.
:2, v/v) as the eluent to furnish compound 3 (1.04 g, 3.2 mmol) in 80% yield as an oil. TLC Rf = 0.2 (20% EtOAc–hexanes). [α]25D = −177.5 (c 1.0, CHCl3). IR (neat): 3138, 2958, 2868, 1623, 1502, 1462, 1365, 1193, 1074, 1023, 868, 687 cm−1. 1H NMR (300 MHz, CDCl3): δ 7.93 (d, J = 5.2 Hz, 1H), 7.38–7.34 (m, 2H), 6.30 (s, 1H), 4.02–3.94 (m, 2H), 3.93–2.84 (m, 2H), 2.69–2.52 (m, 1H), 1.98–1.85 (m, 2H), 1.83–1.66 (m, 1H), 1.60–1.47 (m, 1H), 1.18 (s, 9H), 1.13 (d, J = 6.8 Hz, 3H). 13C NMR (75 MHz, CDCl3): δ 172.8, 143.2, 139.9, 127.6, 108.6, 107.4, 64.7, 56.4, 37.1, 29.7, 27.6, 22.4, 17.1. MS (ESI): 328 [M + H]+. HRMS (ESI): m/z calcd for C16H26O4NS 328.1577, found 328.1576.
:1 v/v, 5 mL) under nitrogen, followed by three drops of methanol. At the first appearance of a green coloration, the reaction mixture was cooled to 5 °C and stirred for a further 2 h. The now orange solution was transferred to a stirred solution of compound 3 (0.16 g, 0.5 mmol) in dry diethyl ether (4 mL) at −78 °C. The resulting mixture was stirred at the same temperature for 20 min, and the reaction was then quenched by the slow addition of methanol (15 mL). Diethyl ether (10 mL) and water (10 mL) were added, the phases were separated, and the aqueous phase was extracted with diethyl ether (2 × 10 mL). The combined extracts were washed with brine (10 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure to afford the crude product, which was purified by column chromatography on silica gel using hexanes–EtOAc (8:2, v/v) as the eluent to furnish compound 18 (0.12 g, 0.36 mmol) in 72% yield as an oil. TLC Rf = 0.1 (20% EtOAc–hexanes). 1H NMR (300 MHz, CDCl3): δ 7.40–7.34 (m, 2H), 6.36 (s, 1H), 5.80 (dd, J = 17.3, 11.3 Hz, 1H), 5.0 (dd, J = 11.3, 1.5 Hz, 1H), 4.96 (dd, J = 17.3, 1.5 Hz, 1H), 4.0–3.96 (m, 2H), 3.90–3.86 (m, 2H), 3.34 (d, J = 9.0 Hz, 1H), 2.89 (d, J = 8.3 Hz, 1H), 2.02–1.95 (m, 1H), 1.97–1.82 (m, 1H), 1.76–1.64 (m, 2H), 1.56–1.43 (m, 1H), 1.25 (s, 9H), 1.02 (s, 3H), 0.09 (s, 3H), 0.84 (d, J = 6.7 Hz, 3H).
:brine (50 mL, 4:1 v/v) with stirring. The resulting white suspension was filtered through a short plug of Celite, and washed with ethyl acetate (20 mL). The combined filtrates were washed with brine (10 mL), dried over Na2SO4, filtered and concentrated under reduced pressure to afford the crude product which was purified by column chromatography on silica gel using hexanes–EtOAc (7:3, v/v) to give the amine 20 (1.25 g, 3.4 mmol) in 85% yield as a yellow color oil. TLC Rf = 0.21 (30% EtOAc–hexanes). [α]25D = −7.9 (c 1.2, CHCl3). IR (neat): 3448, 2931, 1626, 1389, 1218, 1048, 760, 703, 613 cm−1. 1H NMR (300 MHz, CDCl3): δ 7.38–7.34 (m, 2H), 6.30 (s, 1H), 5.81–5.71 (m, 1H), 5.17–5.11 (m, 2H), 4.0–3.96 (m, 2H), 3.90–3.86 (m, 2H), 3.22–3.17 (m, 2H), 2.41–2.30 (m, 1H), 2.23–2.15 (m, 1H), 2.02–1.95 (m, 1H), 1.84–1.77 (m, 1H), 1.76–1.59 (m, 2H), 1.58–1.49 (m, 1H), 1.18 (s, 9H), 0.87 (d, J = 6.7 Hz, 3H). 13C NMR (75 MHz, CDCl3): δ 143.2, 139.9, 134.7, 127.6, 119.0, 108.6, 107.6, 64.6, 58.3, 55.7, 37.4, 36.0, 26.3, 24.2, 22.7, 14.8. MS (ESI): 370 [M + H]+. HRMS (ESI): m/z calcd for C19H32O4NS 370.2047, found 370.2052.
:3, v/v) to give the cross-metathesis product 2 (0.46 g, 1.1 mmol) in 78% yield as an oil. TLC Rf = 0.3 (30% EtOAc–hexanes). [α]25D = −21.5 (c 1.0, CHCl3). IR (neat): 3448, 2922, 2852, 1713, 1461, 1214, 1122, 1028, 926, 748, 667 cm−1. 1H NMR (300 MHz, CDCl3): δ 7.38–7.33 (m, 2H), 6.30 (s, 1H), 5.09 (t, J = 6.8 Hz, 1H), 4.02–3.98 (m, 2H), 3.92–3.88 (m, 2H), 3.16–3.08 (m, 2H), 2.26–2.12 (m, 2H), 2.03–1.92 (m, 1H), 1.84–1.41 (m, 4H), 1.72 (s, 3H), 1.63 (s, 3H), 1.18 (s, 9H), 0.87 (d, J = 6.4 Hz, 3H). 13C NMR (75 MHz, CDCl3): δ 143.2, 139.9, 135.4, 127.6, 120.1, 108.6, 107.7, 64.7, 59.3, 55.5, 37.5, 35.8, 30.1, 26.3, 25.9, 22.6, 18.0, 14.8. MS (ESI): 354 [M + H]+. HRMS (ESI): m/z calcd for C19H32O3NS 354.2111, found 354.2112. Note: the mass is for the ketone resulting from the hydrolysis of the dioxolane.
:5, v/v) afforded nupharamine (70 mg, 0.28 mmol) in 70% yield as an oil. TLC Rf = 0.3 (5% MeOH–dichloromethane). [α]25D = −37.5 (c 0.7, CHCl3). {lit.8 [α]22D = −38.7 (c 0.75, CHCl3)}. IR (neat): 3444, 3019, 2923, 2855, 1636, 1461, 1376, 1218, 1079, 769 cm−1. 1H NMR (300 MHz, CDCl3): δ 7.36–7.33 (m, 2H), 6.42 (s, 1H), 3.62 (dd, J = 11.5, 2.2 Hz, 1H), 2.42–2.37 (m, 1H), 1.9–1.72 (m, 4H), 1.6–1.42 (m, 4H), 1.26–1.13 (m, 2H), 1.21 (s, 3H), 1.19 (s, 3H), 0.9 (d, J = 6.4 Hz, 3H). MS (ESI): 252 [M + H]+. HRMS (ESI): m/z calcd for C15H26O2N 252.1958, found 252.1966.
Acknowledgements
S. Rajendar is thankful to CSIR for SRF fellowship. S.R. acknowledges funding from DST and CSIR, New Delhi as a part of XII five year plan programme under the title ORIGIN (CSC-108).Notes and references
- (a) R. T. Lalonde, C. F. Wong and K. C. Das, J. Am. Chem. Soc., 1972, 94, 8522 CrossRef CAS; (b) B. Maurer and G. Ohloof, Helv. Chim. Acta, 1976, 59, 1169 CrossRef CAS PubMed.
- Y. Arata and T. Ohashi, Yakugaku Zasshi, 1957, 77, 792 CAS.
- T. Ohashi, Yakugaku Zasshi, 1959, 79, 729 CAS.
- (a) J. T. Wrobel, in The Alkaloids, ed. R. H. F. Manske, Academic Press, New York, 1967, vol. 9, pp. 441–465 Search PubMed; (b) J. Cybuski and J. T. Wrobel, in The Alkaloids, ed. A. Brossi, Academic Press, New York, 1989, vol. 35, pp. 215–257 Search PubMed; (c) H. Matsuda, H. Shimoda and M. Yoshikawa, Bioorg. Med. Chem., 2001, 9, 1031 CrossRef CAS.
- (a) Y. Arata and T. Ohashi, Yakugaku Zasshi, 1957, 77, 236 CAS; (b) M. Kokate, I. Kawasaki, S. Matsutani, S. Kusumoto and T. Kaneko, Bull. Chem. Soc. Jpn., 1962, 35, 698 CrossRef; (c) C. F. Wong, E. Auer and R. T. Lalonde, J. Org. Chem., 1970, 35, 517 CrossRef CAS; (d) H. Matsuda, T. Morikawa, M. Oda, Y. Asao and M. Yoshikawa, Bioorg. Med. Chem. Lett., 2003, 13, 4445 CrossRef CAS PubMed; (e) H. Matsuda, K. Yoshida, K. Miyagawa, Y. Nemoto, Y. Asao and M. Yoshikawa, Bioorg. Med. Chem. Lett., 2006, 16, 1567 CrossRef CAS PubMed.
- (a) Y. Shishido and C. Kibayashi, Tetrahedron Lett., 1991, 32, 4325 CrossRef; (b) J. Barluenga, F. Aznar, C. Ribas and C. Valdes, J. Org. Chem., 1999, 64, 3736 CrossRef CAS.
- T. Honda, F. Ishikawa and S. Yamane, J. Chem. Soc., Chem. Commun., 1994, 499 RSC.
- S. Blechert and J. Gebauer, Synlett, 2005, 2826 CrossRef.
- F. A. Davis and M. Santhanaraman, J. Org. Chem., 2006, 71, 4222 CrossRef CAS PubMed.
- R. W. Bates and C. J. Lim, Synlett, 2010, 866 CrossRef CAS.
- P. Grisenti, P. Ferraboschi, A. Manzocchi and E. Santaniello, Tetrahedron, 1992, 18, 3827 Search PubMed. The reaction progress was monitored by HPLC. The title compound had physical characteristics in excellent agreement with the known literature compound. S. Raghavan and S. Rajendar, Org. Biomol. Chem., 2015, 13, 5044 Search PubMed . For the preparation of the enantiomer of 11 see: R. Baker, M. J. O'Mahony and C. J. Swain, J. Chem. Soc., Perkin Trans. 1, 1987, 1623 RSC.
- I. Paterson and I. Fleming, Tetrahedron Lett., 1979, 23, 993 CrossRef.
- S. Raghavan, V. Vinoth Kumar and L. Raju Chowhan, Synlett, 2010, 1807 CrossRef CAS.
- Compound 6 was obtained by a three step sequence (i) 3-furaldehyde was treated with methyl magnesium iodide to furnish the secondary alcohol; (ii) oxidation with IBX in DMSO yielded 3-acetylfuran; (iii) the silyl enol ether 6 was prepared from 3-acetylfuran and tert-butyldimethylchlorosilane under standard conditions in the presence of ZnCl2/Et3N in benzene; see: A. Benıtez, F. Ruth Herrera, M. Romero and F. X. Talamas, J. Org. Chem., 1996, 61, 1487 CrossRef.
- The configuration of the newly created stereocenter of the major isomer was not established since it was to be destroyed subsequently.
- S. A. Snyder and E. J. Corey, J. Am. Chem. Soc., 2006, 128, 740 CrossRef CAS PubMed.
- R. K. Boeckman, M. Ricodel, R. Ferreira, L. H. Mitchell, P. Shao, M. J. Neeb and Y. Fang, Tetrahedron, 2011, 67, 9787 CrossRef CAS PubMed.
- (a) D. B. Dess and J. C. Martin, J. Org. Chem., 1983, 48, 4155 CrossRef CAS; (b) D. B. Dess and J. C. Martin, J. Am. Chem. Soc., 1991, 113, 7277 CrossRef CAS.
- D. J. Weix and J. A. Ellman, Org. Lett., 2003, 5, 1317 CrossRef CAS PubMed.
- Prenyl lithium was prepared by reductive cleavage of phenyl prenyl ether with lithium; see: A. J. Birch, J. E. T. Corrie and G. S. R. Subba Rao, Aust. J. Chem., 1970, 23, 1811 CrossRef CAS.
- The configuration at C2 was assigned based on the precedent.
- J. C. Gonzalez-Gomez, M. Medjahdi, F. Foubelo and M. Yus, J. Org. Chem., 2010, 75, 6308 CrossRef CAS PubMed.
- A. K. Chatterjee, D. P. Sanders and R. H. Grubbs, Org. Lett., 2002, 4, 1939 CrossRef CAS PubMed.
- J. W. Evans and J. A. Ellman, J. Org. Chem., 2003, 68, 9948 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ob01750e |
| This journal is © The Royal Society of Chemistry 2016 |