
Tumor cell-specific photothermal killing by SELEX-derived DNA aptamer-targeted gold nanorods†
Ramya
Chandrasekaran
a,
Alexander Sheng Wei
Lee
b,
Lim Wei
Yap
a,
David A.
Jans
b,
Kylie M.
Wagstaff‡
*b and
Wenlong
Cheng‡
*a
aDepartment of Chemical Engineering, Monash University, VIC 3800 Australia. E-mail: wenlong.cheng@monash.edu
bDepartment of Biochemistry and Molecular Biology, Monash University, VIC3800, Australia. E-mail: kylie.wagstaff@monash.edu
First published on 30th November 2015
Abstract
Despite widespread availability of cytotoxic chemotherapeutic agents, the killing of tumour cells without affecting healthy surrounding tissue remains elusive, although recent developments in terms of plasmonic nanoparticles capable of photothermal killing have some promise. Here we describe novel DNA aptamer-tethered gold nanorods (GNRs) that act as efficient photothermal therapeutics against tumour cells, but not their isogenic normal cell counterparts. A modified Cell-SELEX process was developed to select a novel DNA aptamer (KW16-13) that specifically recognised and was internalised by cells of the MCF10CA1h human breast ductal carcinoma line but not by those of its isogenic normal counterpart (MCF10A). GNRs conjugated to KW16-13 were readily internalized by the MCF10CA1h tumour cells with minimal uptake by MCF10A normal cells. Upon near infrared (NIR) light irradiation, tumour cell death of >96%, could be effected, compared to <1% in the normal cells or cells incubated with GNRs alone, our KW16-13 aptamer-targeted GNRs thus showing >71-fold tumor cell death than GNRs-targeted with a previously described aptamer. This demonstrates the significant potential for aptamer functionalised-GNRs to be used effective and above all selective anti-cancer photothermal therapeutics.
Introduction
Breast cancer is the second most prevalent cancer worldwide with more than 1.7 million new cases diagnosed worldwide in 2012 (WHO). Although numerous chemotherapeutic agents are available for the treatment of breast cancer, none of them are truly specific for tumour cells, instead resulting in significant and unwanted toxic side-effects. A potential approach is photothermal therapy,2 which involves using light to elevate the localised temperature of the tumour tissue to 42–45 °C, resulting in cell death.3 Since this type of non-specific heating damages the tissue as a whole, including the normal tissue surrounding the tumour cells, nanotechnology approaches have been applied to photothermal therapy for cancer treatment, with varying degrees of success.4 Nanoparticles are particularly attractive for this application due to their ability to accumulate in the poor lymph vessel architecture commonly associated with tumour tissues.5,6 Nanoparticles with a high surface to volume ratio can also be functionalised with targeting ligands to enhance cell surface binding.7 When irradiated with light, the nanoparticles can kill the cells in the area, but this damages the entire tissue and results in unwanted side effects.Plasmonic nanoparticles capable of photothermal killing have great promise as therapeutics through their chemical inertness, high biocompatibility and fine-tunability of structural parameters.8 In particular, gold nanorods (GNRs) have particular advantages in that they can exhibit a strong broadband longitudinal surface plasmon absorption (LSPR) in the range of 600 to 1100 nm.9–12 The LSPR band can be fine-tuned simply by varying the aspect ratio (length of long to short sides) of the GNR to reside in the tissue-transparent Near Infra-Red (NIR) region.13 Importantly, GNRs sustain a higher absorption cross section at NIR frequencies per unit volume compared to nanospheres or nanoshells,1 making them highly attractive possibilities for treating deep tissue cancers9 with minimal damage to healthy tissue.
Due to the nature of tumour cells and the fact that they originate from normal cells, one of the major challenges to cancer therapy is to specifically target tumour cells with minimal damage to the surrounding healthy normal tissue. To actively target cancers, various biomolecules including peptides and antibodies14–22 have been used to functionalise GNRs for targeted therapies,23,24 but none of these approaches have proved truly tumour specific, as the target molecule to which they are directed on the tumour are also usually expressed on normal tissue, although to a lesser extent. DNA aptamers25–27 which are short single-stranded pieces of DNA or RNA that fold into 3-dimensional structures and bind to target molecules based on their shape, represent an exciting alternative, with advantages such as facile scale-up by chemical synthesis, ease of conjugation to GNRs and a relative absence of immunogenic effects.28–30 Although aptamer-GNR conjugates have previously been used in drug delivery/cancer therapy to target cancer cells with some success19,22,24,31–33 these studies compared the photothermal killing of Apt-GNRs on various tumour cell lines without a normal counterpart cell comparison. Here we utilise the powerful MCF10 breast cancer isogenic cell series, where a normal human breast cell line (MCF10A) has been used to generate various tumour cell lines, resulting in a series of normal/tumour cells on an identical genetic background, enabling true comparison of the specificity of treatments for tumorigenic cells. This study represents the first report of isogenic tumour and normal cells being used for direct comparison of the tumour cell specific photothermal killing effect of Apt-GNRs.
In this study we describe the generation of a new tumour-specific aptamer (KW16-13)-GNR complex targeted against MCF10CA1h breast cancer cells and not MCF10A normal breast cells from an identical genetic background.34,35 KW16-13 demonstrates strong binding to and internalisation by MCF10CA1h cells as well as two other isogenic breast cancer lines, but not to the isogenic normal MCF10A cell, thus acting as a targeting molecule able to differentiate specifically between tumor and normal cells. The KW16-13-GNRs constructs could act as highly efficient photothermal agents exhibiting strong tumour cell killing following laser irradiation, with >70-fold specificity for tumour compared to normal cells (Scheme 1). This demonstrates the significant potential for aptamer coupled-GNRs to be used for safe, effective and above all selective anti-cancer therapy.
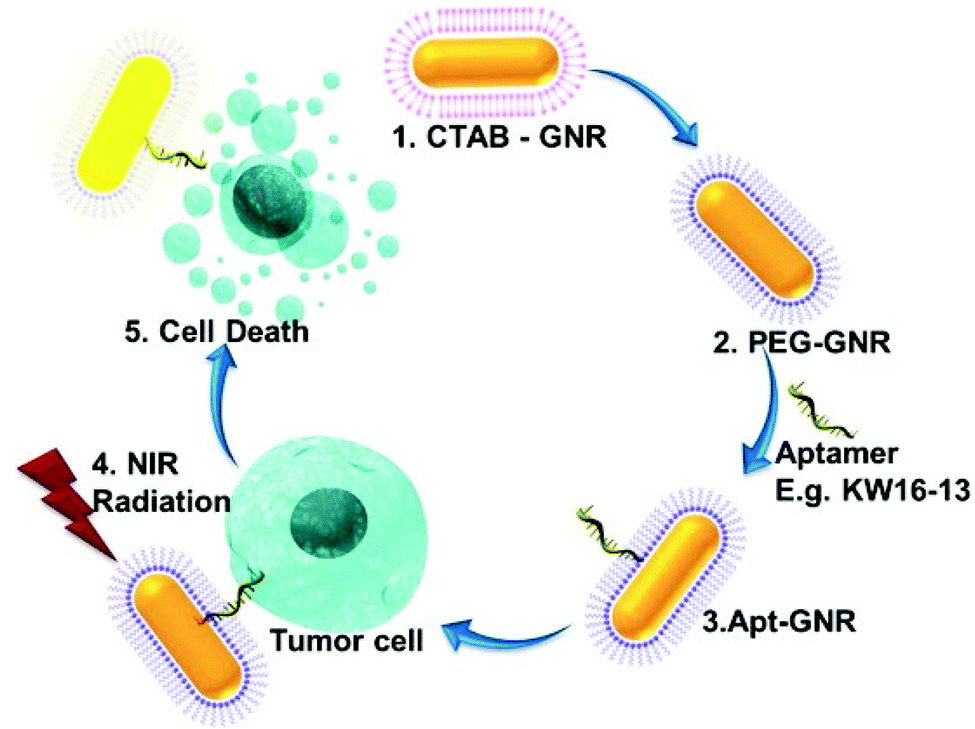 | ||
| Scheme 1 Schematic representation of the synthesis and use of Apt-GNRs for targeted photothermal therapy. CTAB-GNRs are synthesised using a seed-mediated approach (1). The CTAB is replaced by PEG (2) to enable aptamer attachment (3). The resultant Apt-GNRs are incubated with tumour cells followed by photothermal ablation in the near infrared range (4), leading to cell death specifically in tumour cells (5). CTAB-cetyl trimethylammonium bromide, PEG-poly ethylene glycol, Apt-aptamer, GNR-gold nanorod, NIR-near infra-red. | ||
Results and discussion
Generation of tumour-specific DNA aptamers by modified Cell-SELEX
Tumour cell-specific targeting aptamers were generated using a series of isogenic cell lines from the tumour progression model, where the lines differ only in their tumorigenic status. The lines are based on the spontaneously immortalized non-tumorigenic MCF10A normal human breast epithelial line, which has been transformed with the activated c-Ha-ras oncogene to generate the pre-malignant MCF10AT line,35 which in turn was used to generate the MCF10CA1h cell line through xenograft implant in mice.34 The MCF10AT cell line generates small nodules in mice, which are representative of benign ductal aggregates and mild hyperplasia with 25% progressing into carcinomas. In contrast, the MCF10CA1h cell line forms well-differentiated carcinomas in 100% of mice and the MCF10Ca1a has a fully malignant and metastatic phenotype and is consistent with a high grade carcinoma.36 This series of cell lines represents a powerful human breast tumour progression model.To generate aptamers specific for tumour cells, a modifed whole-cell SELEX procedure29 was used (Scheme 2). Aptamers were targeted against the MCF10Ca1h tumour cells, with the MCF10A isogenic normal cells used for negative counter-selection. After each round of positive/negative selection, the remaining aptamer pool was amplified by PCR prior to the next round. Simultaneously, aptamer selection was monitored using a 5′-FITC-labelled primer to generate fluorescent aptamer pools by PCR. Following separation and removal of the non-fluorescent reverse strands the ssDNA pools were refolded and incubated with either MCF10A normal or MCF10Ca1h tumour cells (4 h, 37 °C). Live cell confocal laser scanning microscopy (CLSM) of the washed samples demonstrated increased specificity of the aptamer pool for the tumour cells with each subsequent round of selection (Fig. 1A). Quantitation of the relative fluorescence levels in each cell type revealed an ∼9-fold increase in tumour-cell selectivity after 16 rounds compared to the unselected random aptamer library (Fig. 1B). After 18 rounds of Cell-SELEX, the tumour cell-selectivity remained constant and the aptamer pool was subcloned and sequenced. The resultant sequences were grouped into families using an iterative clustal X analysis process (see Experimental section) and the 6 most abundant aptamer species synthesised for individual analysis.
 | ||
| Scheme 2 Schematic representation of the Cell-SELEX approach to derive tumour cell-specific targeting aptamers. A ssDNA DNA aptamer library (1) is incubated with the MCF10CA1h target cells (2) for positive selection. The bound aptamers are extracted from the cell surface (3) after vigorous washing and incubated with the MCF10A control normal cell (4) for negative selection. The unbound aptamers (5) are then amplified by PCR (6) and the positive/negative selection rounds are repeated until a constant selected aptamer pool is generated, typically 18–20 rounds of SELEX. The resultant pool is then subcloned (7) and sequenced (8). | ||
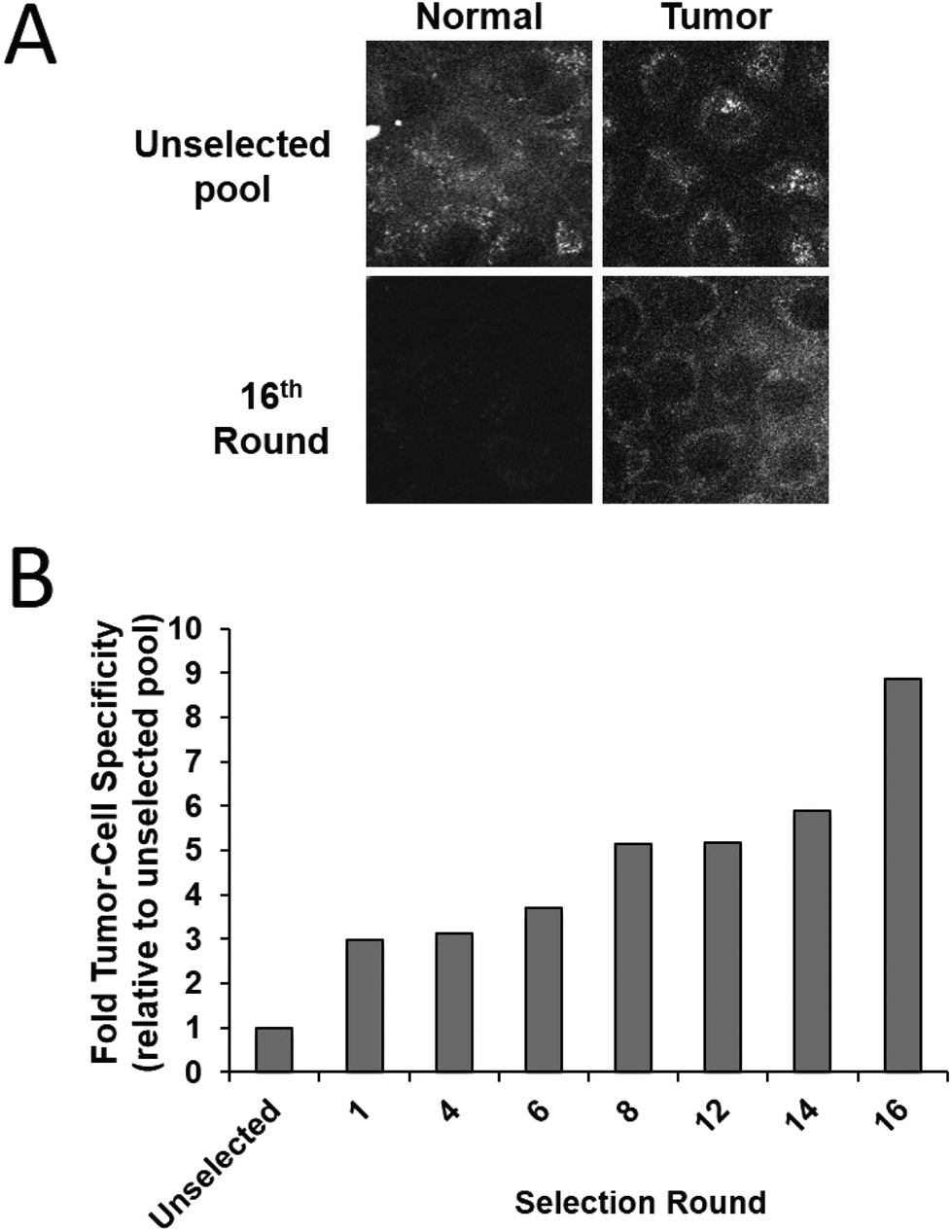 | ||
| Fig. 1 Aptamers selected by the Cell-SELEX process show tumour cell-specific targeting. A. Fluorescently labelled aptamer pools, generated by PCR using 5′-FAM-tagged primers after each round of Cell-SELEX (e.g. the unselected pool and the 16th round of selection), were incubated with MCF10A normal or MCF10Ca1h tumour cells for 4 hours at 37 °C, washed and imaged live by CLSM. B. The specific cellular fluorescence for each sample was determined by image analysis from digitised images such as those in (A). The data is represented as fold tumour-cell specificity, relative to the unselected pool for each round. | ||
DNA aptamers bind to and are internalised by the target tumour but not isogenic normal cells
Single stranded DNA oligonucleotides corresponding to the 6 selected aptamer sequences were synthesised with a fluorescent 5′-FAM tag (Integrated DNA Technologies). Prior to use, the aptamers were refolded and then incubated with the MCF10 tumour progression series cell lines for 6 h to examine tumour cell selectivity. After rigorous washing the cells were imaged live by CLSM (Fig. 2 and not shown), with two aptamers, KW16-16 and KW16-13, demonstrating the greatest level of selectivity for tumour cells, with little to no binding to the normal MCF10A cells observed for either aptamer (Fig. 2A). As a positive control, an aptamer from the literature, KMF2-1a,37 originally targeted against the MCF10AT1 cell line (see above),34 was tested in parallel. Similar to KW16-16 and -13, FAM-KMF2-1a showed increased binding to the more advanced tumour lines (Fig. 2), but noticeable binding to the MCF10A normal cells was also observed, indicative of reduced tumour cell selectivity (Fig. 2A), particularly in comparison to KW16-13. This demonstrates that aptamers generated in similar fashion can have dramatic differences in tumour cell selectivity presumably dependent upon their unique interacting partners on the cell surface and importantly that KW16-13 has robust tumour cell selectivity and is therefore a strong candidate for use in future therapeutic applications. | ||
| Fig. 2 KW16-16 and KW16-13 aptamers specifically bind to and are internalised by breast cancer cells. A. 5′-FITC-labelled KW16-16, KW16-13 and the KMF2-1a control aptamer were incubated with MCF10A normal and the increasingly tumorigenic isogenic MCF10AT, MCF10CA1h and MCF10Ca1a cells for 4 h at 37 °C, washed and imaged live by CLSM. B. The mean specific fluorescence of each sample was determined from digitised images such as those in (A). Results represent mean ± SEM (n > 50), for a single typical experiment from a series of two similar experiments. C. Fold-tumour cell selectivity, relative to MCF10A normal cells was determined from (B). | ||
To further examine the cell selectivity of the generated aptamers and therefore their potential utility in therapeutic applications, KW16-13, KW16-16 and KMF2-1a were tested against a panel of other human cell lines, which included the transformed embryonic kidney 293 T line, the A549 lung adenocarcinoma line and the HeLa cervical cancer line (ESI Fig. S1†). No staining above background was observed in any of the samples, with the noticeable exception of KMF2-1a, which demonstrated significant cytoplasmic uptake in HeLa cells only. This further confirms the limited specificity of the KMF2-1a aptamer for its targeted line and validates the KW16-13 and -16 aptamers as breast cancer specific, positioning them as excellent candidates for further examination. As KW16-13 showed higher tumour selectivity than the other aptamers tested, it was selected for further examination in this study.
Synthesis and characterization of KW16-13-GNRs
GNRs coated with cetyltrimethylammonium bromide (CTAB) were synthesized using a seed-mediated approach13 and imaged by transmission electron microscopy (TEM; Fig. 3, inset), the GNRs found to be a highly uniform and reproducible population with a calculated aspect ratio of ±2.602. UV visible spectra analysis was performed to investigate the optical properties of the GNRs (Fig. 3), with two main plasmon resonances evident, corresponding to a transverse peak around 518 nm and a strong longitudinal peak (LSPR) at 682 nm. In order to connect aptamers to the surface, the GNRs must first be PEGylated to remove the CTAB layer and provide reactive groups to which the thiol-aptamers can subsequently be attached. UV visible spectra analysis (Fig. 3) demonstrated a small shift in LSPR (to 678 nm), confirming the replacement of CTAB by PEG. PEG coating of the GNRs was further confirmed using Fourier Transformer Infra-Red (FTIR) analysis (ESI Fig. S2†). A further shift to 675 nm was observed after reaction with thiolated-KW16-13, confirming aptamer coating. GNR functionalization with KW16-13 was further established using agarose gel electrophoresis (ESI Fig. S3†). Similar results were observed for GNRs coated with the KMF2-1a (not shown). Apts were coated onto GNRs at a ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, which was found to be optimal in our small scale initial testing.
:1, which was found to be optimal in our small scale initial testing.
 | ||
| Fig. 3 Integrity of CTAB-, PEG- and Apt-GNRs as indicated by UV spectral analysis. UV visible spectra were determined for each of the stages of aptamer synthesis as described in the Experimental section. The inset shows a typical TEM image of KW16-13-GNRs. | ||
KW16-13-GNRs specifically recognise tumour cells
The light scattering properties of the GNRs were exploited to confirm that the KW16-13-GNRs retain tumour cell-specific targeting using dark field microscopy. Under a narrow beam of white light, GNRs will scatter light at a different wavelength depending upon their size and due to the surface plasmon resonance of the GNRs generated in this study the scattered light is in the visible spectra. MCF10A normal and MCF10CA1h cells were incubated without or with 100 μl of either PEG-GNRs or KW16-13GNRs for 4 h, fixed with 4% paraformaldehyde and then analysed by dark field microscopy. Compared to control, the PEG-GNRs demonstrated a small amount of binding/uptake in both the normal and tumour cells indicative of low-level non-specific uptake (Fig. 4). In contrast, the KW16-13-GNRs were internalised strongly by the MCF10CA1h cells, with few binding to the MCF10A normal cells. These results demonstrate that the KW16-13-GNRs retain tumour cell specificity and are excellent candidates for investigation as anti-tumour therapeutics. | ||
| Fig. 4 Tumor cell specific binding by KW16-13-GNRs. Normal (MCF10A) and tumour (MCF10CA1h) cells were incubated without (control) or with PEG-, KW16-13-, or KMF2-1a-GNRs for 4 h at 37 °C, washed and fixed with 4% PFA, and imaged by dark field microscopy using a 100× objective. Arrows indicate representative sites of GNR association with cells (not all GNR binding sites are indicated). | ||
KW16-13-GNRs effect specific photothermal killing of tumour but not normal cells
To examine the photothermal cell killing properties of the GNR complexes, both MCF10A normal and MCF10CA1h tumour cells were incubated with either PEG-, KW16-13- or KMF2-1a-GNRs for 4 h at 37 °C, prior to 600 mW irradiation for 5 min using an 808 nm laser as described in the Experimental section. This wavelength overlaps with the LSPR of the GNRs used in this study and also corresponds with the region at which tissue has the lowest absorption, which results in the maximum penetration of light and is therefore the most suitable for photothermal therapy. The photothermal properties of the GNRs were examined using an infrared thermometer which demonstrated a rapid and consistent rise in temperature to >45 °C within 5 min (ESI Fig. S4†). Cells were stained with Trypan blue to identify dead cells, imaged under bright field microscopy at 4× (see 4× enlarged – Fig. 5A) and scored for % cell death in the irradiated area (Fig. 5B). The photothermal therapy had an almost immediate effect, consistent with previous reports.35 Additionally, we observed that extending the trypan blue incubation period beyond 5 min, led to cell detachment from the coverslip, hampering quantitation. This is evident in Fig. 5A, where some of the irradiated cells in the KW16-13GNR tumor sample have detached, leaving a visible blank area in the center of the irradiated zone. | ||
| Fig. 5 Tumor cell-specific photothermal killing by KW16-13-GNRs as measured by Trypan blue exclusion. A. Normal (MCF10A) and tumour (MCF10CA1h) cells were incubated with the indicated Apt-GNR complexes for 4 h at 37 °C, washed and irradiated with 600 mw NIR light for 5 min. Trypan blue was added and the samples imaged under bright field microscopy using a 4× objective B. Trypan blue positive (dead) cells were scored for each sample. Results represent the mean ± SEM (n > 120), averaged from a series of 3 similar experiments. C. Fold tumour cell specificity, relative to MCF10A cells, was calculated from the results of (B). | ||
Both tumour and normal control cells showed no visible effects after irradiation treatment alone, consistent with previous reports that short exposure to laser irradiation in this wavelength is not detrimental to tissue,9 as cellular organelles absorb light at very low levels in the NIR region. Cells incubated with PEG-GNRs alone prior to irradiation displayed very low levels of cellular death (<1%), which was however similar in both tumour and normal cells, indicative of a low level of non-specific uptake of the GNRs by the cells.
Both the KW16-13- and the KMF2-1a-GNRs were able to mediate photothermal cell killing in response to NIR irradiation, but the effects varied greatly in terms of both efficiency and tumour cell specificity between the two complexes. While the KW16-13-GNRs induced >95% cell death in the irradiated area, the KM2-1a-GNRs produced only 3–6% cellular death under the same conditions (Fig. 5). Importantly, the KW16-13-GNRs also displayed a pronounced tumour cell specificity, causing death in only 1% of normal cells (consistent with background levels), resulting in ∼74-fold more tumour cell death than normal cells. This was in contrast to the KMF2-1a-GNRs, which showed only a 2.5-fold enhancement in tumour cell killing compared to normal cells (Fig. 5C). The 808 nm laser used in this study was chosen as the tissue penetration properties of this wavelength make it the ideal choice for progression towards preclinical studies. However, the GNRs used in this study have a maximal absorption in the 700 nm range, but even with the 808 nm laser we have achieved almost 100% tumour cell death, demonstrating the effectiveness of the photothermal approach. Future studies will concentrate on tuning the plasmon resonance of the GNRs to be optimal at 808 nm, potentially increasing the specificity and effectiveness of the treatment, enabling shorter irradiation treatments.
To further validate active targeting of tumor cells by the KW16-13-GNRs, flow cytometry (FACS) based cell viability assays were conducted by irradiating the cells with the 808 nm laser after incubation with the GNRs as explained in the Experimental section. The cells were then stained with propidium iodide21 and analysed by flow cytometry (Fig. 6A). Consistent with the experiments above, the control cells remained 100% viable and the PEG-GNRs showed ∼4-6% cell death due to low-level non-specific uptake in either line. Although KW16-13-GNRs and KMF2-1a-GNRs both initiated tumor cell specific death, the efficiency of KW16-13-GNRs (<71%) was much higher than that of the KMF2-1a-GNRs (<37%). Significantly, normal cells treated with KW16-13-GNRs did not show any significant cell death beyond the non-specific level observed for PEG-GNRs, whereas the KMF2-1a-GNRs had a significant detrimental effect (>18% cell death) confirming the specificity of KW16-13-GNRs (Fig. 6B).
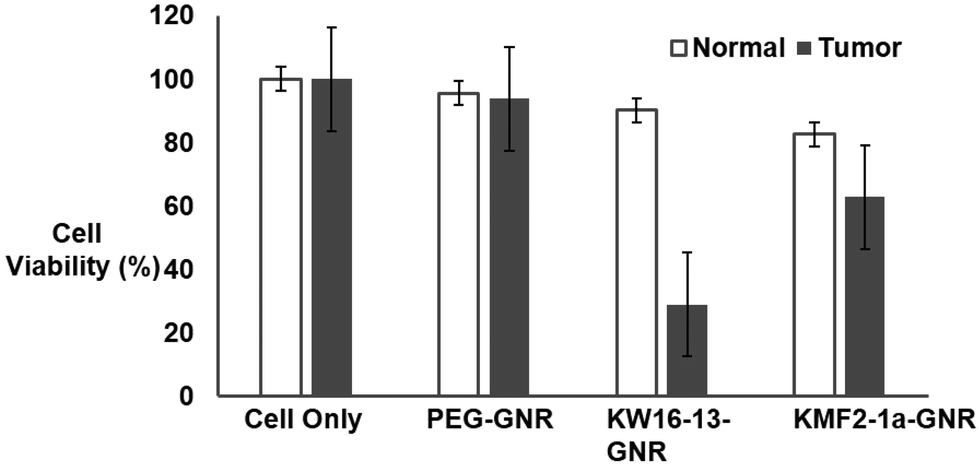 | ||
| Fig. 6 Tumor cell-specific photothermal killing by KW16-13-GNRs as measured by FACS analysis/PI staining. Normal (MCF10A) and tumour (MCF10CA1h) cells were incubated with the indicated Apt-GNR complexes for 4 h at 37 °C, washed and irradiated with 600 mw NIR light for 5 min. Propidium Iodide was added and the samples analysed for viability by flow cytometry. Results represent the mean ± SEM (n = 10000), averaged from a series of 2 similar experiments. | ||
Taken together this data demonstrates that the KW16-13-functionalised GNRs are able to mediate efficient and above all highly specific tumour selective cell killing and are therefore highly suitable candidates for development as anti-tumour agents.
Experimental section
Chemicals
Cetyltrimethylammonium bromide (CTAB), gold(III) chloride hydrates (HAuCl4), silver nitrate (AgNO3), ascorbic acid, sodium borohydride (NaBH4), tris(2-carboxyethyl) phosphine) (TCEP) and paraformaldehyde (PFA) were all purchased from Sigma Aldrich. Monomethoxy poly-(ethylene glycol) thiol (mPEG-SH; MW 5000 Da) was purchased from Polymer science, Inc. Trypan blue solution and propidium iodide stain were purchased from Thermo Fisher Scientific Inc.100g, 5 min to recover aptamer sequences from the supernatant. For the first round of selection only, the entire selected pool was pre-amplified by PCR (due to the low number of individual DNA sequences present), using a standard forward primer and a biotin-tagged reverse primer. The pool was then amplified by standard PCR (again using a standard forward and biotin-tagged reverse primer) and the resulting double-stranded aptamers were separated using streptavidin-coated beads and column centrifugation to remove the unwanted biotin-tagged reverse sequences from the aptamer sequence (see PCR and strand separation). The single stranded aptamer sequences were then precipitated and used for the next round of SELEX. Prior to each subsequent round of SELEX, the selected aptamer pool from the previous round was denatured/flash cooled as previously to generate folded ssDNA. Subsequent rounds of SELEX were performed as per the first round except the recovered aptamer sequences underwent a counter-selection step against MCF10A cells as follows: aptamers recovered from the supernatant of boiled MCF10CA1h cells (post-incubation) were incubated with a confluent 10 cm dish of MCF10A cells pre-washed twice with wash buffer, 1 h, RT with gentle agitation. Unbound aptamers recovered in the supernatant (selected pool). PCR amplification and strand separation proceeded as per first pool. From the third round of SELEX onwards, the culture dish for both positive and negative selection was reduced to 6 cm to decrease non-specific binding.
Conclusions
We have successfully developed and validated a DNA aptamer that is highly specific for breast cancer cells, with no observable binding to isogenic normal cells. When coupled to the surface of GNRs tuned for plasmonic resonance in the NIR range, they serve as highly effective tumour targeting moieties that efficiently deliver GNRs to tumour cells, resulting in >95% tumour cell death after NIR irradiation. The high tumour-cell specificity exhibited by the novel GNRs, under the direction of the KW16-13 aptamer makes them exciting candidates for development as novel anti-tumour therapeutics, to exert maximal specific tumour cell death that is safe, effective and minimally invasive.Acknowledgements
The authors would like to thank Dr John Zhu, Alex Fulcher and Andrew Fryga for assistance with the cytoviva, bright field microscopy and flow cytometry respectively. Also the authors would like to thank Dr Bin Su for his help with the scheme. This research was financially supported under the Australian Research Council's Discovery projects funding schemes DP120100170 and Monash Interdisciplinary projects. This work was performed in part at the Melbourne Centre for Nanofabrication (MCN) in the Victorian Node of the Australian National Fabrication Facility (ANFF). The bright field image analysis and TEM imaging were conducted at the Monash Micro Imaging facility and Monash Centre for Electron microscopy, respectively. FACS analysis was performed at the Flow Core Facility, Monash.References
- S. Kessentini and D. Barchiesi, Biomed. Opt. Express., 2012, 3, 590–604 CrossRef PubMed.
- J. van der Zee, Ann. Oncol., 2002, 13, 1173–1184 CrossRef CAS PubMed.
- C. Christophi, A. Winkworth, V. Muralihdaran and P. Evans, Surg. Oncol., 1998, 7, 83–90 CrossRef CAS PubMed.
- J. Fang and Y. C. Chen, Curr. Pharm. Des., 2013, 19, 6622–6634 CrossRef CAS PubMed.
- H. Hashizume, P. Baluk, S. Morikawa, J. W. McLean, G. Thurston, S. Roberge, R. K. Jain and D. M. McDonald, Am. J. Pathol., 2000, 156, 1363–1380 CrossRef CAS PubMed.
- G. Khalid, Drug Discovery Today: Technol., 2012, 9, 161–166 CrossRef PubMed.
- M. E. Davis, Z. G. Chen and D. M. Shin, Nat. Rev. Drug Discovery, 2008, 7, 771–782 CrossRef CAS PubMed.
- S. J. Tan, M. J. Campolongo, D. Luo and W. L. Cheng, Nat. Nanotechnol., 2011, 6, 268–276 CrossRef CAS PubMed.
- A. M. Alkilany, L. B. Thompson, S. P. Boulos, P. N. Sisco and C. J. Murphy, Adv. Drug Delivery Rev., 2012, 64, 190–199 CrossRef CAS PubMed.
- Z. Y. Ma, H. X. Xia, Y. P. Liu, B. Liu, W. Chen and Y. D. Zhao, Chin. Sci. Bull., 2013, 58, 2530–2536 CrossRef CAS.
- Z. Zhang, J. Wang and C. Chen, Theranostics, 2013, 3, 223–238 CrossRef CAS PubMed.
- W. Xiong, R. Mazid, L. W. Yap, X. Li and W. Cheng, Nanoscale, 2014, 6, 14388–14393 RSC.
- B. Nikoobakht and M. A. El-Sayed, Chem. Mater., 2003, 15, 1957–1962 CrossRef CAS.
- K. C. Black, J. Yi, J. G. Rivera, D. C. Zelasko-Leon and P. B. Messersmith, Nanomedicine, 2013, 8, 17–28 CrossRef CAS PubMed.
- S. Charan, K. Sanjiv, N. Singh, F. Chien, Y. Chen, N. N. Nergui and P. Chen, Bioconjugate Chem., 2012, 23, 2173–2182 CrossRef CAS PubMed.
- H. N. Green, D. V. Martyshkin, C. M. Rodenburg, E. L. Rosenthal and S. B. Mirov, J. Nanotechnol., 2011, 2011, 1–7 CrossRef.
- X. Huang, I. H. El-Sayed, W. Qian and M. A. El-Sayed, J. Am. Chem. Soc., 2006, 128, 2115–2120 CrossRef CAS PubMed.
- D. K. Kirui, S. Krishnan, A. D. Strickland and C. A. Batt, Macromol. Biosci., 2011, 11, 779–788 CrossRef CAS PubMed.
- E. Lee, Y. Hong, J. Choi, S. Haam, J. S. Suh, Y. M. Huh and J. Yang, Nanotechnology, 2012, 23, 465101 CrossRef PubMed.
- A. V. Liopo, W. M. Conjusteau and A. A. Oraevsky, Progress in Biomedical Optics and Imaging-Proc. SPIE, 2012, 822.
- D. Pissuwan, S. M. Valenzuela, M. C. Killingsworth, X. Xu and M. B. Cortie, J. Nanopart. Res., 2007, 9, 1109–1124 CrossRef CAS.
- C. S. Rejiya, J. Kumar, V. Raji, M. Vibin and A. Abraham, Pharmacol. Res., 2012, 65, 261–269 CrossRef CAS PubMed.
- H. Liao and J. H. Hafner, Chem. Mater., 2005, 17, 4636–4641 CrossRef CAS.
- C. Gui and D. X. Cui, Cancer Biol. Med., 2012, 9, 221–233 CAS.
- J. Choi, Y. Park, E. B. Choi, H. Kim, D. J. Kim, Y. Hong, S. Ryu, J. H. Lee, J. S. Suh, J. Yang, Y. M. Huh and S. Haam, J. Biomed. Opt., 2013, 19 Search PubMed.
- Y.-F. Huang, H.-T. Chang and W. Tan, Anal. Chem., 2008, 80, 567–572 CrossRef CAS PubMed.
- J. Wang, K. Sefah, M. B. Altman, T. Chen, M. You, Z. Zhao, C. Z. Huang and W. Tan, Chem. – Asian J., 2013, 8, 2417–2422 CrossRef CAS PubMed.
- A. D. Keefe, S. Pai and A. Ellington, Nat. Rev. Drug Discovery., 2010, 9, 537–550 CrossRef CAS PubMed.
- K. Sefah, D. Shangguan, X. Xiong, M. B. O'Donoghue and W. Tan, Nat. Protoc., 2010, 5, 1169–1185 CrossRef CAS PubMed.
- W. Tan, M. J. Donovan and J. Jiang, Chem. Rev., 2013, 113, 2842–2862 CrossRef CAS PubMed.
- J. Wang, G. Zhu, M. You, E. Song, M. I. Shukoor, K. Zhang, M. B. Altman, Y. Chen, Z. Zhu, C. Z. Huang and W. Tan, ACS Nano, 2012, 6, 5070–5077 CrossRef CAS PubMed.
- Y.-F. Huang, K. Sefah, S. Bamrungsap, H.-T. Chang and W. Tan, Langmuir, 2008, 24, 11860–11865 CrossRef CAS PubMed.
- Z. Xiao, C. Ji, J. Shi, E. M. Pridgen, J. Frieder, J. Wu and O. C. Farokhzad, Angew. Chem., Int. Ed., 2012, 51, 11853–11857 CrossRef CAS PubMed.
- P. J. Dawson, S. R. Wolman, L. Tait, G. H. Heppner and F. R. Miller, Am. J. Pathol., 1996, 148, 313–319 CAS.
- H. D. Soule, T. M. Maloney, S. R. Wolman, W. D. Peterson Jr., R. Brenz, C. M. McGrath, J. Russo, R. J. Pauley, R. F. Jones and S. C. Brooks, Cancer Res., 1990, 50, 6075–6086 CAS.
- S. J. Santner, P. J. Dawson, L. Tait, H. D. Soule, J. Eliason, A. N. Mohamed, S. R. Wolman, G. H. Heppner and F. R. Miller, Breast Cancer Res. Treat., 2001, 65, 101–110 CrossRef CAS PubMed.
- K. Zhang, K. Sefah, L. Tang, Z. Zhao, G. Zhu, M. Ye, W. Sun, S. Goodison and W. Tan, ChemMedChem, 2012, 7, 79–84 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Confocal images of aptamer incubated with 293T, A549 or HeLa cells, FTIR spectra of PEG-GNR, gel electrophoresis of Apt-GNR and time dependent temperature curve of GNRs. See DOI: 10.1039/c5nr07831h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |