
Ordered self-assembly of amphipathic graphene nanosheets into three-dimensional layered architectures†
Qile
Fang
,
Xufeng
Zhou
*,
Wei
Deng
and
Zhaoping
Liu
*
Ningbo Institute of Materials Technology and Engineering, Chinese Academy of Sciences, Ningbo, Zhejiang 315201, P. R. China. E-mail: liuzp@nimte.ac.cn; zhouxf@nimte.ac.cn
First published on 23rd November 2015
Abstract
A novel layered graphene-based architecture is achieved via an ordered self-assembly process. Amphipathic graphene nanosheets are joined horizontally into large sheets via edge splicing, and a cross-linking agent of poly(vinyl alcohol) bridges them into integrated three-dimensional monoliths with tunable interlayer spacing. This layered architecture possesses highly ordered and favorable microchannels for molecular transfer.
The superior properties of graphene nanomaterials have been recognised in various fields, and the assembly of graphene nanosheets into three-dimensional (3D) hierarchical architectures opens up a broader avenue for applications of this “miracle material”.1–4 The 3D macrostructures not only maintain the intrinsic features of the individual nano-blocks (graphene nanosheets), but also possess new collective physiochemical properties, which are remarkably different from both the individual building blocks and their bulk material (graphite).5–7 More importantly, such “bottom-up” nanotechnology propels the development of device processing of graphene nanomaterials, which will be more manageable for practical use.8–10
Folding, scrolling and bending of graphene nanosheets can occur in different micro-environments or force fields,11 which renders various assembly modes for them in 3D architectures, subsequently dominating their performance. Graphene oxide (GO) is the main precursor for graphene-based 3D assembly (different from the method of chemical vapor deposition12,13), because the abundant oxygen-containing groups on GO nanosheets result in a highly hydrophilic property and homogeneous aqueous suspensions.14–16 So far, most of their resulting 3D monoliths present disordered porous structures,17–19 and in such circumstances, the oxygenous functional groups at both the planes and edges of the GO nanosheets have the same probability of contacting the neighboring individuals. Only a few works have reported that oriented or ordered microstructures can be achieved for 3D graphene-based architectures. Qiu et al. have fabricated ordered porous graphene monoliths upon directional crystal growth by freeze casting, but a randomly oriented porous structure was obtained if the GO dispersion was directly frozen without pre-reduction, which means only the proper marriage of graphene chemistry with ice physics can lead to such ordered porous structures.20 Besides, some other works demonstrated that modified graphene sheets mixed with the cross-linking agent poly(vinyl alcohol) (PVA) can form ordered or oriented microstructures along the freezing direction.21,22 Obviously, ice template-directed assembly is the primary method for such synthesis of ordered 3D graphene-based architectures whose precursor (GO) must be pre-treated appropriately.23 Different from the template-directed assembly, a self-assembly method without structural directing templates usually leads to a disordered structure, such as the most common method of hydrothermal-induced self-assembly, in which GO building blocks with chemical and structural homogeneity always induce disordered self-assembly.17
Ordered microstructures can realize directional transfer processes within the 3D graphene-based architectures, at either the basal plane of the graphene (electron or other charge carriers) or in the inner channel of the 3D space (ions, molecules, etc.), which can be easily applied to the intentional design of ordered microstructures for diverse applications. In particular, ordered microstructures with open and penetrating channels can also decrease the transfer resistance and provide a barrier-free path. Although GO can exhibit nematic and lamellar liquid crystallinity in aqueous dispersions at a certain mass concentration, displaying anisotropy,24,25 their ordered assembly into 3D solid monoliths remains challenging and the rare successful examples have all relied on template-directing. Here, we report a unique self-assembly approach, without an ice template, for preparing ordered 3D graphene-based architectures with a layered microstructure. Amphipathic graphene nanosheets (hydrophilic edges and hydrophobic basal planes) are prepared as the precursors, which carry out a two-level assembly along two directions with the assistance of PVA polymers to construct a 3D layered monolith, whose interlayer spacing is uniform and can be readily adjusted by varying the concentration of PVA.
The amphipathic graphene nanosheets were realized by sulfonation of reduced GO sheets, which has been reported previously and is illustrated in Fig. 1a. Starting from GO nanosheets, their functional groups were regulated via three successive steps, pre-reduction, sulfonation and post-reduction26,27 (please refer to the Experimental section in the ESI†for details). The product of sulfated reduced GO (S-RGO) displayed strong hydrophilicity. It is stable in aqueous solution even after standing for 1 week (Fig. 1b), which guarantees a homogeneous dispersion of the functionalized GO precursor for 3D assembly. Because of the double reduction, S-RGO also exhibits a certain lipophilicity, so it can absorb toluene and be fully immersed in it (Fig. S1, ESI†). The AFM image in Fig. 1c shows that the thickness of the sheet edge area was ∼1.0 nm, while the inside area was measured to be ∼2.0 nm, indicating that this sheet was a stack of two single-layers. The wrinkled paper-like texture of the S-RGO sheet can be observed in the TEM image (Fig. 1d), where the inset SAED pattern presents the typical hexagonal crystalline structure of graphene and coincides well with the pattern of monolayer reduced GO.28,29 This means that the obtained S-RGO mainly consists of monolayer and/or bi-layer sheets.
 | ||
| Fig. 1 Synthesis and properties of S-RGO. (a) Schematic illustration of the preparation of S-RGO. (b) Photograph showing the synthesized S-RGO aqueous solution after standing for 1 week. (c) AFM image of S-RGO. (d) TEM image of S-RGO, and the corresponding SAED pattern (inset graph). (e) Raman spectra of GO and S-RGO. (f) FTIR spectra of GO and S-RGO. (g) XRD patterns of S-RGO, GO and graphite powder. | ||
After pre-reduction and post-reduction, more conjugated structures have been restored in the modified nanosheets, which can be confirmed by the Raman spectra in Fig. 1e. The intensity ratio of the D band centered at 1348 cm−1 (K point phonons of A1g symmetry associated with structural defects) to the G band centered at 1585 cm−1 (zone center phonons of E2g symmetry observed for sp2 carbon domains) increases from 0.912 of GO to 1.099 of S-RGO,30,31 because the average size of the sp2 carbon domains decreases with the increasing number of smaller graphitic domains formed during the double reduction process.32,33 Besides the change on the basal graphitic carbons, it can be seen from the FTIR spectra in Fig. 1f that the peaks at 1720 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and 1054 cm−1 (C–O) are attenuated significantly in the S-RGO sample, implying that most of the original oxygen-containing groups on the GO (both at edges and basal planes) were removed, especially carboxyl groups. Meanwhile, sulfonic acid groups were grafted onto the nanosheets during the sulfonation process based on diazonium functionalization, whose characteristic vibrations at 1179 cm−1, 1124 cm−1 and 1035 cm−1 (two νS–O and one νs–phenyl) can be obviously seen in the FTIR spectrum.26,34 Although some researchers thought that diazonium chemistry-based functionalization attacks both the edge and the basal plane of graphene,35 it's noteworthy that S-RGO possesses a similar interlayer spacing (0.33 nm, derived from the XRD diffraction peak at 2θ = 26.65° in Fig. 1g) to that of graphite (0.34 nm, 2θ = 26.06°), rather than GO (0.70 nm, 2θ = 12.57°). This result not only confirms the effective elimination of most oxygen-containing functional groups on the GO plane by NaBH4 and hydrazine reduction, but also suggests the grafting of sulfonic acid groups on the edges of the graphene sheets rather than on the planes, which has also been confirmed by other studies.31,36 Therefore, the obtained S-RGO is an amphipathic nanosheet with hydrophilic edges and hydrophobic basal planes, which can enrich the properties of graphene-based materials and extend their potential applications.
O) and 1054 cm−1 (C–O) are attenuated significantly in the S-RGO sample, implying that most of the original oxygen-containing groups on the GO (both at edges and basal planes) were removed, especially carboxyl groups. Meanwhile, sulfonic acid groups were grafted onto the nanosheets during the sulfonation process based on diazonium functionalization, whose characteristic vibrations at 1179 cm−1, 1124 cm−1 and 1035 cm−1 (two νS–O and one νs–phenyl) can be obviously seen in the FTIR spectrum.26,34 Although some researchers thought that diazonium chemistry-based functionalization attacks both the edge and the basal plane of graphene,35 it's noteworthy that S-RGO possesses a similar interlayer spacing (0.33 nm, derived from the XRD diffraction peak at 2θ = 26.65° in Fig. 1g) to that of graphite (0.34 nm, 2θ = 26.06°), rather than GO (0.70 nm, 2θ = 12.57°). This result not only confirms the effective elimination of most oxygen-containing functional groups on the GO plane by NaBH4 and hydrazine reduction, but also suggests the grafting of sulfonic acid groups on the edges of the graphene sheets rather than on the planes, which has also been confirmed by other studies.31,36 Therefore, the obtained S-RGO is an amphipathic nanosheet with hydrophilic edges and hydrophobic basal planes, which can enrich the properties of graphene-based materials and extend their potential applications.
PVA polymers have been applied as an excellent cross-linking agent for the 3D assembly of GO. Their –OH rich structure can bridge the oxygen-containing groups on individual GO sheets to form 3D networks.37,38 Herein, a stable hydrogel without volume shrinkage was formed after hydrothermal treatment of a mixed aqueous suspension of S-RGO and PVA, and the corresponding aerogel was obtained by subsequent freeze-drying, as shown in Fig. 2a and b. However, pure S-RGO dispersion can't gelate in the same conditions, as is shown in Fig. S2 (ESI†). Structural evaluation of the 3D S-RGO + PVA monolith was conducted using SEM (shown in Fig. 2c–f). Of particular interest is that a large-scale ordered structure can be observed, which is in sharp contrast to that of the randomly oriented microstructure of the aerogels prepared from the GO and PVA mixture in our experiments (the gelation process and microscopic structures of the corresponding RGO + PVA monolith are provided in Fig. S3, ESI†) and in previous reports.37,38 The SEM image (Fig. 2c) of one small block breaking off from the 3D S-RGO + PVA aerogel presents a well-organized alignment and continuous layered structure, and most notably has a uniform orientation at the millimeter scale. In a magnified SEM image, even the interlayer spacing is quite remarkable, which looks like a stack of paper when viewed from the side (Fig. 2d). What's more, every layer of high-continuity can also be observed by one layer peeling off from the monolith (Fig. 2e). Considering all the components in this reaction system, each layer with such a large area is undoubtedly composed of numerous S-RGO nanosheets that self-assemble along a 2D plane due to their unique amphipathic properties. From the cross-sectional view of the layers in Fig. 2f–h, with the increase of magnification, it can be clearly seen that the interlayer spaces are filled with numerous filaments. We speculate that the filaments are PVA which adhere to the layer surfaces and bridge two adjacent layers to finally form a bulk composite with uniform interlayer spacing. As far as we know, such organized layered structures at the millimeter scale have not been reported for 3D graphene-based macrostructures, and more microscopic layered structures are provided in Fig. S4 (ESI†).
 | ||
Fig. 2 (a), (b) Photographs of hydrogel and corresponding aerogel composed of S-RGO and PVA with a S-RGO/PVA weight ratio of 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. (c–h) SEM images of the S-RGO + PVA aerogel at different magnifications. :1. (c–h) SEM images of the S-RGO + PVA aerogel at different magnifications. | ||
The ordered assembly principle of the 3D S-RGO + PVA architecture was further studied. It can be seen from the SEM images in Fig. 3a that the directly freeze-dried product of the S-RGO dispersion after hydrothermal treatment without adding PVA is composed of pieces of lamella. As a result, it can't form a gel or construct a stable 3D monolith. However, a large number of S-RGO nanosheets piece together through their edges to form large sheets with lateral sizes >100 μm (Fig. 3b). Although this is not as large as the interconnected sheets in the 3D layered structure with the addition of PVA (Fig. 2), the above results evidently prove the spontaneous assembly of S-RGO sheets within the 2D plane in aqueous solutions. A previous report suggested that fluorinated GO nanosheets tended to cross-link via the edges rather than the planes to form large interconnected sheets if the fluorinated GO has more functional groups at the edges and C–F bonds in the plane.39 The similar surface chemistry of S-RGO could be used to explain the phenomenon observed in this study, that the amphipathic S-RGO possesses most hydrophilic groups (sulfonic acid groups) at the edges and the majority of the original oxygen-containing groups in the plane are reduced. We need to point out that this parallel-splicing large sheet might be stacked with several S-RGO nanosheets via π–π interactions derived from their hydrophobic planes, as the large sheet and its side are clearly visible in the low magnification SEM image. With the assistance of PVA, these parallel-splicing S-RGO sheets were assembled into a layered 3D macrostructure. Certainly, PVA also assists the long range cross-linking of S-RGO nanosheets, as the dimension of the S-RGO layers in the ordered 3D layered architecture can reach the millimeter scale in the presence of PVA. Giving insight into the layered structure, one layer can be integrally peeled off from the 3D macrostructure and there appears to be seamless splicing along the horizontal direction of every layer (Fig. 3c). The S element from –SO3H of S-RGO is distributed evenly on the large layers (Fig. S5, ESI†). If given a certain external force, such as pressing the monolith by hand, the layers would deform into wavy structures but still be joined together to remain as an intact monolith (Fig. 3d), revealing that the joined layer has a certain tenacity and the connection between S-RGO sheets in each layer is quite strong.
 | ||
| Fig. 3 (a), (b) SEM images of the directly freeze-dried product of the S-RGO dispersion (after hydrothermal treatment) without PVA. (c) SEM image of one layer peeled off from the 3D layered S-RGO + PVA monolith. (d) SEM image of the deformed layers in 3D S-RGO + PVA monolith under an external force. | ||
High temperature annealing was adopted for the S-RGO + PVA monolith in inert gas, which gave a pyrolytic product without a macroscopical shape change. According to the comparison images in Fig. 4a and b, apparently most of the adherent interlinings between the self-assembled S-RGO layers disappeared after annealing, which can be ascribed to the thermal decomposition of PVA whose melting temperature (pure polymer) with a 98–99% hydrolysis degree is 226 °C.40 Thermogravimetric analysis also supports that a remarkable weight loss around 200 °C can be observed in the S-RGO + PVA monolith compared with the net S-RGO (Fig. S6, ESI†). The Raman spectra in Fig. S7 (ESI†) also confirm the removal of PVA, in which the characteristic vibrational peak at 1134 cm−1, an indicator for the degree of crystallinity of the PVA polymer,40 vanishes completely in the annealed S-RGO + PVA sample. These results provide strong evidence to support our proposal that PVA filaments play a bridging role in the interlayer. At a high temperature of 800 °C, non-carbon elements in the PVA chain were removed and some of the carbon elements converted to amorphous carbons remaining in interlayer as a skeleton,38 which can explain why the 3D S-RGO + PVA monolith still kept its macrostructure after annealing. Beyond all doubt, PVA plays a critical role in the ordered self-assembly and structural construction of the 3D S-RGO + PVA macrostructure, especially in the linkage of each parallel-splicing S-RGO layer. UV-vis spectroscopy was used to monitor the interaction between PVA and S-RGO. In the solution state before the hydrothermal treatment, the addition of PVA brought a 3 nm red-shift of the band at 257 nm of the S-RGO dispersion which is attributed to π–π* transitions of CC bonds (Fig. S8, ESI†),28,41 reflecting that the PVA polymers interact with the aromatic matrix of S-RGO once connected with the S-RGO nanosheets in the aqueous solution, and subsequently guide the large parallel-splicing of S-RGO sheets to self-assemble face to face along the vertical direction during the hydrothermal process. What's more, the varied amount of PVA added could lead to different interlayer distances. The sample in Fig. 4c (mass ratio of S-RGO:PVA = 4:1) contained 20 w/w% PVA and the one in Fig. 4d (mass ratio of S-RGO:PVA = 1:1) was 50 w/w%. Obviously, less PVA results in smaller interlayer spacing (5–10 μm), compared to the 10–15 μm spacing of the 1:1 3D S-RGO + PVA sample. Hence, the layered structure of this ordered material can be easily adjusted by altering the precursor proportions.
 | ||
| Fig. 4 (a), (b) SEM images of the S-RGO + PVA aerogel before and after annealing. (c), (d) The cross-sectional view of 3D layered S-RGO + PVA monoliths with different PVA amounts, (c) the mass ratio of S-RGO to PVA is 4:1 and (d) the mass ratio of S-RGO to PVA is 1:1. | ||
It is quite clear that ice template-directed formation of ordered or oriented graphene-based architectures, which has been reported in previous works,20,21 can be excluded in this study, since there was no directed freezing involved in our sample preparation. Instead, it's a complete self-assembly process. Fig. 5 gives the corresponding schematic illustration of the ordered self-assembly of layered architectures based on S-RGO and PVA. It involves two-level assembly processes along two directions. The linkage between marginal sulfonic groups makes amphipathic graphene nanosheets join horizontally into large flat sheets (layers) up to millimeter scale (two-dimensional assembly), whereas in the vertical direction, PVA polymers bridge two adjacent layers into an integrated 3D structure (three-dimensional assembly). Certainly, the two-level assemblies are carried out simultaneously in the hydrothermal process, and the synergetic effects between the two processes should also be taken into account. In the disordered self-assembly based on GO precursors, hydrogen bonding between the functional groups and π–π interactions between graphitic regions (or with other additives containing aromatic rings) are usually thought to be the main driving forces to form a 3D network,17,42,43 however, the specific driving forces in this ordered self-assembly system are still not quite clear, and more research is needed to investigate the mechanisms.
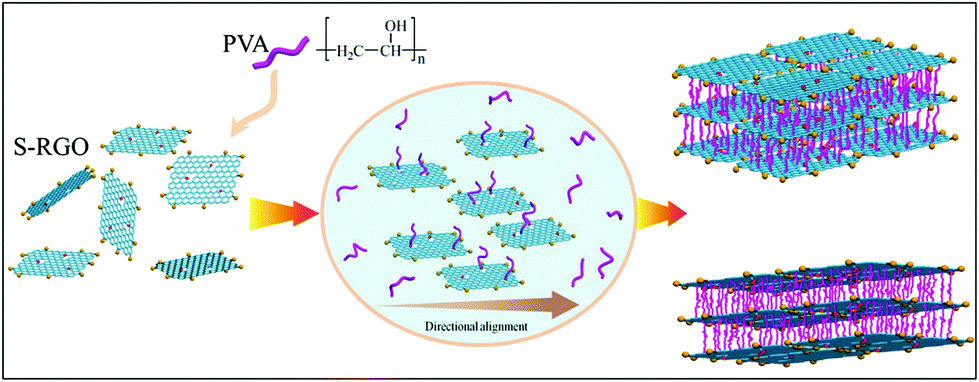 | ||
| Fig. 5 Schematic illustration of the ordered self-assembly of S-RGO nanosheets with PVA. | ||
The N2 adsorption–desorption isotherms of the ordered S-RGO + PVA and disordered RGO + PVA aerogels are displayed in Fig. 6a. Actually, ordered self-assembly made the S-RGO + PVA aerogel possess a lower specific surface area (49.06 m2 g−1, vs. 212.48 m2 g−1 of the RGO + PVA aerogel), which might be attributed to the π–π stacking of S-RGO sheets derived from their hydrophobic planes as mentioned above. We have compared the adsorption kinetics of dye molecules (methylene blue, MB) on these two types of aerogels which have excluded the difference of their volume density (ESI†), and the results in Fig. 6b show that the interior channel direction of the 3D graphene-based architectures significantly affects the transfer process of ions or molecules within the monolith, especially for macromolecules. The pseudo-second-order model fits the data best (R2 = 0.9917 and 0.9934) out of the three commonly used kinetic fitting models (their regression parameters are listed in Table S1, ESI†), which firstly declares that the adsorption of MB on aerogels belongs to intragranular diffusion and it is thought to be the main rate-determining step.44 Although the S-RGO + PVA aerogel has a lower specific surface area, it exhibits a higher adsorption capacity for MB (411.85 mg g−1, vs. 381.61 mg g−1 of the RGO + PVA aerogel), which is mainly due to the increase of available adsorption sites per unit area. Both the oxygen-containing groups and the aromatic matrix are effective sites for MB adsorption. After reduction from GO to S-RGO, most sp2 carbons were recovered, giving rise to enhanced π–π interactions between S-RGO and MB. It is of great importance that the S-RGO + PVA aerogel with an ordered layered microstructure possesses a significantly high rate constant K2 (3.0369 × 10−4 mg g−1 h−1), which is almost an order of magnitude higher than that of the RGO + PVA aerogel (K2 = 4.1997 × 10−5 mg g−1 h−1) with a disordered pore structure. The advantage of S-RGO + PVA aerogels with ordered architectures is similar to another type of popular materials, ordered mesoporous materials, whose ordered structures contribute to their application potential as catalysts, adsorbents and in medical diagnosis.45 Although the pore size of the 3D graphene-based architectures prepared in this study belongs to the macropore region, the arrangement of individual graphene building blocks still has a crucial influence on the transfer process within monolith, and the ordered layered structure is beneficial for macromolecule transfer.
 | ||
| Fig. 6 (a) N2 adsorption–desorption isotherms of S-RGO + PVA and RGO + PVA aerogels; (b) time-dependent adsorption of MB on S-RGO + PVA and RGO + PVA aerogels, and three kinetic models are fitted for the curves. The initial concentration of MB is 50 mg L−1 and the solid-aqueous ratio is 20 mg/150 mL. | ||
In summary, we have developed a novel layered 3D graphene-based architecture via an ordered self-assembly method. GO nanosheets firstly need to be modified by a sulfonation process to regulate the functional groups on their planes and edges, obtaining amphipathic nanosheets (S-RGO), which can be joined horizontally into large flat sheets up to the millimeter scale via edge group splicing. Under the guidance of PVA, the parallel-splicing S-RGO sheets arrange face to face and construct a layered 3D monolith whose interlayer spacing is artificially controllable. These highly ordered interior channels of the graphene-based monolith have been demonstrated as a favorable microstructure for molecular transfer. This type of architectural material may have a promising future for high-performance applications in the fields of environmental purification, energy, catalysis, microelectronics and medical diagnosis.
Acknowledgements
This work was kindly supported by the National Natural Science Foundation of China (Grant No. 21201173 and 51502312), Key Research Program of the Chinese Academy of Sciences (Grant No. KGZD-EW-T08), and China Postdoctoral Science Foundation (Grant No. 2015M570528).References
- A. K. Geim, Science, 2009, 324, 1530 CrossRef CAS PubMed.
- K. S. Novoselov, V. I. Fal'ko, L. Colombo, P. R. Gellert, M. G. Schwab and K. Kim, Nature, 2012, 490, 192 CrossRef CAS PubMed.
- Q. Fang and B. Chen, Chem. Eng. J., 2015, 264, 753 CrossRef CAS.
- Z. Li, Z. Liu, H. Sun and C. Gao, Chem. Rev., 2015, 115, 7046 CrossRef CAS PubMed.
- Y. Gao and Z. Tang, Small, 2011, 7, 2133 CrossRef CAS PubMed.
- Z. Wu, Y. Sun, Y. Tan, S. Yang, X. Feng and K. Mullen, J. Am. Chem. Soc., 2012, 134, 19532 CrossRef CAS PubMed.
- Y. Wu, N. Yi, L. Huang, T. Zhang, S. Fang, H. Chang, N. Li, J. Oh, J. A. Lee, M. Kozlov, A. C. Chipara, H. Terrones, P. Xiao, G. Long, Y. Huang, F. Zhang, L. Zhang, X. Lepro, C. Haines, M. D. Lima, N. P. Lopez, L. P. Rajukumar, A. L. Elias, S. Feng, S. J. Kim, N. T. Narayanan, P. M. Ajayan, M. Terrones, A. Aliev, P. Chu, Z. Zhang, R. H. Baughman and Y. Chen, Nat. Commun., 2015, 6, 6141 CrossRef CAS PubMed.
- S. Nardecchia, D. Carriazo, M. L. Ferrer, M. C. Gutierrez and F. del Monte, Chem. Soc. Rev., 2013, 42, 794 RSC.
- M. Yang, N. Zhang, M. Pagliaro and Y. Xu, Chem. Soc. Rev., 2014, 43, 8240 RSC.
- N. Zhang, M. Yang, S. Liu, Y. Sun and Y. Xu, Chem. Rev., 2015, 115, 10307 CrossRef CAS PubMed.
- R. L. D. Whitby, ACS Nano, 2014, 8, 9733 CrossRef CAS PubMed.
- Z. Chen, W. Ren, L. Gao, B. Liu, S. Pei and H. Cheng, Nat. Mater., 2011, 10, 424 CrossRef CAS PubMed.
- F. Yavari, Z. Chen, A. V. Thomas, W. Ren, H. Cheng and N. Koratkar, Sci. Rep., 2011, 1, 116 Search PubMed.
- W. Gao, L. B. Alemany, L. J. Ci and P. M. Ajayan, Nat. Chem., 2009, 1, 403 CrossRef CAS PubMed.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228 RSC.
- X. Cao, Z. Yin and H. Zhang, Energy Environ. Sci., 2014, 7, 1850 CAS.
- Y. Xu, K. Sheng, C. Li and G. Shi, ACS Nano, 2010, 4, 4324 CrossRef CAS PubMed.
- H. Huang, P. Chen, X. Zhang, Y. Lu and W. Zhan, Small, 2013, 9, 1397 CrossRef CAS PubMed.
- Y. Zhao, J. Liu, Y. Hu, H. Cheng, C. Hu, C. Jiang, L. Jiang, A. Cao and L. Qu, Adv. Mater., 2013, 25, 591 CrossRef CAS PubMed.
- L. Qiu, J. Z. Liu, L. Y. Chang, Y. Wu and D. Li, Nat. Commun., 2012, 3, 1241 CrossRef PubMed.
- J. L. Vickery, A. J. Patil and S. Mann, Adv. Mater., 2009, 21, 2180 CrossRef CAS.
- Y. Huang, D. Wu, J. Jiang, Y. Mai, F. Zhang, H. Pan and X. Feng, Nano Energy, 2015, 12, 287 CrossRef CAS.
- L. Estevez, A. Kelarakis, Q. Gong, E. H. Da'as and E. P. Giannelis, J. Am. Chem. Soc., 2011, 133, 6122 CrossRef CAS PubMed.
- Z. Xu and C. Gao, ACS Nano, 2011, 5, 2908 CrossRef CAS PubMed.
- Z. Xu and C. Gao, Acc. Chem. Res., 2014, 47, 1267 CrossRef CAS PubMed.
- Y. Si and E. T. Samulski, Nano Lett., 2008, 8, 1679 CrossRef CAS PubMed.
- J. R. Lomeda, C. D. Doyle, D. V. Kosynkin, W. Hwang and J. M. Tour, J. Am. Chem. Soc., 2008, 130, 16201 CrossRef CAS PubMed.
- X. Qi, K. Pu, X. Zhou, H. Li, B. Liu, F. Boey, W. Huang and H. Zhang, Small, 2010, 6, 663 CrossRef CAS PubMed.
- Y. Hernandez, V. Nicolosi, M. Lotya, F. M. Blighe, Z. Sun, S. De, I. T. McGovern, B. Holland, M. Byrne, Y. K. Gun'Ko, J. J. Boland, P. Niraj, G. Duesberg, S. Krishnamurthy, R. Goodhue, J. Hutchison, V. Scardaci, A. C. Ferrari and J. N. Coleman, Nat. Nanotechnol., 2008, 3, 563 CrossRef CAS PubMed.
- V. Chandra, J. Park, Y. Chun, J. W. Lee, I. Hwang and K. S. Kim, ACS Nano, 2010, 4, 3979 CrossRef CAS PubMed.
- G. Zhao, L. Jiang, Y. He, J. Li, H. Dong, X. Wang and W. Hu, Adv. Mater., 2011, 23, 3959 CrossRef CAS PubMed.
- J. Chen, Y. Li, L. Huang, N. Jia, C. Li and G. Shi, Adv. Mater., 2015, 27, 3654 CrossRef CAS PubMed.
- J. Chen, Y. Li, L. Huang, C. Li and G. Shi, Carbon, 2015, 81, 826 CrossRef CAS.
- L. Zhou, X. Lin, T. Huang and A. Yu, J. Mater. Chem. A, 2014, 2, 5117 CAS.
- Y. Zhu, D. K. James and J. M. Tour, Adv. Mater., 2012, 24, 4924 CrossRef CAS PubMed.
- J. W. Burress, S. Gadipelli, J. Ford, J. M. Simmons, W. Zhou and T. Yildirim, Angew. Chem., Int. Ed., 2010, 49, 8902 CrossRef CAS PubMed.
- J. E. Kim and H. S. Lee, Polymer, 2014, 55, 287 CrossRef CAS.
- Y. Tao, D. Kong, C. Zhang, W. Lv, M. Wang, B. Li, Z. Huang, F. Kang and Q. Yang, Carbon, 2014, 69, 169 CrossRef CAS.
- P. M. Sudeep, T. N. Narayanan, A. Ganesan, M. M. Shaijumon, H. Yang, S. Ozden, P. K. Patra, M. Pasquali, R. Vajtai, S. Ganguli, A. K. Roy, M. R. Anantharaman and P. M. Ajayan, ACS Nano, 2013, 7, 7034 CrossRef CAS PubMed.
- C. Yang and Y. Lee, Thin Solid Films, 2009, 517, 4735 CrossRef CAS.
- D. Li, M. B. Muller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101 CrossRef CAS PubMed.
- M. A. Worsley, S. O. Kucheyev, H. E. Mason, M. D. Merrill, B. P. Mayer, J. Lewicki, C. A. Valdez, M. E. Suss, M. Stadermann, P. J. Pauzauskie, J. H. Satcher, J. Biener and T. F. Baumann, Chem. Commun., 2012, 48, 8428 RSC.
- X. Wu, J. Zhou, W. Xing, G. Wang, H. Cui, S. Zhuo, Q. Xue, Z. Yan and S. Z. Qiao, J. Mater. Chem., 2012, 22, 23186 RSC.
- Q. Fang and B. Chen, J. Mater. Chem. A, 2014, 2, 8941 CAS.
- M. E. Davis, Nature, 2002, 417, 813 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5nr06432e |
| This journal is © The Royal Society of Chemistry 2016 |