
A stimuli-responsive nanoparticulate system using poly(ethylenimine)-graft-polysorbate for controlled protein release
Wing-Fu
Lai
*a and
Ho Cheung
Shum
*ab
aDepartment of Mechanical Engineering, The University of Hong Kong, Hong Kong Special Administrative Region, China. E-mail: rori0610@graduate.hku.hk; ashum@hku.hk
bHKU-Shenzhen Institute of Research and Innovation, Shenzhen, Guangdong, China
First published on 23rd November 2015
Abstract
Proteins have emerged as an important class of therapeutic agents due to their high specificity in their physiological actions. Over the years, diverse protein carriers have been developed; however, some concerns, such as the relatively low loading efficiency and release sustainability, have limited the efficiency of protein delivery. This study reports the use of hydrogel nanoparticles based on a novel copolymer, poly(ethylenimine)-graft-polysorbate (PEIP), as effective protein carriers. The copolymer is fabricated by grafting poly(ethylenimine) (PEI) with polysorbate 20 using carbonyldiimidazole chemistry. Its cytotoxicity is much lower than that of unmodified PEI in RGC5 and HEK293 cells. In comparison with nanoparticles formed by unmodified PEI, our nanoparticles are not only more efficient in cellular internalization, as indicated by the 5- to 6-fold reduction in the time they take to cause 90% of cells to exhibit intracellular fluorescence, but also give a protein loading efficiency as high as 70–90%. These, together with the salt-responsiveness of the nanoparticles in protein release and the retention of the activity of the loaded protein, suggest that PEIP and its hydrogel nanoparticles warrant further development as protein carriers for therapeutic applications.
1. Introduction
Proteins have emerged as an important class of therapeutic agents because of their higher specificity in their physiological actions as compared to conventional drugs.1,2 However, protein pharmaceuticals generally have poor stability, rendering their therapeutic activity highly susceptible to loss caused by proteolysis, aggregation, unfolding and denaturation.3,4 In addition, protein pharmaceuticals, especially for long-term conditions, may have to be administered periodically for desired therapeutic effects, resulting in fluctuations in drug levels and frequent drug administration.5 Over the years, different carriers have been developed to enhance the delivery efficiency and release sustainability of protein pharmaceuticals. For instance, chitosan-coated mesoporous silicon microparticles have been investigated for their capacity to load proteins by adsorption.6 Their protein release sustainability is found to be higher compared to their uncoated counterparts. More recently, HIV-1 envelope glycoprotein (CN54gp140) has been delivered intravaginally with liposomes that are incorporated into the hydroxyethyl cellulose (HEC) aqueous gel.7 The mucoadhesive strength of the liposomal gel formulation offers a stable and practical dosage form for vaginal immunization. Despite the potential of different protein carriers reported in the literature,8,9 wide applications of many of these systems are hindered by various limitations, such as high cytotoxicity, low protein loading capacity and poor protein release sustainability.To address these limitations, we propose a novel nanoparticulate hydrogel system that carries fragile protein drugs by physical entrapment and allows sustained release. By physical entrapment in which chemical or photochemical triggering is not involved, possible structural changes introduced into the loaded protein can be minimized.10,11 The hydrogel nanoparticulate system developed in this study is prepared by utilizing a copolymer namely PEIP, which is synthesized by grafting polysorbate 20 (PS) onto poly(ethylenimine) (PEI) and can stabilize alginate (Alg) pre-gel nuclei into individual sponge-like nanoparticles. PS belongs to a class of fatty acid esters that have been reported to stabilize nanoparticles in a colloidal system,12–15 and is extensively used in the food industry as dispersants, emulsifiers and stabilizers.16 Earlier studies have also shown that PS enhances the intracellular uptake of epirubicin in human colon adenocarcinoma Caco-2 cells, and increases mucosal-to-serosal absorption of epirubicin in the rat jejunum and ileum.17,18 With the stabilizing property and drug-absorption-enhancing activity of PS, the hydrogel nanoparticles generated by the PS-containing PEIP should have good stability and be effective in intracellular protein delivery.
2. Experimental section
2.1. Materials
PEI (Mw = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da) was purchased from Aladdin (Shanghai, China). Dimethyl sulfoxide (DMSO), alginic acid (sodium salt from brown algae), triethylamine (Et3N), 1,1′-carbonyldiimidazole (CDI), calcium chloride (CaCl2), sodium chloride (NaCl) and various other chemicals were purchased from Sigma–Aldrich (St Louis, MO, USA). They were of reagent grade and were used without further purification. Dulbecco's Modified Eagle's Medium (DMEM; Gibco, Grand Island), penicillin G–streptomycin sulfate (Life Technologies Corporation, USA) and fetal bovine serum (FBS, Hangzhou Sijiqing Biological Engineering Materials Co, China) were used as the cell culture media. Trypsin–EDTA (0.25% trypsin–EDTA) was obtained from Invitrogen.
000 Da) was purchased from Aladdin (Shanghai, China). Dimethyl sulfoxide (DMSO), alginic acid (sodium salt from brown algae), triethylamine (Et3N), 1,1′-carbonyldiimidazole (CDI), calcium chloride (CaCl2), sodium chloride (NaCl) and various other chemicals were purchased from Sigma–Aldrich (St Louis, MO, USA). They were of reagent grade and were used without further purification. Dulbecco's Modified Eagle's Medium (DMEM; Gibco, Grand Island), penicillin G–streptomycin sulfate (Life Technologies Corporation, USA) and fetal bovine serum (FBS, Hangzhou Sijiqing Biological Engineering Materials Co, China) were used as the cell culture media. Trypsin–EDTA (0.25% trypsin–EDTA) was obtained from Invitrogen.
2.2. Synthesis and structural characterization of PEIP
0.1 g of CDI was dissolved in 1 mL of degassed DMSO, and was mixed with a DMSO solution of PS (0.02 g mL−1). Et3N was added into the reaction mixture to reach a final concentration of 4.5 μL mL−1. The reaction was carried out for 3 hours in the dark under an inert nitrogen atmosphere. The activated PS was then added into a DMSO solution of PEI (0.33 g mL−1). The reaction was conducted for 16 hours in the dark under an inert nitrogen atmosphere. The crude reaction mixture was dialyzed against triply distilled water at 4 °C for 3 days with a molecular weight cutoff of 12 kDa. Afterwards, the solution was lyophilized for 2 days to obtain PEIP.PS, PEI and PEIP were structurally characterized using proton nuclear magnetic resonance (1H-NMR). In brief, 20 mg of PS, PEI and PEIP were solubilized in 0.7 mL of deuterium oxide (D2O). 1H-NMR spectra were recorded on an NMR spectrometer (500 MHz; Bruker Corporation, Germany). In addition to 1H-NMR, Fourier-transform infrared (FT-IR) spectroscopy was performed using an FT-IR spectrometer (Spectrum 2000, Perkin Elmer) under ambient conditions. The potassium bromide (KBr) disk technique was used for analysis. Spectra were obtained at a resolution of 2 cm−1, and were reported as an average of 16 scans.
2.3. Generation of Alg/PEI and Alg/PEIP nanoparticles
6 mg of CaCl2 was dissolved in 6 mL of distilled water. The solution was added dropwise into 45 mL of the Alg solution (0.6 mg mL−1). After 30 min, 8 mL of the PEI or PEIP solution, at a concentration of 0.68 mg mL−1, was added into the solution mixture under constant stirring at 600 rpm. The resulting mixture was first left under ambient conditions for 30 min, and then at 4 °C for 16 hours. The nanoparticles were retrieved by centrifugation for 40 min at 4 °C at a relative centrifugal force of 10000g, where g is the gravitational constant.
2.4. Physical characterization of nanoparticles
The z-average diameters and ζ potentials of Alg/PEI and Alg/PEIP nanoparticles were examined using dynamic light scattering (DLS) (Zetasizer 3000, Malvern Instruments, UK). The z-average diameters of the nanoparticles were calculated using the non-negative least squares (NNLS) method. Data were reported as an average of 10 measurements. The surface morphology of Alg/PEI and Alg/PEIP nanoparticles was observed using scanning electron microscopy (SEM) and transmission electron microscopy (TEM). SEM analysis was performed by first sputter-coating the nanoparticles with gold, followed by observation of the surface morphology of the nanoparticles using a JEOL JSM-6380 (Japan) microscope. The elemental composition of the surface of the nanoparticles was further characterized using the X-ray energy dispersive spectroscopy (EDS) system attached to the microscope. To perform TEM analysis, phosphotungstic acid (PTA) was added into the nanoparticle-containing solution at a final concentration of 0.1% (w/v). A drop of the solution was then placed on a 200-mesh carbon-coated copper grid. After evaporation of the solvent under ambient conditions for 5 min, images were taken using a Philips CM100 transmission electron microscope operated at an acceleration voltage of 100 kV.2.5. Evaluation of cellular uptake of nanoparticles
RGC5 cells were cultured in DMEM containing 10% FBS, 100 UI mL−1 penicillin, 100 μg mL−1 streptomycin, and 2 mM L-glutamine. 24 hours before the assay, cells were seeded in a 24-well plate at a density of 1 × 105 cells per well, and were incubated under a humidified atmosphere of 5% CO2 at 37 °C. During the experiment, an aqueous solution of fluorescein-labelled hydrogel nanoparticles, which were prepared using the same method as described in section 2.3 but the Alg solution used for nanoparticle generation contained 0.4% (w/v) sodium fluorescein, was added into each well. The cells were incubated at 37 °C for different time intervals. Afterwards, the medium was removed. The cells were rinsed with phosphate-buffered saline (PBS) before 0.5 mL of fresh culture medium was added. The cellular uptake of the nanoparticles was monitored using fluorescence microscopy. The percentage of fluorescence-positive cells was determined as previously reported.19 The experiment was replicated thrice.2.6. Cytotoxicity assay
HEK293 and RGC5 cells were cultured in DMEM containing 10% FBS, 100 UI mL−1 penicillin, 100 μg mL−1 streptomycin, and 2 mM L-glutamine. 24 hours before the assay, cells were seeded in a 96-well plate at a density of 5000 cells per well, and incubated under a humidified atmosphere of 5% CO2 at 37 °C. During the experiment, the growth medium was replaced with 100 μL of cell culture medium containing the polymer (PEI or PEIP) or the hydrogel nanoparticles, at a desired concentration. After 5-hour incubation at 37 °C, the medium was replaced with fresh growth medium. The CellTiter 96 AQueous non-radioactive cell proliferation assay (MTS assay; Promega Corp., Madison, WI) was then performed either immediately or after 24 hours of post-treatment incubation, according to the manufacturer's instructions. Cell viability (%) in each well was determined by dividing the absorbance value (A490) of the test well by the A490 value of the control well, followed by a multiplication of the quotient by 100%.2.7. Determination of the protein loading efficiency
Three protein models were adapted in this study: (i) bovine serum albumin (BSA); (ii) Bacillus licheniformis α-amylase (BLA); and (iii) chicken egg-white lysozyme (LYS). To prepare the protein-loaded nanoparticles, the protein was first mixed thoroughly with the Alg solution at a concentration of 4 mg mL−1. Nanoparticles of Alg/PEI and Alg/PEIP were then generated using the same method as described in section 2.3, and were obtained by centrifugation at a relative centrifugal force of 10000g at 4 °C for 40 min. The total concentration of the unloaded protein in the supernatant was determined using the Bradford reagent (Sigma-Aldrich, Missouri, USA). The loading efficiency was calculated using the following equation as previously reported:20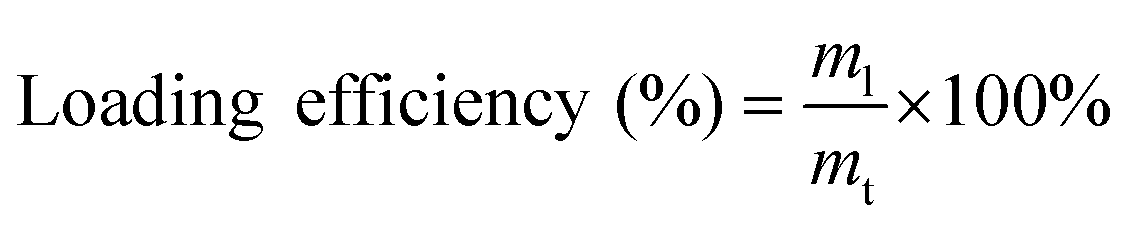 | (1) |
2.8. Drug release evaluation
The protein-loaded nanoparticles were mixed with 2 mL of distilled water or physiological saline (0.9% NaCl, pH 7.4) as the releasing medium, and were incubated at 37 °C and 5% CO2 with saturated humidity. At a pre-set time interval, 100 μL of the releasing medium was removed for testing, and was replaced with 100 μL of the medium. The amount of the protein released from the nanoparticles was determined using the Bradford reagent (Sigma-Aldrich, Missouri, USA). The cumulative protein release was calculated using the following equation as previously reported:4 | (2) |
2.9. Determination of the protein activity
BLA-loaded nanoparticles were retrieved by centrifugation at a relative centrifugal force of 10000g at 4 °C for 40 min. The resulting pellet was crushed thoroughly with a mortar and pestle in PBS. After removing the debris by filtration, the amount of BLA extracted from the nanoparticles was determined using the Bradford reagent (Sigma-Aldrich, Missouri, USA). 0.5 mL of the solution containing the extracted BLA was added into 2 mL of the substrate solution, which was prepared by dissolving 100 mg of soluble starch and 100 mg of povidone–iodine in 50 mL of distilled water. The absorbance at 580 nm was measured using a UV-visible spectrophotometer (Pharmacia, USA) at pre-set time intervals. The activity of the extracted BLA was determined based on the initial linear portion of the graph constructed.
The same method as described above was adopted to extract LYS from nanoparticles, and to determine the activities of the extracted LYS. For the latter, 100 μL of the solution containing the extracted LYS was added into a cuvette, followed by addition of 1 mL of a 0.01% (w/v) Micrococcus lysodeikticus cell suspension. The absorbance at 450 nm was measured to determine the activity of the extracted LYS.
2.10. Statistical analysis
All data were expressed as the means ± standard deviation (SD). Student's t-test was performed to assess the statistical significance. Differences with a p-value <0.05 were considered to be statistically significant.3. Results and discussion
3.1. Polymer synthesis and structural characterization
This study employs a coupling reagent-mediated approach to copolymerize PEI with PS. PS has been widely used as a solubilizing agent for poorly water-soluble compounds.21 It can also enhance drug permeation across tight-junctions,22–25 and can effectively stabilize nanoparticles from aggregation.12–15 After the incorporation of PS into PEI, the copolymer formed is expected to have high potential for drug delivery applications. During the copolymerization process, the hydroxyl groups of PS molecules are activated by CDI to form active imidazolyl carbamate intermediates, which are then attacked by primary amine groups of PEI to form urethane linkages, with imidazole being released as a by-product (Fig. 1). As urethane linkages can be hydrolyzed by the acidic environment in lysosomes after internalization,26 or enzymatically by cytochrome P450 and esterases,27,28 they should be highly biodegradable and may be used to elicit the intracellular release of loaded drugs during drug delivery. | ||
| Fig. 1 The synthetic scheme of PEIP. | ||
The success of PEI/PS graft copolymerization is verified using 1H-NMR (Fig. 2A). The representative signals of PEI (–CH2CH2N– protons) in D2O resonate between 2.5 and 2.8 ppm. These signals of PEI are observed in the spectrum of PEIP but not in the spectrum of PS. The presence of representative signals at 0.9 ppm (H3), 1.3 ppm (H2) and 2.3 ppm (H1) in the spectrum of PEIP further suggests the successful incorporation of PS into PEI. The molar ratio of PS to PEI in PEIP is calculated based on the proton integral values of the 1H NMR spectrum: 0.9 ppm (H3 from PS) and 2.5–2.8 ppm (CH2 of PEI). It is approximated to be 1:3.
 | ||
| Fig. 2 Structural characterization of PEIP. (A) 1H-NMR of (i) PEI, (ii) PS and (iii) PEIP. (B) FT-IR spectra of PS, PEI and PEIP. | ||
Grafting of PS onto PEI is further verified using FT-IR in Fig. 2B. The spectrum of PS exhibits characteristic signals at 1100 cm−1 and 2900 cm−1, which can be assigned to the vibrations of C–O and C–H bonds, respectively. These peaks are present in the spectrum of PEIP. In addition, a band is observed in the spectrum of PEI at 1580 cm−1, which comes from the bending vibration of NH2. This band appears in the spectrum of PEIP but not in the spectrum of PS, which contains no amine group.
3.2. Nanoparticle formation and morphological analysis
Alg is an anionic polysaccharide of (1–4)-linked β-D-mannuronic acid and α-L-guluronic acid. It has low toxicity, high biocompatibility and high biodegradability.29–35 During nanoparticle fabrication, Alg first interacts with Ca2+ to form pre-gel nuclei under constant stirring.36 Subsequent addition of an aqueous PEIP solution leads to the formation of polyelectrolyte complexes (PEC) via electrostatic interactions, thereby restricting further co-operative binding between Alg and Ca2+, and stabilizing the pre-gel nuclei into sponge-like Alg/PEIP nanoparticles.37 The schematic representation of the formation of nanoparticles is shown in Fig. 3A. | ||
| Fig. 3 (A) Schematic representation of the formation of nanoparticles between Alg and the polycation. (B, C) Representative electron micrographs of (B) Alg/PEI and (C) Alg/PEIP nanoparticles taken by (i) SEM and (ii) TEM. The scale bar represents 200 nm. | ||
The morphology of the nanoparticles is examined using SEM and TEM (Fig. 3B and C). The SEM micrographs show that the nanoparticles formed by both PEIP and PEI are almost spherical, with some creases being seen on the surface of the nanoparticles. The creases are formed possibly as a result of the shrinkage of the hydrogel matrix when nanoparticles are exposed to air and the subsequent partial drying of the nanoparticles through evaporation.
3.3. Compositional and physical characterization of nanoparticles
The elemental composition of the nanoparticles is studied using EDS. The weight percentages of nitrogen in Alg/PEI and Alg/PEIP nanoparticles are around 15% and 17%, respectively (Fig. 4A). The significantly higher weight percentage of nitrogen in Alg/PEIP is partially attributed to the highly branched structure of PEIP, which may interact more effectively and stably with Alg during polyelectrolyte complexation. The enhanced polyelectrolyte complexation due to the branched structure of the polymer has been reported in the PEI/DNA system,38–40 in which PEI with a higher degree of branching is found to be more effective in interacting with the negatively charged DNA molecules during complex formation. | ||
| Fig. 4 (A) The weight percentages of different elements in Alg/PEI and Alg/PEIP nanoparticles as determined using EDS. Data are presented as the means ± SD of triplicate experiments. (B) The size distribution profiles of Alg/PEI and Alg/PEIP nanoparticles as determined using SEM. (C) The z-average diameters of Alg/PEIP and Alg/PEI nanoparticles as measured using DLS. Data are reported as an average of 10 measurements, and are presented as the means ± SD. (D) ζ potentials of Alg/PEI and Alg/PEIP nanoparticles as measured using DLS. Data are reported as an average of 10 measurements, and are presented as the means ± SD. | ||
Particle size is an important parameter determining the delivery efficiency of micro- and nanoparticulate drug delivery systems,41 because changes in the particle size may lead to changes in the diffusion length and surface area for molecules to diffuse out. Based on the SEM micrographs obtained, the size distribution profiles of the Alg/PEI and Alg/PEIP nanoparticles are analyzed, and shown in Fig. 4B. The average diameters of Alg/PEI and Alg/PEIP are also approximated using SEM, and are found to be around 185 nm and 53 nm, respectively. This is consistent with the estimation made by DLS (Fig. 4C), in which the z-average diameter of Alg/PEIP is found to be around 70 nm, and is approximately three times smaller than that of Alg/PEI (201 nm). For the nanoparticles to be applicable to systemic administration of protein drugs, their stability in the presence of serum is required. While the size of Alg/PEI nanoparticles increases from around 200 nm to over 500 nm in the presence of 10% FBS, the change in the size of Alg/PEIP nanoparticles is not statistically significant. The smaller size of Alg/PEIP, both in the presence and absence of FBS, is partially explained by the highly branched structure of PEIP caused by graft copolymerization. This branched structure may not only facilitate more stable interactions between the polycation and Alg during polyelectrolyte complexation, but may also restrict the mobility of Alg in the nanoparticles more effectively. Apart from this, PS is a class of fatty acid esters, which have been used as stabilizers in colloidal systems12–15 and food products.16 The stabilizing property of PS in PEIP may, therefore, further resist nanoparticle aggregation and hence improve the stability of hydrogel nanoparticles.
The ζ potentials of Alg/PEI and Alg/PEIP nanoparticles are negative (Fig. 4D) probably because the amount of the cationic polymer added is not sufficient to compensate for the negative charge of Alg. The surface of the nanoparticles is therefore only partially coated with PEI or PEIP. Relative to Alg/PEI nanoparticles, Alg/PEIP nanoparticles have a less negative ζ potential. This supports our hypothesis that in comparison with PEI, PEIP interacts more stably with Alg. In the presence of 10% FBS, the ζ potentials of Alg/PEI and Alg/PEIP nanoparticles become more negative, probably due to the adsorption of serum proteins onto the nanoparticles. Despite this, the ζ potential of Alg/PEIP nanoparticles is still less negative than that of Alg/PEI.
3.4. Cellular internalization of nanoparticles
For a nanoparticulate system to deliver proteins into cells effectively, one of the hurdles to overcome is the plasma membrane, which functions as a selectively permeable barrier to govern the transport of substances into and out of the cell.42 As retinal ganglion cells (RGCs) are one of the cell types that are difficult to be transfected with a high efficiency,43 RGC-5 is selected in this study as a cell model to examine the capacity of the hydrogel nanoparticles to pass through the plasma membrane. Our results show that 90% of cells exhibit intracellular fluorescence within only one hour of treatment with fluorescein-labelled Alg/PEIP nanoparticles. The length of time required by Alg/PEIP for reaching such a level of intracellular fluorescence is 5–6 times shorter than that required by Alg/PEI (Fig. 5). | ||
| Fig. 5 (A) Fluorescence and bright-field images of RGC5 cells incubated with fluorescein-labelled Alg/PEI and Alg/PEIP nanoparticles for different time intervals. The scale bar represents 200 nm. (B) Percentages of fluorescence-positive cells after incubation with fluorescein-labelled Alg/PEI and Alg/PEIP nanoparticles for different periods of time. Data are presented as the means ± SD of triplicate experiments. | ||
The higher cellular internalization efficiency of Alg/PEIP may be attributed to the ability of PS to fluidize the plasma membrane by disrupting the lipid arrangements in the membrane44,45 and hence to increase the cellular permeability.46–48 Moreover, the smaller size and less negative ζ potential of Alg/PEIP nanoparticles may further facilitate binding of the nanoparticles to the cell surface for cellular internalization.49 All these properties may collectively contribute to enhancing the efficiency of cellular uptake.
3.5. Cytotoxic evaluation of nanoparticles
Apart from the efficiency of cellular uptake, another important factor to be considered is the toxicity of the protein delivery system. The toxicity of PEIP and unmodified PEI, as well as their corresponding hydrogel nanoparticles formed with Alg, is evaluated in RGC-5 cells using the MTS assay. Moreover, HEK293 cells are adopted to assess the potential nephrotoxicity of the hydrogel nanoparticles. This cell line is selected because it is one of the commonly used cell lines in drug toxicology studies,50 particularly in assessing the effect of a drug candidate on the renal system.51From our results, it is seen that PEIP and Alg/PEIP nanoparticles are less cytotoxic than unmodified PEI and Alg/PEI nanoparticles, respectively (Fig. 6A and B). This is presumably related to the loss of some of the free amine groups of PEI during the fabrication of PEIP, leading to a slight reduction in the charge density of the resulting polymer. As a high charge density is known to be associated with the toxicity of a polycation,52–54 the slightly lower charge density of PEIP may account for its lower cytotoxicity. The lower charge density of PEIP in comparison with PEI is supported by the acid–base titration profiles shown in Fig. 6C. Earlier studies have demonstrated that the pH buffering capacity is associated with the amine content of polycations such as poly(amidoamine) (PAMAM) dendrimers55 and PEI.56 Compared to PEI, PEIP has a slightly lower capacity to buffer changes in pH (Fig. 6C). This suggests that PEIP may have a lower content of free primary amine groups, which are converted to amide groups during the synthesis of PEIP.
 | ||
| Fig. 6 (A, B) Cytotoxicity of PEI, PEIP, Alg/PEI and Alg/PEIP. The cytotoxicity assay is performed in (A) HEK293 and (B) RGC5 cells after 5-hour treatment with different polymer and nanoparticle concentrations, (i) with or (ii) without the subsequent 24-hour post-treatment incubation. Data are presented as the means ± SD of triplicate experiments. (C) Buffering capacity profiles of PEI and PEIP. Data are presented as the means ± SD of triplicate experiments. | ||
The lower charge density of PEIP may impact not only its buffering capacity but also its complexation capacity.57 However, unlike the buffering capacity which depends predominantly on the percentage of protonable amine groups present,58,59 the process of polyelectrolyte complexation is influenced by many other factors, including the polycation/polyanion ratio, degree of branching of the polymer, pH and temperature.38–40,60 Based on the molar ratio of PS to PEI in PEIP as estimated by NMR (Fig. 2A), the reduction in the percentage of protonable amine groups during PEIP synthesis is approximated to be less than 1%. Together with the highly branched structure of PEIP, the compromising effect due to the lower charge density of PEIP on polyelectrolyte complexation is expected to be negligible. Such a hypothesis is supported by the higher nitrogen content and less negative ζ potential of the Alg/PEIP nanoparticles as compared to those of Alg/PEI nanoparticles, according to the EDS and DLS analyses in Fig. 4.
3.6. Protein loading into nanoparticles
BSA, BLA and LYS are used as protein models for loading into hydrogel nanoparticles. These proteins have molecular weights of the same order of magnitude,61–63 but have different isoelectric points. The isoelectric points of BSA, BLA and LYS are at around pH 5, 7 and 11, respectively.61–63 The net charges of BSA, BLA and LYS at the physiological pH, therefore, are negative, neutral and positive, respectively. These protein models can provide information on how the net charge of a protein affects the fabrication and properties of the protein-loaded hydrogel nanoparticles.Due to the charge of the loaded protein, the polycation/polyanion mixing ratio may be altered during the process of nanoparticle fabrication. The mixing ratio is critical for determining not only the intensity of interactions between different polyelectrolytes but also the degree of self-aggregation of the polyelectrolytes during complexation.64,65 In this study, we observe that an alternation in the polycation/polyanion mixing ratio during protein loading results in a change in the size of the nanoparticles formed. While Alg/PEI and Alg/PEIP nanoparticles have sizes in the nano-size range (approximately 100–800 nm) after being loaded with BLA and LYS, the sizes of BSA-loaded Alg/PEI and Alg/PEIP particles are on the micron scale (around 1–3 μm) (Fig. 7A). This may be associated with the disrupted balance between polycations and polyanions during BSA loading. BSA has a negative net charge. When it is added into the Alg/PEI or Alg/PEIP systems, the amount of polyanions substantially outweighs the amount of polycations present. As BSA may compete with Alg for PEI and PEIP molecules, this hinders the effective formation of Alg/PEI and Alg/PEIP nanoparticles, resulting in the formation of hydrogel particles with a larger size. In addition to the size, the ζ potential of the protein-loaded hydrogel nanoparticles is largely affected by the charge of the loaded protein. When the negatively charged BSA is loaded, the ζ potentials of the resulting Alg/PEIP and Alg/PEI nanoparticles are −18.19 mV and −21.38 mV, respectively, as shown in Fig. 7B. However, when the protein is changed to LYS, which is positively charged at the physiological pH, the ζ potentials of the resulting nanoparticles become positive (11.16 mV for Alg/PEIP and 5.59 mV for Alg/PEI).
 | ||
| Fig. 7 (A) The z-average diameters of Alg/PEI and Alg/PEIP nanoparticles loaded with different proteins. Data are reported as an average of 10 measurements, and are presented as the means ± SD. (B) ζ potentials of Alg/PEI and Alg/PEIP nanoparticles loaded with different proteins. Data are reported as an average of 10 measurements, and are presented as the means ± SD. (C) The protein loading efficiencies of Alg/PEI and Alg/PEIP. Data are presented as the means ± SD of triplicate experiments. | ||
Finally, the protein loading efficiencies of Alg/PEI and Alg/PEIP nanoparticles are compared. The loading efficiency of Alg/PEIP is higher than that of Alg/PEI in all the protein models tested, as shown in Fig. 7C. The lower loading efficiency of Alg/PEI reflects a greater loss of proteins during the loading process. The higher protein loading efficiency of Alg/PEIP is partly attributed to the highly cross-linked structure of PEIP, which presumably plays a role in enhancing the entrapment of proteins.
3.7. Sustained and controlled release of proteins from nanoparticles
While a high loading efficiency is important, the ability of a carrier to prolong protein release is also required to extend the duration of therapeutic effects and reduce the frequency of administration.66–71 Our results demonstrate that the release of proteins from both Alg/PEI and Alg/PEIP nanoparticles is responsive to the ionic strength of the surrounding medium (Fig. 8A). In distilled water, the release of proteins from the nanoparticles is negligible over a period of 30 days, with the percentage of cumulative release being around 5% or less. However, in physiological saline, which is used to simulate the ionic strength of the plasma as previously reported,72 the release of proteins is greatly enhanced. This may be because complex coacervation is driven by a combination of attractive electrostatic forces and entropically-favorable molecular rearrangements.73,74 An increase in the ionic strength of the solution may decrease the entropic driving force for complex formation.74–76 This ultimately destabilizes the structure of the hydrogel nanoparticles formed, and enhances the release of the loaded protein. In fact, the salt-responsiveness of polyelectrolyte complexation has already been demonstrated in other oppositely charged polyelectrolyte systems (including the gelatin–acacia,77 heparin–BSA,78 and heparin–insulin systems78) in which the stability and formation of the polyelectrolyte complexes are found to be reduced upon addition of microions. In this study, we take advantage of the sensitivity of the polyelectrolyte complexation process to the ionic strength to generate salt-responsive hydrogel nanoparticles. The salt-responsiveness of the hydrogel nanoparticles is favorable in the practical sense because it implies that the protein-loaded hydrogel nanoparticles can be stable for a prolonged period of time during storage in distilled water, and the protein is released only when the nanoparticles are injected systemically into the blood circulation. | ||
| Fig. 8 (A) The release profiles of protein-loaded Alg/PEI and Alg/PEIP nanoparticles at 37 °C in (i) distilled water and (ii) physiological saline. Protein-loaded Alg/PEI and Alg/PEIP nanoparticles are designated as P/Alg/PEI and P/Alg/PEIP, respectively, where P represents the name of the protein. Data are presented as the means ± SD of triplicate experiments. (B) Time-dependent changes (i) in the content of starch–iodine complexes after addition of the extracted BLA, and (ii) in the content of Micrococcus lysodeikticus after addition of the extracted LYS. The proteins extracted from the protein-loaded Alg/PEI and Alg/PEIP nanoparticles are designated as Alg/PEI/P and Alg/PEIP/P, respectively, where P represents the name of the protein. Data are presented as the means ± SD of triplicate experiments. (C) Protein activities of the extracted (i) BLA and (ii) LYS. The proteins extracted from the protein-loaded Alg/PEI and Alg/PEIP nanoparticles are designated as Alg/PEI/P and Alg/PEIP/P, respectively, where P represents the name of the protein. Data are presented as the means ± SD of triplicate experiments. | ||
During the protein release studies, the release rates from Alg/PEI and Alg/PEIP nanoparticles are more rapid at the beginning and slow down subsequently. This may be because when the nanoparticles are first subjected to the medium, a relatively high protein concentration gradient is established between the inside and outside of the nanoparticles, causing the protein near the surface of the nanoparticles to diffuse out of the matrix easily. In comparison with those nanoparticles formed by PEI, Alg/PEIP nanoparticles show a significantly higher protein release sustainability over the entire testing period. This is partly attributed to the higher molecular weight and cross-linking density of PEIP relative to PEI. The latter results in a higher degree of chain entanglement, lower polymer mobility and subsequently a lower rate of diffusion.
Over the entire period of the release studies, the percentages of the cumulative release of proteins from Alg/PEI and Alg/PEIP nanoparticles are around 6–10% for BLA and 15–30% for both BSA and LYS. Such high sustainability of protein release is comparable to that achieved by some reported hydrogel systems,79–81 such as the one formed by UV-initiated photopolymerization of poly(ethylene glycol) diacrylate (PEG-DA) which reaches a cumulative release of only 15–80% over a period of 9 months.79 In fact, for a hydrogel system to be used practically for protein delivery, it should not only deliver proteins in a sustained and controlled manner, but should also maintain the activity of the loaded protein.82 Unlike photopolymerized hydrogels in which exposure of the protein to UV may compromise the protein stability,83 the mild conditions for polyelectrolyte complexation can better maintain the activity of the protein to be loaded. The maintenance of the protein activity in our hydrogel nanoparticles is evidenced by the high activity level (over 80%) of BLA extracted from Alg/PEI or Alg/PEIP nanoparticles, as quantified by monitoring the enzymatic activity of BLA based on the rate of reduction in the content of starch–iodine complexes (Fig. 8B). A similarly high activity of LYS is also observed after the protein loading process, as revealed by the high rate of lysis of Micrococcus lysodeikticus in the presence of LYS extracted from the hydrogel nanoparticles. This indicates that the activity of the loaded protein has not been compromised by the fabrication process of the hydrogel nanoparticles, or by the interactions between the protein and the hydrogel components during the process.
4. Conclusion
The development of carriers for prolonged protein release and efficient delivery is essential for the effective administration of protein drugs. In this work, we generate PEIP for the fabrication of Alg/PEIP nanoparticles, which are stabilized against aggregation and exhibit a high cellular internalization efficiency. Apart from this, the highly branched structure of PEIP enhances the protein release sustainability of the hydrogel nanoparticles by increasing the degree of chain entanglement, thereby decreasing the diffusion rate of the loaded protein. These, along with the salt-responsiveness of the nanoparticles and the retention of the activity of the loaded protein, make PEIP and its hydrogel nanoparticles promising candidates to be further explored for future protein delivery applications.Acknowledgements
The authors would like to acknowledge Prof. Wai-Kin Chan's group at the Department of Chemistry in the University of Hong Kong for NMR support, and Prof. Min Wang's group at the Department of Mechanical Engineering in the University of Hong Kong for assistance with DLS and UV-Vis measurements. We would also like to thank Donald Mak, To-Man Mok, Frankie Yu-Fai Chan, Amy Sui-Ling Wong, Yau-Kei Chan, Samuel Kwun-Hei Sy and Matthew Yuk-Heng Tang for support and comments on the experimental studies as well as on the manuscript. This research was supported by the Early Career Scheme (HKU 707712P) and the General Research Fund (HKU 719813E and 17304514) from the Research Grants Council of Hong Kong, the General Program (21476189/ B060201) and Young Scholar's Program (NSFC51206138/E0605) from the National Natural Science Foundation of China, and the small project funding (201409176157) from the University of Hong Kong.References
- B. F. Choonara, Y. E. Choonara, P. Kumar, D. Bijukumar, L. C. du Toit and V. Pillay, Biotechnol. Adv., 2014, 32, 1269–1282 CrossRef CAS PubMed.
- M. George and T. E. Abraham, J. Controlled Release, 2006, 114, 1–14 CrossRef CAS PubMed.
- M. H. Smith and L. A. Lyon, Macromolecules, 2011, 44, 8154–8160 CrossRef CAS PubMed.
- J. K. Li, N. Wang and X. S. Wu, J. Pharm. Sci., 1997, 86, 891–895 CrossRef CAS PubMed.
- C. T. Laurencin and R. Langer, Clin. Lab. Med., 1987, 7, 301–323 CAS.
- E. Pastor, E. Matveeva, A. Valle-Gallego, F. M. Goycoolea and M. Garcia-Fuentes, Colloids Surf., B, 2011, 88, 601–609 CrossRef CAS PubMed.
- P. N. Gupta, A. Pattani, R. M. Curran, V. L. Kett, G. P. Andrews, R. J. Morrow, A. D. Woolfson and R. K. Malcolm, Eur. J. Pharm. Sci., 2012, 46, 315–322 CrossRef CAS PubMed.
- S. Salmaso and P. Caliceti, Int. J. Pharm., 2013, 440, 111–123 CrossRef CAS PubMed.
- W. R. Gombotz and D. K. Pettit, Bioconjugate Chem., 1995, 6, 332–351 CrossRef CAS PubMed.
- X. L. Zhang, J. Huang, P. R. Chang, J. L. Li, Y. M. Chen, D. X. Wang, J. H. Yu and J. H. Chen, Polymer, 2010, 51, 4398–4407 CrossRef CAS.
- D. Sarkar, J. Photochem. Photobiol., A, 2013, 252, 194–202 CrossRef CAS.
- T. Ren, N. Xu, C. Cao, W. Yuan, X. Yu, J. Chen and J. Ren, J. Biomater. Sci., Polym. Ed., 2009, 20, 1369–1380 CrossRef CAS PubMed.
- C. Y. Lin, C. J. Yu, Y. H. Lin and W. L. Tseng, Anal. Chem., 2010, 82, 6830–6837 CrossRef CAS PubMed.
- T. Helgason, T. S. Awad, K. Kristbergsson, D. J. McClements and J. Weiss, J. Colloid Interface Sci., 2009, 334, 75–81 CrossRef CAS PubMed.
- J. Duy, L. B. Connell, W. Eck, S. D. Collins and R. L. Smith, J. Nanopart. Res., 2010, 12, 2363–2369 CrossRef CAS.
- M. Ruiz-Pena, R. Oropesa-Nunez, T. Pons, S. R. W. Louro and A. Perez-Gramatges, Colloids Surf., B, 2010, 75, 282–289 CrossRef CAS PubMed.
- Y. I. Lo, J. Controlled Release, 2003, 90, 37–48 CrossRef CAS PubMed.
- Y. L. Lo, C. Y. Hsu and J. D. Huang, Anticancer Res., 1998, 18, 3005–3009 CAS.
- W. F. Lai and M. C. Lin, PLoS One, 2015, 10, e0126367 CrossRef PubMed.
- E. K. Park, S. B. Lee and Y. M. Lee, Biomaterials, 2005, 26, 1053–1061 CrossRef CAS PubMed.
- C. H. Hsu, Z. R. Cui, R. J. Mumper and M. Jay, AAPS PharmSciTech, 2003, 4, 24–35 CrossRef PubMed.
- B. Vora, A. J. Khopade and N. K. Jain, J. Controlled Release, 1998, 54, 149–165 CrossRef CAS PubMed.
- P. P. Sarpotdar and J. L. Zatz, Drug Dev. Ind. Pharm., 1987, 13, 15–37 CrossRef CAS.
- R. N. Alyautdin, E. B. Tezikov, P. Ramge, D. A. Kharkevich, D. J. Begley and J. Kreuter, J. Microencapsulation, 1998, 15, 67–74 CrossRef CAS PubMed.
- R. N. Alyautdin, V. E. Petrov, K. Langer, A. Berthold, D. A. Kharkevich and J. Kreuter, Pharm. Res., 1997, 14, 325–328 CrossRef CAS.
- D. Bohme and A. G. Beck-Sickinger, J. Pept. Sci., 2015, 21, 186–200 CrossRef PubMed.
- S. W. Cha, H. K. Gu, K. P. Lee, M. H. Lee, S. S. Han and T. C. Jeong, Toxicol. Lett., 2000, 115, 173–181 CrossRef CAS PubMed.
- L. H. Patterson, S. R. McKeown, T. Robson, R. Gallagher, S. M. Raleigh and S. Orr, Anticancer Drug Des., 1999, 14, 473–486 CAS.
- N. Zhang, J. H. Li, W. F. Jiang, C. H. Ren, J. S. Li, J. Y. Xin and K. Li, Int. J. Pharm., 2010, 393, 212–218 CrossRef CAS PubMed.
- Z. Yu, H. J. Li, L. M. Zhang, Z. H. Zhu and L. Q. Yang, Int. J. Pharm., 2014, 473, 501–509 CrossRef CAS PubMed.
- T. Wang and N. Y. He, Nanoscale, 2010, 2, 230–239 RSC.
- J. D. Wan, Polymers, 2012, 4, 1084–1108 CrossRef CAS.
- S. Seiffert, Angew. Chem., Int. Ed., 2013, 52, 11462–11468 CrossRef CAS PubMed.
- H. Guo, Q. Y. Lai, W. Wang, Y. K. Wu, C. N. A. Zhang, Y. Liu and Z. Yuan, Int. J. Pharm., 2013, 451, 1–11 CrossRef CAS PubMed.
- A. P. Bagre, K. Jain and N. K. Jain, Int. J. Pharm., 2013, 456, 31–40 CrossRef CAS PubMed.
- M. Rajaonarivony, C. Vauthier, G. Couarraze, F. Puisieux and P. Couvreur, J. Pharm. Sci., 1993, 82, 912–917 CrossRef CAS PubMed.
- S. J. De and D. Robinson, J. Controlled Release, 2003, 89, 101–112 CrossRef CAS PubMed.
- D. D. Dunlap, A. Maggi, M. R. Soria and L. Monaco, Nucleic Acids Res., 1997, 25, 3095–3101 CrossRef CAS PubMed.
- W. T. Godbey, K. K. Wu and A. G. Mikos, J. Controlled Release, 1999, 60, 149–160 CrossRef CAS PubMed.
- O. Boussif, F. Lezoualch, M. A. Zanta, M. D. Mergny, D. Scherman, B. Demeneix and J. P. Behr, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 7297–7301 CrossRef CAS.
- N. Wang and X. S. Wu, Pharm. Dev. Technol., 1997, 2, 135–142 CrossRef CAS PubMed.
- W. F. Lai and H. S. Jung, PLoS One, 2014, 9, e100258 CrossRef PubMed.
- P. Challa, C. Luna, P. B. Liton, B. S. Chamblin, J. K. Wakefield, R. Ramabhadran, D. L. Epstein and P. Gonzalez, Mol. Vision, 2005, 11, 425–430 CAS.
- G. Terzis, G. Georgiadis, E. Vassiliadou and P. Manta, Eur. J. Appl. Physiol., 2003, 90, 10–15 CrossRef CAS PubMed.
- P. K. Dudeja, K. M. Anderson, J. S. Harris, L. Buckingham and J. S. Coon, Arch. Biochem. Biophys., 1995, 319, 309–315 CrossRef CAS PubMed.
- V. Sakthivel and H. U. Shingadia, Nat. Environ. Pollut. Technol., 2007, 6, 121–126 CAS.
- S. H. Yang, J. J. Liu, Y. Q. Chen and J. H. Jiang, Biomed. Pharmacother., 2012, 66, 187–194 CrossRef CAS PubMed.
- T. W. Kim, Y. J. Kim, H. Chung, I. C. Kwon, H. C. Sung and S. Y. Jeong, J. Controlled Release, 2002, 82, 455–465 CrossRef CAS PubMed.
- S. M. Zou, P. Erbacher, J. S. Remy and J. P. Behr, J. Gene Med., 2000, 2, 128–134 CrossRef CAS PubMed.
- C. Kwon, Y. Choi, D. Jeong, J. G. Kim, J. M. Choi, S. Chun, S. Park and S. Jung, J. Inclusion Phenom. Macrocyclic Chem., 2012, 74, 325–333 CrossRef CAS.
- G. Hettiarachchi, D. Nguyen, J. Wu, D. Lucas, D. Ma, L. Isaacs and V. Briken, PLoS One, 2010, 5 CrossRef CAS PubMed , e10514.
- C. Plank, K. Zatloukal, M. Cotten, K. Mechtler and E. Wagner, Bioconjugate Chem., 1992, 3, 533–539 CrossRef CAS PubMed.
- W. F. Lai, Expert Rev. Med. Devices, 2011, 8, 173–185 CrossRef CAS PubMed.
- J. G. R. Elferink, Inflammation, 1985, 9, 321–331 CrossRef CAS PubMed.
- C. L. Waite, S. M. Sparks, K. E. Uhrich and C. M. Roth, BMC Biotechnol., 2009, 9, 38 CrossRef PubMed.
- S. Nimesh, Curr. Clin. Pharmacol., 2012, 7, 121–130 CrossRef CAS PubMed.
- M. L. Forrest, G. E. Meister, J. T. Koerber and D. W. Pack, Pharm. Res., 2004, 21, 365–371 CrossRef CAS.
- C. Lin, C. J. Blaauboer, M. M. Timoneda, M. C. Lok, M. van Steenbergen, W. E. Hennink, Z. Y. Zhong, J. Feijen and J. F. J. Engbersen, J. Controlled Release, 2008, 126, 166–174 CrossRef CAS PubMed.
- C. Lin, Z. Y. Zhong, M. C. Lok, X. L. Jiang, W. E. Hennink, J. Feijen and J. F. J. Engbersen, J. Controlled Release, 2006, 116, 130–137 CrossRef CAS PubMed.
- D. Priftis and M. Tirrell, Soft Matter, 2012, 8, 9396–9405 RSC.
- R. H. C. Strang, Biochem. Educ., 1984, 12, 57–59 CrossRef CAS.
- M. D. Rao, A. Purnima, D. V. Ramesh and C. Ayyanna, World J. Microbiol. Biotechnol., 2002, 18, 547–550 CrossRef.
- Q. S. Shi, Y. Zhou and Y. Sun, Biotechnol. Prog., 2005, 21, 516–523 CrossRef CAS PubMed.
- A. Q. Ye, Int. J. Food Sci. Technol., 2008, 43, 406–415 CrossRef CAS.
- J. X. Xiao, H. Y. Yu and J. A. Yang, Food Chem., 2011, 125, 1267–1272 CrossRef CAS.
- J. E. Oh, Y. S. Nam, K. H. Lee and T. G. Park, J. Controlled Release, 1999, 57, 269–280 CrossRef CAS PubMed.
- Y. S. Nam, J. Y. Park, S. H. Han and I. S. Chang, Biotechnol. Lett., 2002, 24, 2093–2098 CrossRef CAS.
- R. C. Luo, Y. Cao, P. Shi and C. H. Chen, Small, 2014, 10, 4886–4894 CrossRef CAS PubMed.
- S. H. Kim, J. W. Kim, D. H. Kim, S. H. Han and D. A. Weitz, Small, 2013, 9, 124–131 CrossRef CAS PubMed.
- V. R. Kearns and R. L. Williams, Expert Rev. Med. Devices, 2009, 6, 277–290 CrossRef CAS PubMed.
- L. Y. Chu, T. Yamaguchi and S. Nakao, Adv. Mater., 2002, 14, 386–389 CrossRef CAS.
- H. L. Davis, N. L. Davis and A. L. Clemons, Ann. N. Y. Acad. Sci., 1967, 141, 310–325 CrossRef CAS PubMed.
- J. T. Overbeek and M. J. Voorn, J. Cell. Physiol. Suppl., 1957, 49, 7–22 CrossRef CAS PubMed ; discussion, 22–26.
- S. L. Perry, Y. Li, D. Priftis, L. Leon and M. Tirrell, Polymers, 2014, 6, 1756–1772 CrossRef CAS.
- J. van der Gucht, E. Spruijt, M. Lemmers and M. A. C. Stuart, J. Colloid Interface Sci., 2011, 361, 407–422 CrossRef CAS PubMed.
- P. M. Biesheuvel and M. A. C. Stuart, Langmuir, 2004, 20, 2785–2791 CrossRef CAS PubMed.
- D. J. Burgess, J. Colloid Interface Sci., 1990, 140, 227–238 CrossRef CAS.
- E. Seyrek, P. L. Dubin, C. Tribet and E. A. Gamble, Biomacromolecules, 2003, 4, 273–282 CrossRef CAS PubMed.
- M. B. Mellott, K. Searcy and M. V. Pishko, Biomaterials, 2001, 22, 929–941 CrossRef CAS PubMed.
- S. R. Van Tomme, B. G. De Geest, K. Braeckmans, S. C. De Smedt, F. Siepmann, J. Siepmann, C. F. van Nostrum and W. E. Hennink, J. Controlled Release, 2005, 110, 67–78 CrossRef CAS PubMed.
- Q. Liu, Q. H. Zuo, R. Guo, A. Hong, C. H. Li, Y. Zhang, L. M. He and W. Xue, J. Bioact. Compat. Polym., 2015, 30, 397–411 CrossRef CAS.
- F. Lee, J. E. Chung and M. Kurisawa, J. Controlled Release, 2009, 134, 186–193 CrossRef CAS PubMed.
- K. Sirkar and M. V. Pishko, Anal. Chem., 1998, 70, 2888–2894 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |