
Preparation of hollow microsphere@onion-like solid nanosphere MoS2 coated by a carbon shell as a stable anode for optimized lithium storage†
Bangjun
Guo
,
Ke
Yu
*,
Haili
Song
,
Honglin
Li
,
Yinghua
Tan
,
Hao
Fu
,
Chao
Li
,
Xiang
Lei
and
Ziqiang
Zhu
Key Laboratory of Polar Materials and Devices (Ministry of Education of China), Department of Electronic Engineering, East China Normal University, Shanghai, 200241, P. R. China. E-mail: yk5188@263.net; Fax: +86-21-54345198; Tel: +86-21-54345198
First published on 16th November 2015
Abstract
A one-step hydrothermal method was successfully used to fabricate hollow microsphere@onion-like solid nanosphere MoS2. Then the as-prepared sS-MoS2 was decorated with a carbon shell using dopamine as a carbon source by a facile route, resulting in hollow microsphere@onion-like solid nanosphere MoS2 decorated with carbon shell (sS-MoS2@C). A synergistic effect was observed for the two-component material, leading to new electrochemical processes for lithium storage, with improved electroconductivity and structural soundness, triggering an ascending capacity upon cycling. The as-prepared sS-MoS2@C exhibits optimized electrochemical behaviour with high specific capacity (1107 mA h g−1 at 100 mA g−1), superior high-rate capability (805 mA h g−1 at 5000 mA g−1) and good cycling stability (91.5% of capacity retained after 100 cycles), suggesting its potential application in high-energy lithium-ion batteries.
Introduction
Stimulated by the widespread usage of portable electronics and future electric vehicles, lithium-ion batteries (LIBs) have been regarded as high-efficiency and environmentally-friendly energy storage for consumer electronics in our daily lives.1 With their applications extending into large-scale energy storage fields, such as power grids and transportation, traditional LIBs that are based on graphite electrode materials are limited by their intrinsically low theoretical capacities and cannot fulfill the increasing demand for energy storage. Therefore, there is an urgent need in the pursuit of high-capacity anode materials.2 Developing high-performance electrode materials is one of the most effective approaches. For example, molybdenum disulfide (MoS2), with a S–Mo–S sandwich layered structure analogous to graphite and a strong ability to host multiple Li ions, delivers a high theoretical specific capacity of 670 mA h g−1.3–5Sheet-like nanostructured MoS2 and its composite materials have been reported as one of the most promising LIBs anode materials.6–10 However, the practical application of sheet-like MoS2 in LIBs has thus far been hindered by the concomitant substantial volume expansion, accompanying the Li+ ion insertion, which generates mechanical strain that causes nanostructure pulverization and loss integrity of the active material, which further leads to the inefficient application of MoS2 for Li storage.11,12 To avoid this intrinsic problem, the design of a new firm structural nanomaterial is desirable. Compared with the sheet-like nanostructures, spherical nanostructures have been proved more structurally stable during the Li+ ion insertion–extraction process in LIBs.13–15 The spherical structure is the basic unit of the nanomaterial, and could accommodate more volume expansion and lower the mechanical strain during the Li+ ion insertion, slowing down the pulverization of the nanostructure. For application as a LIBs anode material, the conversion reaction of MoS2 during Li+ ion discharge–charge process results in the formation of metal Mo and Li2S. Once Li2S is formed, it becomes the active component, and its electrochemistry dominates in the subsequent cycles. As the Li2S possesses a low conductivity and poor cyclability, the capacity of the MoS2 anode material often fades quickly upon cycling.9,16 Thus, without subsidiary protection, bare MoS2 nanostructures cannot maintain their original morphology and high conductivity as a result of repeated lithiation–delithiation.17 An efficient approach to address this issue is known as the nano-composite method, which aims to improve the structural stability through employing a second component such as carbon as a protective coating shell.18 Benefiting from their large specific surface areas and porous architectures, covered nanocarbon shells serve two main functions: firstly, to buffer the volume variation of the active MoS2 and prevent the aggregation of pulverized active material; secondly, to protect the MoS2 against direct contact with the electrolyte which may result in the formation of an unstable SEI film which would lower the conductivity.19,20 Recently, many efforts have been devoted to preparing MoS2/carbon architectures assembled from nano-scaled building blocks. These architectures can effectively increase the electrolyte–electrode contact area, buffer the volume changes of electrodes and shorten the diffusion distance of ions.21–23 Nonetheless, the preparation of uniform hierarchical MoS2 micro@nano sphere decorated with carbon shell for application in LIBs has been rarely reported.
Based on this concept, we herein report the synthesis of a novel nanocomposite, in which hollow microsphere@onion-like solid nanosphere MoS2 (sS-MoS2) was synthesised by a facile one-step hydrothermal method without any surfactant, and followed by a decoration with a carbon shell using dopamine as a carbon source (sS-MoS2@C). In serving as an anode material for LIBs, the sS-MoS2 core acts as a host for Li storage, in which the onion-like solid nanosphere MoS2 principally provides efficient storage for Li+ ions and the hollow microsphere MoS2 primarily enhances the special surface area. Meanwhile, the carbon shell helps to protect the MoS2 against direct contact with the electrolyte and promotes a rapid charge-transfer reaction by providing an efficient electron pathway for fast lithiation–delithiation. The cooperation of the two active components in sS-MoS2@C brings about a favourable synergistic effect, leading to a higher capacity than those of the single components, resulting in an optimized cycling stability and outstanding rate behaviour for application as an anode material for LIBs. The as-prepared material was also applied as an anode material in a coin cell which could easily power a light emitting diode (LED).
Experimental
Synthesis of hollow microsphere@onion-like solid nanosphere MoS2
Three-dimensional hollow microsphere@onion-like solid nanosphere MoS2 was prepared in a one-step hydrothermal process. In a typical procedure, 0.2 g sodium molybdate and 0.4 g thioacetamide were dissolved in 60 mL deionized water in a beaker with magnetic stirring for 10 min to obtain a transparent solution. Then 0.4 g oxalic acid was added into the above solution to adjust the pH value and the mixture was stirred continuously for 30 min. The resulting uniformly mixed solution was transferred into a 100 mL Teflon-lined stainless steel autoclave and hydrothermally treated at 200 °C for 24 h in an electric oven, followed by natural cooling to room temperature. After cooling to room temperature, the as-prepared samples were washed extensively with deionized water several times and ultrasonically treated for 30 min to collect the black precipitate, which was then dried at 60 °C for 5 h in air.Preparation of sS-MoS2@C
The as-prepared sS-MoS2 was decorated with carbon shell using dopamine as a carbon source. Typically, 300 mg as-prepared sS-MoS2 was dispersed in Tris-buffer (50 mL, 10 mM, pH = 8.5) by ultrasonication for 1 h to form a suspension. After that, 50 mg dopamine was added to the mixture under stirring. The mixture was subjected to continuous magnetic stirring at room temperature for 24 h. Afterwards, the precipitate, sS-MoS2@poly-dopamine, was collected by centrifugation and then washed several times with deionized water before being dried at 60 °C for 6 h. The obtained sS-MoS2@poly-dopamine was then annealed at 800 °C for 5 h under Ar to convert the poly-dopamine into carbon. The obtained composite was named as sS-MoS2@C-1. In order to tune the coating thickness of the carbon shell, sample sS-MoS2@C-2 was prepared similarly to sS-MoS2@C-1, but the initial concentration of dopamine was changed from 50 mg to 100 mg.Characterization
The morphology and structure of the samples were investigated by a field emission SEM (JEOL-JSM-6700F) operated at 15 kV and TEM (JEOL-2010) at 200 kV. TGA was carried out on a METTLER TOLEDO TGA/SDTA851e System under air flow. Surface area determination was performed by the Brunauer–Emmett–Teller (BET) method. XRD patterns were collected on a Bruker D8 Advance diffractometer with Cu Kα radiation (λ = 1.5418 Å). Raman spectral experiments were carried out using a Jobin-Yvon LabRAM HR 800 micro-Raman spectrometer. X-ray photoelectron spectra (XPS) were collected on a Kratos Axis ULTRA X-ray photoelectron spectrometer using a monochromatic Al Kα X-ray source and a hemispherical electron energy analyzer.Electrochemical measurements
All the electrochemical tests were carried out in a two-electrode coin-cell (CR 2032) which was assembled in an argon–filled glovebox (O2 and H2O levels <0.1 ppm). The as-prepared sS-MoS2@C and sS-MoS2 were used as a working electrode, lithium foil as counter and reference electrodes, a polypropylene film (Celgard 2400) as a separator, and 1 mol L−1 LiPF6 dissolved in a mixture of ethylene carbonate (EC) and dimethyl carbonate (DMC) (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in volume) as an electrolyte. The working electrodes were prepared by a slurry tape casting procedure. The slurry consisted of 70 wt% active materials, 20 wt% carbon black and 10 wt% polyvinylidene fluoride (PVDF) dissolved in N-methyl-2-pyrrolidinone (NMP). The slurry was tape-cast on the copper foil, and dried under vacuum at 60 °C for 24 h and cut into circular pieces before use. Cyclic voltammetry (CV) profiles (0.01–3.0 V, 0.1 mV s−1) were obtained on an electrochemical workstation (CHI 660D, Shanghai, China). Galvanostatic discharge–charge cycles of the cells were conducted between 0.01 and 3.00 V on a LAND CT-2001A battery cycler (Wuhan, China) at room temperature. Electrochemical impedance spectroscopy (EIS) was measured on the coin cell using an electrochemical workstation (Autolab PGSTAT302N, Netherlands) with an amplitude of 0.5 mV over the frequency range from 200 kHz to 0.01 Hz.
:1 in volume) as an electrolyte. The working electrodes were prepared by a slurry tape casting procedure. The slurry consisted of 70 wt% active materials, 20 wt% carbon black and 10 wt% polyvinylidene fluoride (PVDF) dissolved in N-methyl-2-pyrrolidinone (NMP). The slurry was tape-cast on the copper foil, and dried under vacuum at 60 °C for 24 h and cut into circular pieces before use. Cyclic voltammetry (CV) profiles (0.01–3.0 V, 0.1 mV s−1) were obtained on an electrochemical workstation (CHI 660D, Shanghai, China). Galvanostatic discharge–charge cycles of the cells were conducted between 0.01 and 3.00 V on a LAND CT-2001A battery cycler (Wuhan, China) at room temperature. Electrochemical impedance spectroscopy (EIS) was measured on the coin cell using an electrochemical workstation (Autolab PGSTAT302N, Netherlands) with an amplitude of 0.5 mV over the frequency range from 200 kHz to 0.01 Hz.
Results and discussion
Scheme 1 illustrates the designed synthesis of hollow microsphere@onion-like solid nanosphere MoS2 decorated with carbon shell. On the basis of our previous research, we reach the conclusion that the two kinds of microsphere and nanosphere MoS2 growth processes are simultaneous.24 First, the formation mechanism of nanospheres is similar to the Ostwald ripening with a fast self-assembly nucleation and a slow evolution process. Second, the growth process of the microspheres was the growth of amorphous granules along with the spherical surfaces. Eventually, nanospheres and microspheres grow into well-dispersed structures.25,26 We proposed a possible growth mechanism for sS-MoS2 as below. At the very beginning of the heating process, numerous amorphous primary MoS2 nanoparticles were formed in stage I. After being heated to a certain temperature, these amorphous primary MoS2 nanoparticles spontaneously aggregated into evacuating solid spheres, as shown in stage II. In stage III, the loosened solid spheres finally crystallized into well layered onion-like structured nanospheres (Fig. S1†); meanwhile, the amorphous primary MoS2 granules which are the source of the ultimate MoS2 microspheres were initially formed at the temperature of about 200 °C. These amorphous primary MoS2 granules were generated during high temperature via the reaction of the remainder solute and in a stronger acidic reaction environment resulting from the increasingly hydrolyzed undecomposed oxalic acid. Because of the factors of stronger acidic solution, high temperature and low concentration these amorphous granules with different surface free energies aggregated into original cambered pieces and grew along with the spherical surface, which resulted in the formation of hollow microspheres (stage IV). At the same time, nanospheres attached on the new cambered surfaces accompanying the growth of hollow MoS2 microspheres. Usually, dopamine was equipped on the surface of sS-MoS2 in the Tris-buffer solution. After overnight magnetic stirring and being dried for 6 h, the sS-MoS2@dopamine were collected (stage V).27 Lastly, the sS-MoS2@dopamine were annealed at 800 °C under Ar atmosphere to get the sS-MoS2@C (stage VI).28,29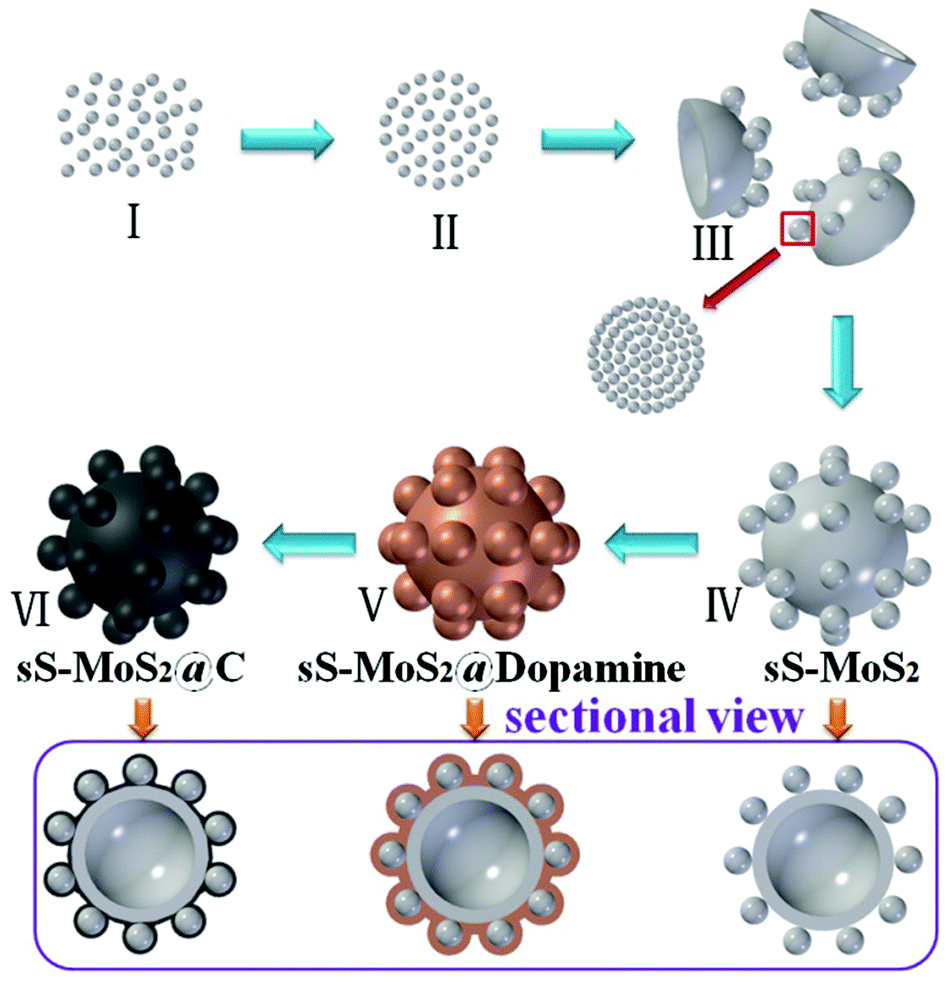 | ||
| Scheme 1 Scheme for the fabrication of sS-MoS2@C. | ||
Scanning electron microscope (SEM) images are shown in Fig. 1. According to Fig. 1a, sS-MoS2 has uniform microsphere@nanosphere morphology with a diameter distribution of about 2 μm. As can be seen from the inset of Fig. 1a, MoS2 nanospheres are distributed harmoniously on the surface of MoS2 microspheres. Fig. 1b shows the outside view of one single sS-MoS2, where the MoS2 nanospheres are well-distributed on the shell with a diameter of 240–250 nm. The inset of Fig. 1b displays an sS-MoS2 with an obvious hole, which reveals the microsphere has a hollow structure. The round arched structure as shown in Fig. 1c further proves this fact. Moreover, Fig. 1c also shows the thickness of the microsphere shell is ∼80 nm. Generally, the sS-MoS2@C inherits the morphology of the sS-MoS2 (Fig. 1d), while it is quite apparent that the surface of sS-MoS2@C is not as smooth as the surface of pure sS-MoS2. More details are shown in Fig. 1e and the inset of Fig. 1d. The morphology of sS-MoS2@C-1 and sS-MoS2@C-2 are demonstrated in Fig. 1e and f, respectively. Notably, the carbon shell thickness of sS-MoS2@C-2 is massive compared to that of sS-MoS2@C-1, which may impede the electron beam penetration into the inner sS-MoS2 core.
 | ||
| Fig. 1 SEM images of bare sS-MoS2 and sS-MoS2@C. (a) Low magnification SEM image of sS-MoS2 and the upper inset is the high magnification SEM image of sS-MoS2. (b) Medium magnification SEM image of sS-MoS2 and the upper inset is a broken sS-MoS2. (c) Medium magnification SEM image of broken sS-MoS2@C. (d) Low magnification SEM image of sS-MoS2@C-1 and the upper inset is the high magnification SEM image of sS-MoS2@C-1. (e) Medium magnification SEM image of sS-MoS2@C-1. (f) Medium magnification SEM image of sS-MoS2@C-2. | ||
Transmission electron microscope (TEM) was further employed to investigate the internal structure of these composite materials. It is noticeable that almost every hollow microsphere is successfully and uniformly fabricated with MoS2 nanospheres (Fig. 2a), which can be attributed to the assistance of oxalic acid and high temperature. Fig. 2b displays a clear view of one sS-MoS2 structure, in which the surface of hollow microsphere and solid nanospheres are all smooth. Fig. 2c is the HRTEM image of the rectangular area in Fig. 2b. The striatures could be observed from the solid nanospheres MoS2, which form the onion-like structure. From the high resolution TEM image (Fig. 2f), the well-resolved interlayer distances with a spacing of 0.62 nm can be clearly distinguished, which corresponds to the (002) plane of MoS2. After dopamine cladding and high temperature annealing treatment, the outer spheres of the MoS2 are almost covered with carbon shells (Fig. 2d). The composite structure was further confirmed by HRTEM. As shown in Fig. 2e, the nanosphere is encapsulated by a self-supporting carbon shell with a uniform thickness of 15–30 nm. The MoS2 core is closely attached to the outside carbon shell, the direct facet-to-facet contact of the MoS2 core and conductive carbon shell makes the core more accessible to both Li+ ions and electrons.
 | ||
| Fig. 2 TEM images of bare sS-MoS2 and sS-MoS2@C. (a) Low magnification TEM image of sS-MoS2. (b) Medium magnification TEM image sS-MoS2. (c) High-resolution TEM image of onion-like solid nanosphere. (d) Medium magnification TEM image of sS-MoS2@C-1. (e) High magnification TEM image of sS-MoS2@C-1. (f) High-resolution TEM image of sS-MoS2. | ||
The crystalline structure of the obtained materials was investigated by X-ray diffraction (XRD) (Fig. 3a). The majority of the diffraction peaks, especially for those with high intensity, can be assigned to the crystalline planes of hexagonal MoS2 phase (JCPDS card no. 37-1492). Especially, the distinct peak at 14.2° characteristic of the (002) crystalline plane for MoS2 suggests the ordered stacking of S–Mo–S layers,30,31 which is in good agreement with the TEM image observed in Fig. 2f. The XRD pattern for the sS-MoS2@C-1 and sS-MoS2@C-2 are respectively shown as the curve (I) and curve (II), and no diffraction peaks from graphitic carbon can be detected, indicating the amorphous nature of the carbon shell.18 The absent peaks of (103) and (105) for sS-MoS2@C could be attributed to the surface strain, induced by the coated carbon shell on the MoS2 spheres surface. Moreover, the broad peak which emerges at 60° is evolved from the (100) peak, which could be explained as the variation of partial grain size from sS-MoS2@C to sS-MoS2. Thirdly, the XRD spectrum shows the crystal structure of compounds. The crystallinity of sS-MoS2 might be decreased during the carbon coating progress and annealing treatment, which brings about these differences from pristine sS-MoS2. Raman spectroscopy was further utilized to characterize the composition of the obtained sS-MoS2, sS-MoS2@C-1 and sS-MoS2@C-2. As shown in Fig. 3b, all three materials was observed two distinct peaks at the bands of 380 and 407 cm−1, which correspond to the E12g vibration mode (the in-layer displacement of Mo and S atoms) and A1g vibration mode (the out-of-layer symmetric displacements of S atoms along the c axis).32–34 In addition, another two notable peaks at the bands of 1357 and 1572 cm−1 were also observed in sS-MoS2@C-1 and sS-MoS2@C-2, which can be assigned to the sp3-hybridized disordered carbon (D-band) and the sp2-hybridized graphitic carbon (G-band).33,34 Moreover, the relative intensity of ID and IG (ID:IG ≈ 0.95) suggests that the quantity of amorphous carbon is almost as many as the quantity of graphitic carbon.
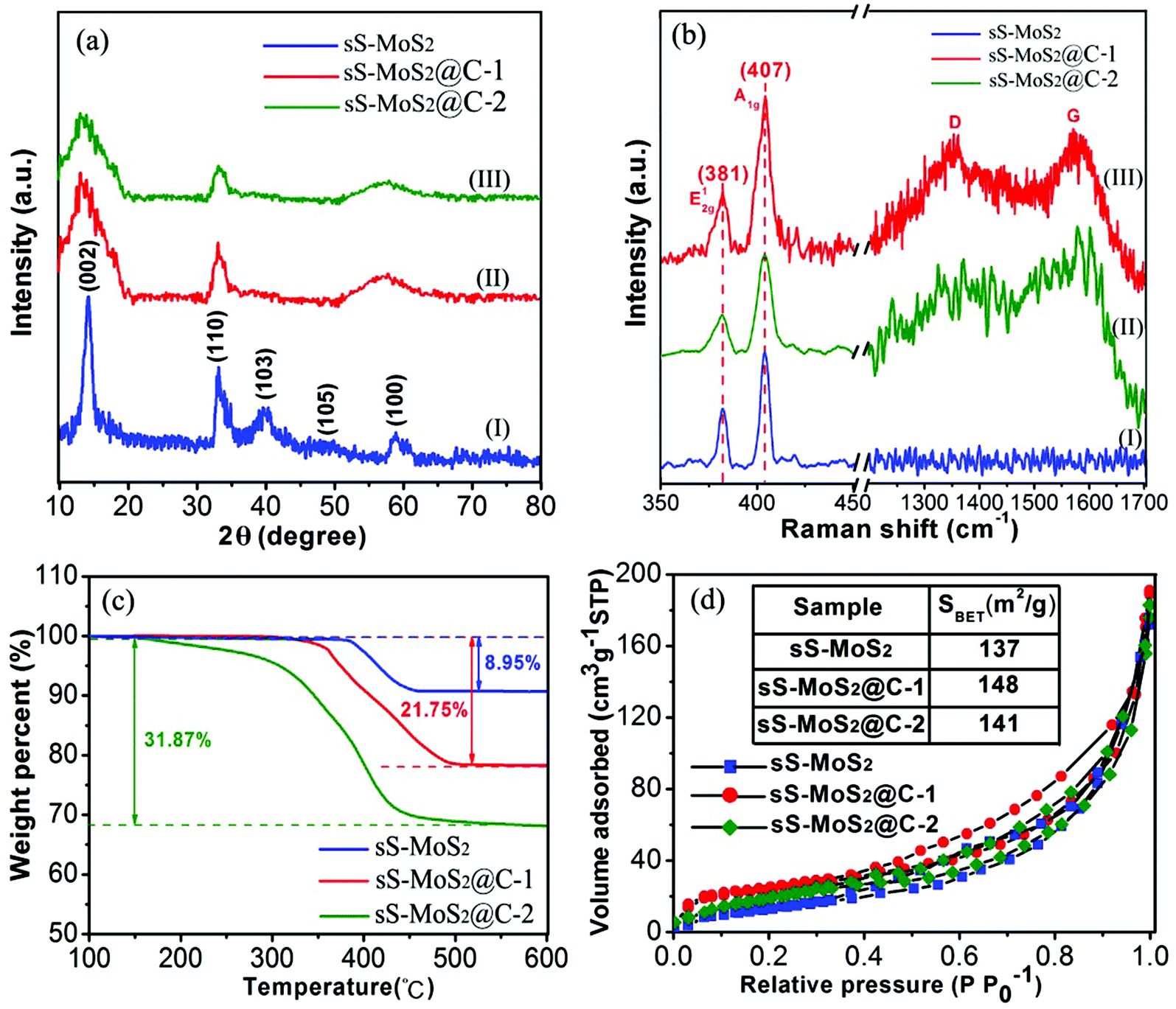 | ||
| Fig. 3 (a) XRD patterns and (b) Raman spectra of bare sS-MoS2, sS-MoS2@C-1 and sS-MoS2@C-2 composites. (c) TG curves of sS-MoS2, sS-MoS2@C-1 and sS-MoS2@C-2 under air atmosphere. (d) N2 adsorption–desorption isotherms of sS-MoS2, sS-MoS2@C-1 and sS-MoS2@C-2. | ||
Thermogravimetric analysis (TGA) was carried out to determine the carbon content of sS-MoS2@C (Fig. 3c). On one hand, for the original sS-MoS2 micro@nano structure, a weight loss (about 8.95%) was observed at approximately 400 °C owing to the oxidative reaction of MoS2 to MoO3 under air atmosphere.6,23 On the other hand, for the sS-MoS2@C the main weight loss between 300 °C and 450 °C can be attributed to the combustion of carbon in air. For the composite materials, weight losses were 21.75% and 31.87%, respectively, corresponding to the content of carbon in sS-MoS2@C-1 and sS-MoS2@C-2 composites which are 12.8% and 22.92%. To study the porous micro@nano structure of sS-MoS2 and sS-MoS2@C, N2 adsorption–desorption isotherms were carried out. It can be seen from Fig. 3d that the Brunauer–Emmett–Teller (BET) surface area of sS-MoS2@C-1 is calculated to be 148 m2 g−1 which is a slight improvement as compared with that of sS-MoS2 (137 m2 g−1) and sS-MoS2@C-2 (141 m2 g−1). Such a high surface area of sS-MoS2@C-1 nanostructure could be attributed to following reasons. Firstly, the hollow MoS2 microspheres provide large interspaces which contribute mainly to the improvement of surface area. Secondly, the amorphous carbon shell also enhances the outer surface area.
In order to confirm the existence and explore the distribution of the carbon shell in the as-formed sS-MoS2@C-1 nanomaterial, energy dispersive X-ray (EDX) mapping analysis and scanning transmission electron microscopy (STEM) were carried out, as shown in Fig. 4. Mapping analyses on a typical microsphere@nanosphere MoS2 decorated with carbon shell reveals the presence of not only the elements Mo and S, but also C. A core–shell structure of sS-MoS2@C-1 is remarkably manifested with a slightly larger circular area of C as compared with that of Mo and S, which proves the existence of the carbon shell on the surface of sS-MoS2. To further characterize the bonding state and chemical nature of the as-prepared sS-MoS2@C nanocomposites, X-ray photoelectron spectroscopy (XPS) was employed. As can be seen from Fig. 5a, the peaks of C, O, S and Mo can be obviously detected. Fig. 5b shows the high-resolution spectrum of Mo 3d, with two peaks being located at 229.5 eV and 232.7 eV, which are attributed to the Mo4+ 3d5/2 and Mo4+ 3d3/2, respectively.35,36 A small S 2s peak is also observed which is located at 226.7 eV, and the peak of oxydic Mo is observed at 235.9 eV. In the peaks of S 2p (Fig. 5c), the peaks at about 162.5 eV and 163.6 eV are related to the S 2p3/2 and S 2p1/2 binding energies, respectively. These surveyed spectrums are in good agreement with the previous studies which can be assigned to the characteristics of MoS2.37–39Fig. 5d shows the binding energy of C 1s at 284.8 eV which is attributed to the amorphous carbon (C–C bond) and the peak at 287.2 eV is assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond. The XPS spectrum also demonstrates the carbon shell covered structure of the sS-MoS2@C composites, providing a prediction of better conductivity for use as anode materials in LIBs.
O bond. The XPS spectrum also demonstrates the carbon shell covered structure of the sS-MoS2@C composites, providing a prediction of better conductivity for use as anode materials in LIBs.
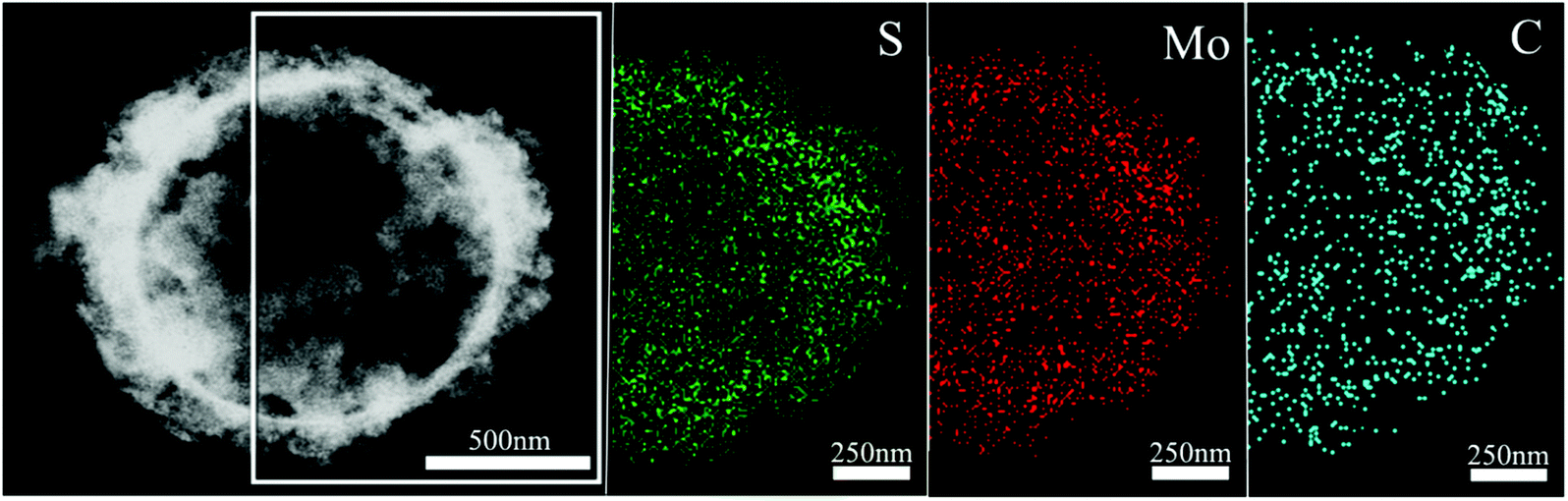 | ||
| Fig. 4 Scanning transmission electron microscopy (STEM) image of sS-MoS2@C−1 microstructure, and the EDX mapping images of S, Mo and C elements. | ||
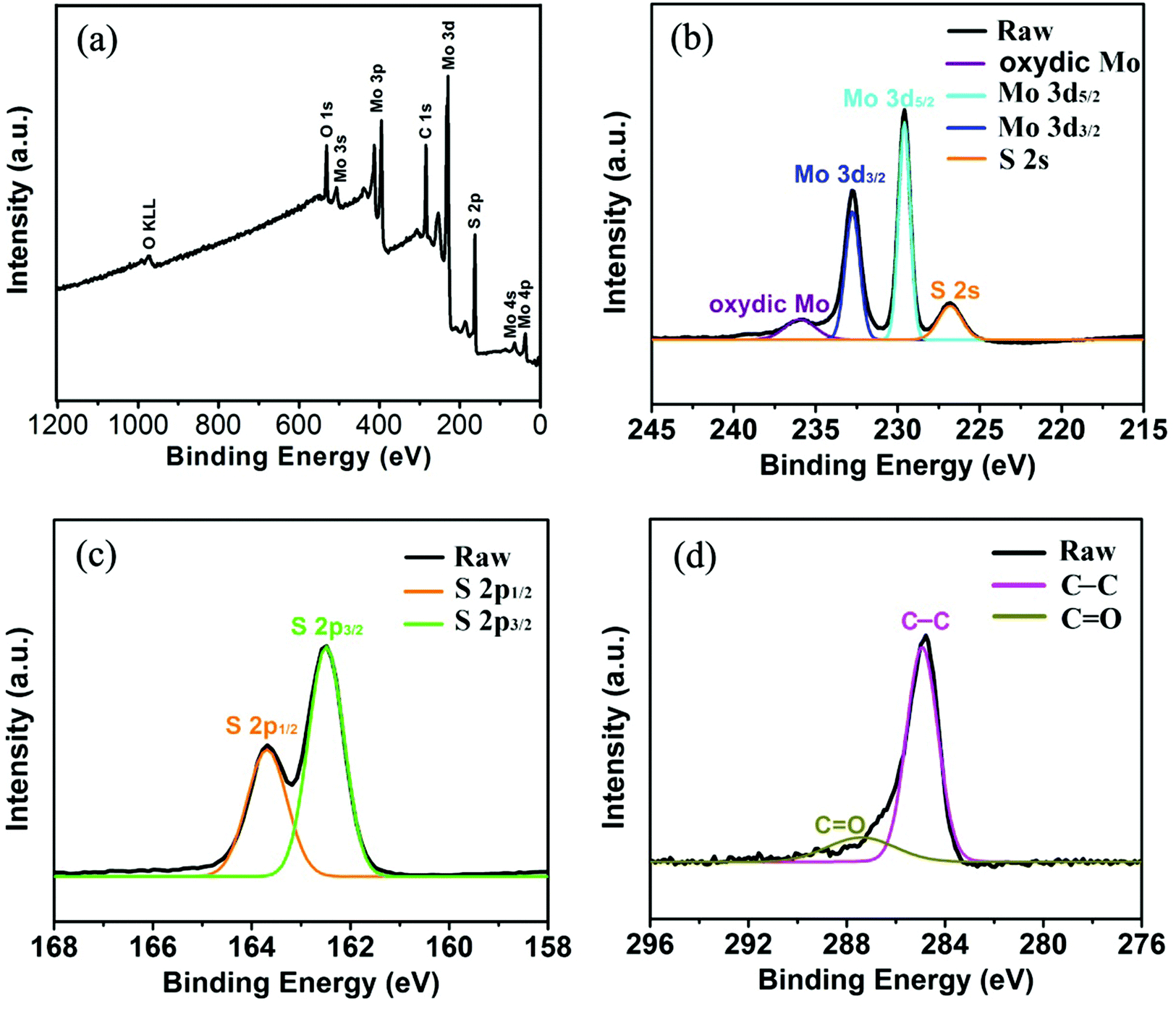 | ||
| Fig. 5 XPS spectra of sS-MoS2@C-1 (a) Survey spectrum. (b) Mo 3d spectrum. (c) S 2p spectrum. (d) C 1s spectrum. | ||
To understand the Li+ storage properties of the sS-MoS2@C-1 nanocomposites, the initial five cyclic voltammetry (CV) sweeps have been examined as shown in Fig. 6a. During the first cathodic process, a reduction peak appears at approximately 0.85 V corresponding to the phase transition from trigonal prismatic to octahedral coordination. The second cathodic peak at about 0.32 V corresponds to the subsequent conversion reaction to form Mo nanoparticles embedded in Li2S, and then the formation of a gel-like polymeric layer resulting from electrochemically driven electrolyte degradation.40,41 In the reverse scan, a ambiguous anodic peak at 1.75 V is due to the incomplete oxidation of Mo metal, and another pronounced peak at 2.38 V can be attributed to the delithiation of Li2S.42,43 In the remainder scans, the intensities of the two cathodic peaks at about 0.85 V and 0.32 V disappear. Meanwhile, two new peaks at 1.05 V and 1.82 V are observed, which indicates the multistep conversion process from S to the formation of Li2S. The cathodic peak at 1.82 V and the anodic peak at 2.38 V constitute a reversible redox couple, which is the main electrochemistry process of this electrode.44 It is also noted that a weak redox couple of the cathodic peak at 0.25 V and the anodic peak at 0.5 V was observed, which could be accounted for by the auxiliary action of electrochemical Li+ storage by the amorphous carbon shell.34,45 Therefore, it is clear that the carbon shell does assist in the electrochemical reaction during discharge–charge. After the first cycle, the electrochemical performance of sS-MoS2@C-1 is mainly dominated by the reversible conversion reaction of sulfur to Li2S. As can be seen from Fig. 6a, the CV performance is very stable and steady, indicating the sS-MoS2@C-1 composite materials have a high reversibility and good stability for the insertion–desertion of Li+. For the bare sS-MoS2 electrode, two redox peaks located at 0.21/0.78 V and 2.48 V in the first cycle correspond to the conversion reaction process: MoS2 + 4Li+ + 4e− ↔ Mo + 2Li2S, which agrees well with the previous lithiation and delithiation profiles of the sS-MoS2@C-1 composites. While in the subsequent cycles, the intensity of the anodic peak changed greatly, suggesting an irreversible conversion reaction during the Li+ insertion–extraction process. It is also worthy to mention that the shape of the peaks for the sS-MoS2@C-1 nanocomposites is more intense and sharper, and the gap between redox peaks is smaller than that for the sS-MoS2, demonstrating that the sS-MoS2@C-1 had a greater efficiency in the redox reaction and lower overall resistance.7,46 These CV behaviours also demonstrate that sS-MoS2@C-1 possesses a much better stability than sS-MoS2 for application as an anode materials in LIBs.
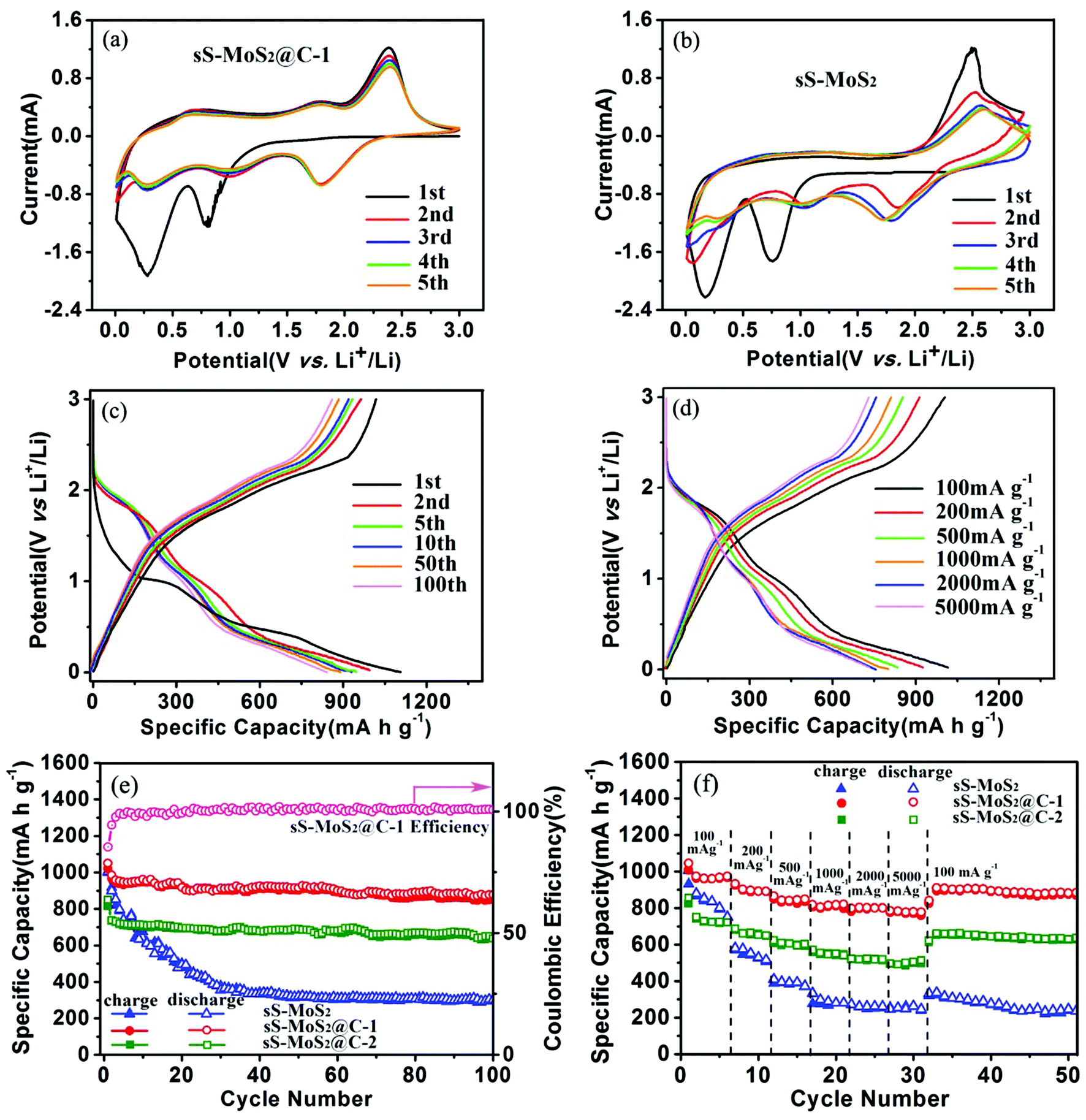 | ||
| Fig. 6 The initial five CV curves of (a) sS-MoS2@C-1 and (b) sS-MoS2 electrodes. (c) Discharge–charge voltage profiles at a current density of 100 mA g−1 of sS-MoS2@C-1. (d) Discharge–charge voltage profiles of sS-MoS2@C-1 at different current densities. (e) Comparative long-term cycling performances of sS-MoS2, sS-MoS2@C-1 and sS-MoS2@C-2 at a current density of 100 mA g−1. (f) Comparative rate performance of sS-MoS2, sS-MoS2@C-1 and sS-MoS2@C-2 at different current densities. | ||
Fig. 6c shows the galvanostatic discharge–charge (GDC) voltage profiles of sS-MoS2@C-1 nanocomposites at 100 mA g−1. During the first discharge, two plateaus appear at approximately 1.0 V and 0.5 V, corresponding to the phase transformation and further decomposition into Li2S and Mo nanoparticles, respectively.16,17 The first discharge process should be associated with the Li+ insertion reaction followed by a conversion reaction. The sloped profile at <0.5 V suggests interfacial Li uptake by the carbon shell.47 In the subsequent charge, the distinct plateau at 2.3 V agrees well with the anodic peak during the CV scan, which is attributed to the oxidation of Li2S.48,49 The subsequent GDC cycles show sloped profiles, with two plateaus at approximately 1.85 V and 2.3 V upon discharge–charge, corresponding to the conversion reaction of sulfur to Li2S. In the first cycle, the discharge and charge capacities of sS-MoS2@C-1 are 1107 mA h g−1 and 1008 mA h g−1, respectively, with a Coulomb Efficiency (CE) of 91%. Moreover, the reversible discharge–charge capacities of the 2nd and 5th cycles are 995–953 mA h g−1 and 947–933 mA h g−1, respectively. As the initial irreversible capacity loss is mainly attributed to the formation of SEI, the CE improvement is largely due to the carbon shell, which inhibits the side reactions of sS-MoS2 by diminishing their unfavourable contact with the electrolyte. After several cycles, the Coulomb Efficiency quickly approaches to 100%, with 10th, 50th, 100th discharge–charge capacities of 930–921 mA h g−1, 896–891 mA h g−1 and 889–886 mA h g−1, respectively. The sS-MoS2@C-1 nanocomposite also exhibits improved cycling and rate performance, as shown in Fig. 6d. Even at high current densities of 1000 mA g−1, 2000 mA g−1 and 5000 mA g−1, the GDC voltage profiles of sS-MoS2@C-1 remain high and stable, with reversible discharge–charge capacities of 825–819 mA h g−1, 811–805 mA h g−1 and 796–791 mA h g−1, respectively.
The cyclic stability and coulombic efficiency of the bare sS-MoS2, sS-MoS2@C-1 and sS-MoS2@C-2 nanocomposites at a current density of 100 mA g−1 are presented in Fig. 6e. It is proved that sS-MoS2@C-1 outperforms the other two samples in terms of cycling stability. The almost completely stable cycling performance of sS-MoS2@C-1 starts from the third cycle. The discharge capacity of the sS-MoS2@C-1 composite stabilizes at about 960 mA h g−1 after three cycles. Even after 100 cycles, a high capacity of 878 mA h g−1 can be maintained, which is 91.5% of the capacity at the third cycle. It is can also be seen from Fig. 6e that the Coulomb Efficiency maintained a high level at about 100% from the third cycle onwards. In sharp contrast, sS-MoS2 shows a much inferior cycling performance. Although it delivers a high initial discharge capacity (998 mA h g−1), the capacity of bare sS-MoS2 prepared in this work decreases continuously within 20 cycles. After 100 cycles, only a low capacity of 302 mA h g−1 is retained. The cycling stability of sS-MoS2@C-2 is somewhat better than that of sS-MoS2, but still inferior to that of sS-MoS2@C-1. The initial discharge capacity is 830 mA h g−1, and the capacity fades to 638 mA h g−1 after 100 cycles. Compared with the high capacity of sS-MoS2@C-1, the relatively lower capacity of sS-MoS2@C-2 could be attributed to the carbon shell being too thick and impeding the high-efficiency transport of Li+. The rate performance of sS-MoS2, sS-MoS2@C-1 and sS-MoS2@C-2 nanocomposites were further investigated by cycling at various discharge–charge current densities ranging from 100–5000 mA g−1 (Fig. 6f). sS-MoS2@C-1 delivers a high capacity of 980 mA h g−1 at 100 mA g−1. As the current density gradually elevates, the capacity decreases slightly. Even at a high current density of 5000 mA g−1, a discharge capacity of 805 mA h g−1 can still be retained, suggesting the excellent high rate capability of the sS-MoS2@C-1 nanocomposite. Remarkably, a stable capacity of 910 mA h g−1 can be attained when the current is reduced to 100 mA g−1. The electrochemical performance of sS-MoS2@C-2 in terms of cycling stability and rate capability is comparable or even superior to that of MoS2-based anodes in previous reports,6,7,23,40 with a high capacity of 526 mA h g−1 at 5000 mA g−1, suggesting the effectiveness of our strategy of introducing the carbon shell. On the contrary, the bare sS-MoS2 delivers much lower capacities at high current densities, and fails quickly for the 20th cycle at 1000 mA g−1. It is obvious that the capacity of bare sS-MoS2 fades rapidly and cannot recover to the initial level, even at 100 mA g−1 after high rate cycling. Considering that all samples have almost the same sS-MoS2 core structure, the difference in capacity should result from the carbon shell. First, the micro@nano spherical structure of the MoS2 core plays the role of buffering the mechanical stresses induced by the volumetric expansion–shrinkage, and preventing the aggregation of the MoS2 cathode material. Second, the carbon shell may serve as a shield and an adsorbent to restrain the discharge products of sulfur from dissolving into the electrolytes. These results show that only with the optimal molar ratio of MoS2 to carbon, the composites can exhibit enhanced Li+ storage properties.
Such high capacity retention of the sS-MoS2@C-1 electrode accounts for its structural stability. To demonstrate its application, we show that one coin cell based on the sS-MoS2@C-1 positive electrode, after being cycled 100 times at 100 mA g−1, can still easily power an LED (3 V, 20 mA), as shown in Fig. 7a. For a better understanding of why the unique sS-MoS2@C architectures possesses such excellent electrochemical performances for lithium storage, we performed electrochemical impedance spectroscopy (EIS) for these three electrodes as shown in Fig. 7b. It can be obviously seen that the diameter of the semicircle for sS-MoS2@C-1 in the high–medium frequency region is the smallest compared to those of sS-MoS2 and sS-MoS2@C-2, which demonstrates that sS-MoS2@C-1 possesses the lowest contact and charge-transfer resistances. The kinetic differences of these electrodes were further investigated using the common R–C equivalent circuit, as shown in the inset of Fig. 7b. The inclined line in the low frequency region is descriptive of the Li+ diffusion impedance, the medium frequency semicircle to the charge transfer resistance Rct and CPE2 of the electrode–electrolyte interface, and the high frequency semicircle could be attributed to resistance Rsf and CPE1 of the SEI film.40,50 The Rct was obtained by fitting data according to the equivalent circuit model that sS-MoS2, sS-MoS2@C-1 and sS-MoS2@C-2 electrodes have charge transfer resistances of 225 Ω, 58 Ω and 75 Ω, respectively. These results clearly validate that the hollow microsphere@onion-like solid nanosphere MoS2 decorated with a carbon shell architecture can not only ensure a high conductivity of the overall electrode, but also largely enhance the electrochemical activity of the sS-MoS2 anode materials during cycling processes. The deliberately designed hollow microsphere@onion-like solid nanosphere MoS2 decorated with a carbon shell confirms that it can achieve excellent cycling performance as well as high capacity and superior rate capability, which could be explained as follows. Firstly, the onion-like MoS2 nanospheres distributed on the surfaces of the hollow MoS2 microspheres with a well arranged layered structure could facilitate the buffering of the mechanical stresses induced by the volumetric expansion–shrinkage, and the uniform spherical structure could prevent the aggregation of the MoS2 layers as per molecular structure topology theory. Secondly, the sS-MoS2 core acts as a host for Li storage, in which onion-like solid nanospheres MoS2 principally provide efficient storage for Li+ ions and the hollow microspheres MoS2 primarily enhance the special surface area. Thirdly, the carbon shell not only enables fast e− transmission, but also provides an unperturbed Li+ supply for the core micro@nano MoS2 spheres, which improves the Li storage kinetics and the stability of the composites. Lastly, the built in carbon shell acts as a shield to stabilize the sS-MoS2 core structure, and protect against direct contact with the electrolyte to restrain the active material from dissolving. Thus, all of the structural advantages of sS-MoS2@C and the synergistic effect of sS-MoS2 and the carbon shell contribute to the stable cycling performance and excellent battery capacity.
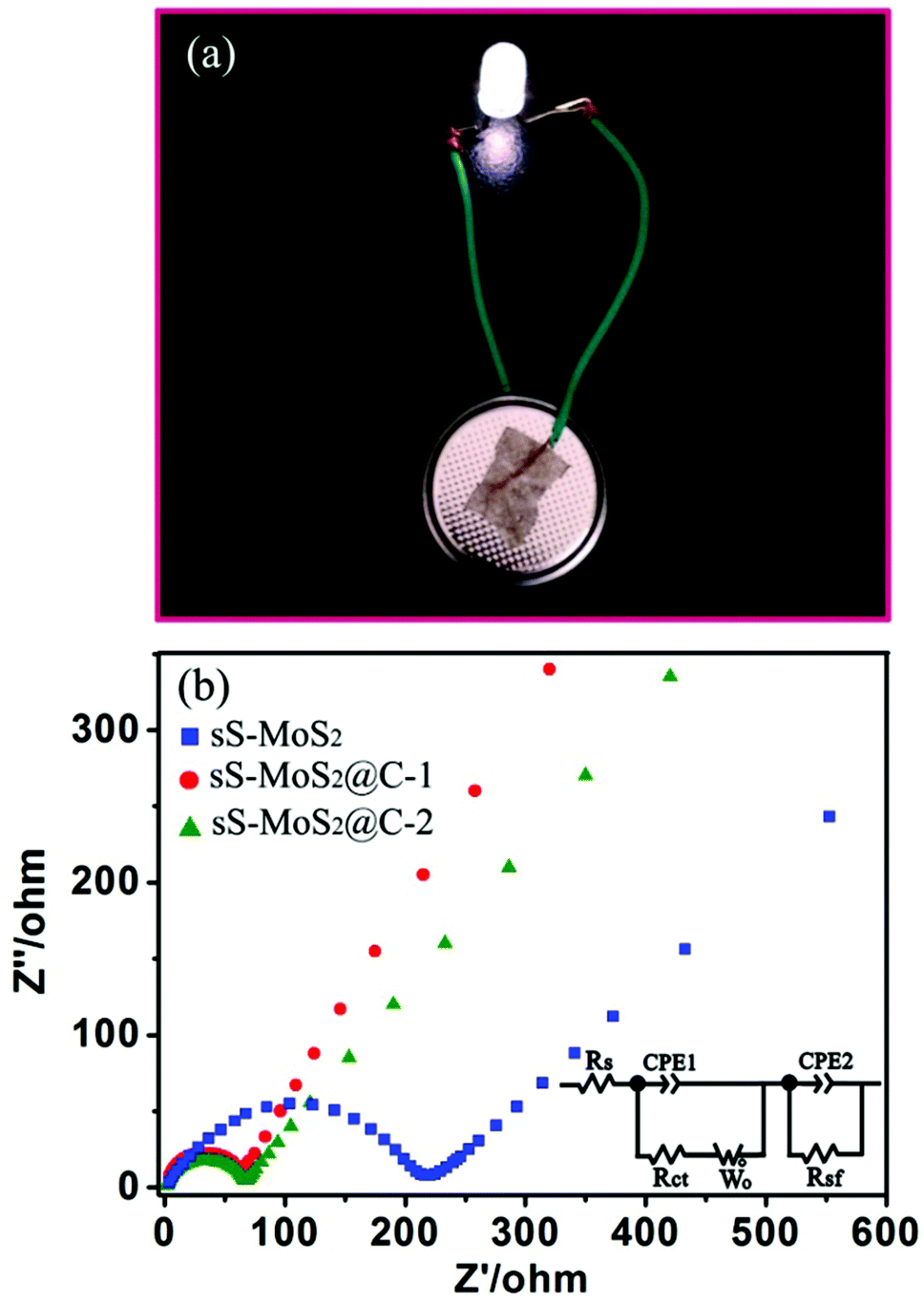 | ||
| Fig. 7 (a) Photograph of an LED powered by one coin cell based on the sS-MoS2@C-1 electrode. (b) Nyquist plots of the sS-MoS2, sS-MoS2@C-1 and sS-MoS2@C-2 electrodes in the frequency range from 200 kHz to 0.01 kHz. | ||
Conclusions
In summary, hollow microsphere@onion-like solid nanosphere MoS2 decorated with a carbon shell structure was prepared, utilizing dopamine as the carbon source. The successfully prepared core–shell sS-MoS2@C nanocomposites have been engineered to accommodate the volume expansion during lithiation and enlarge the electric conductivity in order to serve as electrode materials. In addition to molybdenum disulfide, our concept for preparation of such micro@nano sphere materials can also be applied to other transition metal dichaldogenides (TMDs) materials, such as WS2, MoSe2 and TaS2. With an optimized thickness of the carbon shell, the sample sS-MoS2@C-1 exhibited high reversible capacity, superior rate performances and excellent cycling stability. Furthermore, this work provides insight in that spherical MoS2 nanostructures covered by a carbon shell have been shown to be promising candidates for high-energy LIBs anode materials.Acknowledgements
The authors acknowledge financial support from the NSF of China (Grant No. 61574055, 61274014, 61474043, 61425004), Innovation Research Project of Shanghai Education Commission (Grant No. 13zz033) and Project of Key Laboratory of Polar Materials and Devices (Grant No. KFKT20140003).Notes and references
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 Search PubMed.
- K. Kang, Y. S. Meng, J. Breger, C. P. Grey and G. Ceder, Science, 2006, 311, 977–980 CrossRef CAS PubMed.
- H. Liu, D. Su, R. Zhou, B. Sun, G. Wang and S. Z. Qiao, Adv. Energy Mater., 2012, 2, 970–975 CrossRef CAS.
- T. Stephenson, Z. Li, B. Olsen and D. Mitlin, Energy Environ. Sci., 2014, 7, 209–231 CAS.
- X. Cao, Y. Shi, W. Shi, X. Rui, Q. Yan, J. Kong and H. Zhang, Small, 2013, 9, 3433–3438 CrossRef CAS PubMed.
- F. Zhou, S. Xin, H. W. Liang, L. T. Song and S. H. Yu, Angew. Chem., Int. Ed., 2014, 53, 11552–11556 CrossRef CAS PubMed.
- S. Hu, W. Chen, J. Zhou, F. Yin, E. Uchaker, Q. F. Zhang and G. Z. Cao, J. Mater. Chem. A, 2014, 2, 7862–7872 CAS.
- U. K. Sen and S. Mitra, ACS Appl. Mater. Interfaces, 2013, 5, 1240–1247 CAS.
- L. C. Yang, S. N. Wang, J. J. Mao, J. W. Deng, Q. S. Gao, Y. Tang and O. G. Schmidt, Adv. Mater., 2013, 25, 1180–1184 CrossRef CAS PubMed.
- L. R. Hu, Y. M. Ren, H. X. Yang and Q. Xu, ACS Appl. Mater. Interfaces, 2014, 6, 14644–14652 CAS.
- S. Ding, J. S. Chen and X. W. Lou, Chem. – Eur. J., 2011, 17, 13142–13145 CrossRef CAS PubMed.
- G. Huang, T. Chen, W. Chen, Z. Wang, K. Chang, L. Ma, F. Huang, D. Chen and J. Y. Lee, Small, 2013, 9, 3693–3703 CrossRef CAS PubMed.
- S. J. Ding, J. S. Chen, Z. Y. Wang, Y. L. Cheah, S. Madhavi, X. Hu and X. W. Lou, J. Mater. Chem., 2011, 21, 1677–1680 RSC.
- L. C. Liu, Q. Fan, C. Z. Sun, X. R. Gu, H. Li, F. Gao, Y. F. Chen and L. Dong, J. Power Sources, 2013, 221, 141–148 CrossRef CAS.
- S. J. Ding, D. Y. Zhang, J. S. Chen and X. W. Lou, Nanoscale, 2012, 4, 95–98 RSC.
- K. Chang and W. Chen, ACS Nano, 2011, 5, 4720–4728 CrossRef CAS PubMed.
- Y. G. Li, H. L. Wang, L. Xie, Y. Y. Liang, G. S. Hong and H. J. Dai, J. Am. Chem. Soc., 2011, 133, 7296–7299 CrossRef CAS PubMed.
- H. W. Zhang, L. Zhou, O. Noonan, D. J. Martin, A. K. Whittaker and C. Z. Yu, Adv. Funct. Mater., 2014, 24, 4337–4342 CrossRef CAS.
- X. W. Lou, C. M. Li and L. A. Archer, Adv. Mater., 2009, 21, 2536–2539 CrossRef CAS.
- J. M. Jeong, B. G. Choi, S. C. Lee, K. G. Lee, Y. B. L. Chang, H. U. Lee, S. Kwon, G. Lee, C. S. Lee and Y. S. Huh, Adv. Mater., 2013, 25, 6250–6255 CrossRef CAS PubMed.
- J. J. Wang, C. Luo, T. Gao, A. Langrock, A. C. Mignerey and C. S. Wang, Small, 2015, 4, 472–480 CrossRef.
- J. M. Jeong, K. G. Lee, S. J. Chang, J. W. Kim, Y. K. Han, S. J. Leea and B. G. Choi, Nanoscale, 2015, 7, 324–329 RSC.
- Z. M. Wan, J. Shao, J. J. Yun, H. Y. Zheng, T. Gao, M. Shen, Q. T. Qu and H. H. Zheng, Small, 2014, 23, 4975–4981 CrossRef PubMed.
- Y. H. Tan, K. Yu, T. Yang, Q. F. Zhang, W. T. Cong, H. H. Yin, Z. L. Zhang, Y. W. Chen and Z. Q. Zhu, J. Mater. Chem. C, 2014, 2, 5422–5430 RSC.
- H. C. Zeng, Curr. Nanosci., 2007, 3, 177–181 CrossRef CAS.
- H. Shi, K. Yu, F. Sun and Z. Q. Zhu, CrystEngComm, 2012, 14, 278–285 RSC.
- R. Liu, S. M. Mahurin, C. Li, R. R. Unocic, J. C. Idrobo, H. J. Gao, S. J. Pennycook and S. Dai, Angew. Chem., Int. Ed., 2011, 50, 6799–6802 CrossRef CAS PubMed.
- C. Lei, F. Han, D. Li, W. C. Li, Q. Sun, X. Q. Zhang and A. H. Lu, Nanoscale, 2013, 5, 1168–1175 RSC.
- H. S. Li, L. F. Shen, K. B. Yin, J. Ji, J. Wang, X. Y. Wang and X. G. Zhang, J. Mater. Chem. A, 2013, 1, 7270–7276 CAS.
- H. Li, Z. X. Wang, L. Q. Chen and X. J. Huang, Adv. Mater., 2009, 21, 4593–4607 CrossRef CAS.
- J. S. Chen and X. W. Lou, J. Power Sources, 2010, 195, 2905–2908 CrossRef CAS.
- Y. Shi, Y. Wang, J. I. Wong, A. Y. S. Tan, C. L. Hsu, L. J. Li, Y. C. Lu and H. Y. Yang, Sci. Rep., 2013, 3, 2169–2176 Search PubMed.
- X. Zhao, C. Hu and M. Cao, Chem. – Asian J., 2013, 8, 2701–2707 CrossRef CAS PubMed.
- Z. Wang, T. Chen, W. Chen, K. Chang, L. Ma, G. Huang, D. Chen and J. Y. Lee, J. Mater. Chem. A, 2013, 1, 2202–2210 CAS.
- J. Zheng, H. Zhang, S. Dong, Y. Liu, C. T. Nai, H. S. Shin, H. Y. Jeong, B. Liu and K. P. Loh, Nat. Commun., 2013, 5, 2995–2999 Search PubMed.
- D. H. Youn, J. W. Jang, J. Y. Kim, J. S. Jang, S. H. Choi and J. S. Lee, Sci. Rep., 2014, 4, 5492–5499 CAS.
- K. K. Liu, W. Zhang, Y. H. Lee, Y. C. Lin, M. T. Chang, C. Y. Su, C. S. Chang, H. Li, Y. Shi and H. Zhang, Nano Lett., 2012, 12, 1538–1544 CrossRef CAS PubMed.
- C. Altavilla, M. Sarno and P. Ciambelli, Chem. Mater., 2011, 23, 3879–3885 CrossRef CAS.
- X. L. Li and Y. D. Li, J. Phys. Chem. B, 2004, 108, 13893–13900 CrossRef CAS.
- B. J. Guo, K. Yu, H. Fu, Q. Q. Hua, R. J. Qi, H. L. Li, H. L. Song, S. Guo and Z. Q. Zhu, J. Mater. Chem. A, 2015, 3, 6392–6402 CAS.
- Q. Wang and J. Li, J. Phys. Chem. C, 2007, 111, 1675–1682 CAS.
- Y. Gong, S. Yang, L. Zhan, L. Ma, R. Vajtai and P. M. Ajayan, Adv. Funct. Mater., 2014, 24, 125–130 CrossRef CAS.
- K. Zhang, H. J. Kim, X. Shi, J. T. Lee, J. M. Choi, M. S. Song and J. H. Park, Inorg. Chem., 2013, 52, 9807–9812 CrossRef CAS PubMed.
- G. He, S. Evers, X. Liang, M. Cuisinier, A. Garsuch and L. F. Nazar, ACS Nano, 2013, 7, 10920–10930 CrossRef CAS PubMed.
- E. Benavente, M. Santa Ana, F. Mendizabal and G. Gonźalez, Coord. Chem. Rev., 2002, 224, 87–109 CrossRef CAS.
- L. Shen, E. Uchaker, X. Zhang and G. Cao, Adv. Mater., 2012, 24, 6502–6506 CrossRef CAS PubMed.
- J. R. Dahn, T. Zheng, Y. Liu and J. S. Xue, Science, 1995, 270, 590–593 CAS.
- Y. X. Yin, S. Xin, Y. G. Guo and L. J. Wan, Angew. Chem., Int. Ed., 2013, 52, 13186–13200 CrossRef CAS PubMed.
- X. P. Fang, C. X. Hua, X. W. Guo, Y. S. Hu, Z. X. Wang, X. P. Gao, F. Wu, J. Z. Wang and L. Q. Chen, Electrochim. Acta, 2012, 81, 155–160 CrossRef CAS.
- J. Wang, J. L. Liu, D. L. Chao, J. X. Yan, J. Y. Lin and Z. X. Shen, Adv. Mater., 2014, 26, 7162–7169 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5nr05595d |
| This journal is © The Royal Society of Chemistry 2016 |