
3D nanochannel electroporation for high-throughput cell transfection with high uniformity and dosage control†
Lingqian
Chang‡
ab,
Paul
Bertani‡
ac,
Daniel
Gallego-Perez
a,
Zhaogang
Yang
a,
Feng
Chen
a,
Chiling
Chiang
d,
Veysi
Malkoc
a,
Tairong
Kuang
a,
Keliang
Gao
a,
L. James
Lee
*abe and
Wu
Lu
*ac
aNational Science and Engineering Center for Affordable Nanoengineering of Polymeric Biomedical Devices, The Ohio State University, Columbus, Ohio 43210, USA. E-mail: lu.173@osu.edu; lee.31@osu.edu
bDepartment of Biomedical Engineering, The Ohio State University, Columbus, Ohio 43210, USA
cDepartment of Electrical and Computer Engineering, The Ohio State University, Columbus, Ohio 43210, USA
dDepartment of Internal Medicine, Ohio State University, Columbus, OH 43209, USA
eDepartment of Chemical and Biomolecular Engineering, The Ohio State University, Columbus, OH 43210, USA
First published on 9th July 2015
Abstract
Of great interest to modern medicine and biomedical research is the ability to inject individual target cells with the desired genes or drug molecules. Some advances in cell electroporation allow for high throughput, high cell viability, or excellent dosage control, yet no platform is available for the combination of all three. In an effort to solve this problem, here we show a “3D nano-channel electroporation (NEP) chip” on a silicon platform designed to meet these three criteria. This NEP chip can simultaneously deliver the desired molecules into 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per cm2 on the top surface of the device. Each 650 nm pore aligns to a cell and can be used to deliver extremely small biological elements to very large plasmids (>10 kbp). When compared to conventional bulk electroporation (BEP), the NEP chip shows a 20 fold improvement in dosage control and uniformity, while still maintaining high cell viability (>90%) even in cells such as cardiac cells which are characteristically difficult to transfect. This high-throughput 3D NEP system provides an innovative and medically valuable platform with uniform and reliable cellular transfection, allowing for a steady supply of healthy, engineered cells.
000 cells per cm2 on the top surface of the device. Each 650 nm pore aligns to a cell and can be used to deliver extremely small biological elements to very large plasmids (>10 kbp). When compared to conventional bulk electroporation (BEP), the NEP chip shows a 20 fold improvement in dosage control and uniformity, while still maintaining high cell viability (>90%) even in cells such as cardiac cells which are characteristically difficult to transfect. This high-throughput 3D NEP system provides an innovative and medically valuable platform with uniform and reliable cellular transfection, allowing for a steady supply of healthy, engineered cells.
Introduction
A wide variety of physical methods have been developed for introducing exogenous materials into cells for different bio-medical purposes.1,2 These strategies avert concerns on oncogenesis and immune rejection caused from viral infection previously used in cell transfection.3 Among all physical delivery methods, microinjection is the most straight forward approach that is capable of delivering cargo into the cytosol using a fine pipette to pierce through a cell membrane.4,5 However, its manual operation highly decreases the practical efficiency and user-friendliness, thereby limiting the applications.5,6 Particle bombardment (gene gun) is a physical–mechanical process which usually causes irreversible membrane break-down due to the interaction between particles and cell membrane.7 Poration (e.g. laser irradiation, sonoporation, electroporation) is characterized as temporal membrane perturbation by external forces focusing on target cells.8–10 To optimize a physical method, two parameters, including poration induced cell-membrane damage and delivery efficiency, are evaluated.11,12 To achieve both criteria, electroporation has turned out to be a more viable approach in both in vitro and in vivo applications.13 Transfection efficiency and cell viability can be balanced by optimization of the local electric field.14 In addition, electroporation offers another advantage over laser irradiation and sonoporation as directly propelling surface charged molecules (e.g. DNA, RNA) into cells under an electric field further facilitates the efficiency of intracellular delivery.15,16While being widely used in gene therapy, wound healing, drug screening, and cell reprogramming recently,17,18 bulk electroporation (BEP), the most common prototype, is usually considered as causing serious cell damage, random transfection, and low efficiency especially to primary cells. In BEP, millions of cells are concentrated in a small space and are shocked by high voltage pulses between a pair of electrodes. Such a system setup is used for its simplicity and operational flexibility, but suffers from poor cell viability as a large portion of cells cannot survive the high voltage pulses. Among the surviving cells, only a small portion could be transfected by successful poration. Meanwhile, the local electrical field varies from cell to cell, causing a totally stochastic process of delivery.19
In recent years, microfluidic-electroporation (MEP) systems have been reported to improve the cell viability by confining a single-cell at a narrow region in which a low voltage (less than 10 V) is used for membrane permeabilization while the cargo is transported into cells via diffusion.20–22 Precise control of the amount of molecules injected into target cells, namely, dosage control, has been desired by biomedical researchers for a long time, as it is important for a variety of biological applications.23 Limited by the channel-scale, the diffusion based delivery in MEP and BEP makes them unable to achieve transfection with dosage control, usually resulting in extremely low efficiency when delivering molecules with large molecular weights (e.g. >10 kbp). None of the aforementioned methods have demonstrated precise delivery of exogenous reagents into living cells with negligible cell damage. Nano-electroporation systems (NEP) are reported recently for single-cell transfection with high efficiency and precise delivery.19,24–26 Different from BEP, NEP offers transient and reversible poration under a high voltage, while instantaneously propelling charged bio-reagents into cells through the nanochannel, which could achieve the benefits of both high cell viability and transfection efficiency. The hallmark of NEP, dosage controllability, makes it unique compared to previously reported counterparts, while illuminating the cell transfection with quality control for medical research and preclinical use. However, all previously-reported NEP is based on a 2D planar design, which is ideal for single-cell study (with less than one hundred cells) each time, but widely losing significance in medical and pre-clinical fields that require a large population of cells. Therefore, a high-throughput nanochannel electroporation chip capable of handling a large number of cells simultaneously is critically needed. Previous approaches used track-etched poly(ethylene terephthalate) or poly carbonate membranes.27–29 However, the random distribution of nanopores in track-etched membranes leads to a non-uniform cell–pore contact, which makes them impossible for uniform transfection and dosage control.
To address these issues, we developed a nanochannel array platform for high-throughput cell electroporation with the advantages of dosage control, high uniformity, while showing significantly higher efficiency of intracellular delivery when using ultra-large molecular weight cargo (>10 kbp plasmids) with high cell viability compared to BEP. A 3D NEP chip, with a uniform and parallel nanochannel array, was batch-produced by projection photolithography and deep reactive ion-etching (DRIE). The 3D NEP system significantly increases the single-cell transfection capability from less than 100 cells per chip (2D NEP) up to more than 40000 cells per cm2 and up to million cells per wafer-scale. We delivered FAM-labeled oligonucleotides into H9C2 cells (mouse cardio-myoblasts), and a working voltage range was identified. Dosage control was demonstrated with a large number of cells. Compared to other high-throughput electroporation systems (BEP and 3D MEP), 3D NEP offers significantly higher transfection uniformity. Moreover, we tested the capability of 3D NEP for the intracellular delivery of a large weight transgenic factor (13 kbp transcriptional plasmid) into MEFs (mouse embryonic fibroblasts). NEP demonstrated significant higher efficiency, cell viability and uniformity as compared to the BEP system. The 3D NEP system provides a medically valuable platform for supplying high-throughput engineered cells with uniform and predictable transfection.
Experimental section
Fabrication of the 3D NEP chip
The fabrication of a nanochannel array chip was based on projection photolithography and DRIE (the detailed protocol is illustrated in Fig. S1†). A silicon wafer (Double Side Polished, 500 μm thickness, (100) orientation) was thinned to the thickness of 250 μm using wet etching (45% KOH (potassium hydroxide) at 80 °C, etch rate: ∼1 μm min−1). A microchannel array (50 μm in diameter) was patterned on the one side using photolithography (EVG 620 aligner, photoresist: SPR220-7, thickness: 10 μm). Using photoresist as a mask, a uniform micro-trench array, with depth ∼230 μm was etched with a DRIE system (Oxford Plasma Lab 100 system). A modified Bosch process (alternative SF6 and C4F8 plasmas) was developed with parameters optimized. A nanopore array (650 nm in diameter, 50 μm pore-to-pore distance) was patterned on the other side using projection photolithography (GCA 6100C Stepper (i-line), photoresist: AZ-5214 E, thickness: 1 μm), followed by a second DRIE process in which the nanochannel array was connected to the microchannel array. The nanopore top surface was used to seed cells in the procedure of 3D NEP. A dielectric layer (Si3N4) was deposited on the side using plasma-enhanced chemical vapor deposition (Plasma Therm 790) for electrical isolation. The nanochannel size of 650 nm is selected to keep minimum damage to cells and at the same time to be able to deliver very large plasmids without nanochannels being potentially blocked during electroporation.System setup
A 3D NEP system was developed in order to obtain high-throughput cell electroporation. Detailed descriptions are given in Results and discussion section as well as illustrated in Fig. 2. The PDMS spacer (thickness 2 mm) was prepared by mixing the pre-polymer and curing agent in a ratio of 10:1 (Sylgard 184, Dow Corning). The support platform, the substrate, and two clamps were made of PMMA and fabricated using a digital micro-milling machine (Mechanical Lab, Department of Chemical Engineering, The Ohio State University). For 3D NEP, a pair of electrodes was connected to the two ports of the power system (Gene Pulser X-cell system, Bio-Rad). The bottom electrode is a microscope slide (25 mm by 75 mm) deposited with 100 nm gold (Denton 502A, e-beam evaporator). A rod electrode (copper) is used as the top electrode.
3D MEP and BEP
In comparison with 3D NEP, a silicon 3D MEP chip was fabricated based on photolithography, wet etching and DRIE. The details of the process were described in previous work.30 For a MEP experiment, the chip was cut into 1 cm × 1 cm, and was mounted on the same system setup shown in Fig. S2.† The electroporation conditions used in 3D MEP experiments are 4 V, 50 ms, 5 pulses. A commercial BEP system (Neon Transfection System, Life technology) was also used in our experiments for a comparative study. The protocols for cell electroporation were used according to the website of the neon system. For propidium iodide (PI) and GFP plasmid delivery, the conditions are: 1350 V, 20 ms, 1 pulse. For OSKM plasmids delivery, the conditions are 1750 V, 20 ms, 1 pulse.Cells and biomolecules
Two types of cells were used in experiments for different purposes. H9C2 (cardiomyoblasts) cells were obtained from ATCC, and were cultured in Dulbecco's Modified Eagle's Medium (Catalog no. 30-2002) with the addition of 10% (v/v) fetus bovine serum (FBS, heat inactivated, catalog no. 26010). Mouse embryonic fibroblasts (MEFs) were isolated from E12.5 embryos, and were cultured in DMEM with addition of 10% FBS and non-essential-amino acids. As a heart myoblast cell line, H9C2 cells are chosen for demonstration because they are typically difficult to be transfected using bulk electroporation. Besides, H9C2 cell line is very valuable as an in vitro model to study the drug metabolizing enzymes in the heart.31 FAM labeled oligonucleotides (FAM-ODN, alpha DNA Co., cat no. 427520, excitation /emitting wavelength, 492/517 nm) were used to test the performance of the 3D NEP system for voltage optimization and dosage control validation. Propidium iodide (PI dye, cat. no. P3566, Invitrogen, excitation/emitting wavelength, 535/617 nm) was delivered for the comparison of the uniformity and efficiency of intracellular delivery. PmaxGFP (3.5 kbp, Cat. no. VSC-1001, Amaxa Nucleofector Technology) was used to check the transfection performance of the delivery of large molecules into living cells. For demonstrating the efficiency of NEP delivering large molecular weight cargos into MEFs cells, OSKM plasmid (OCT-4, SOX2, KCL-1, and c-MYC transcription factors, and RFP encoded on a pCAG plasmid backbone, Addgene) was used.Cell loading, release, and viability
In NEP experiments, two types of adherent cells (H9C2 and MEFs) were trypsinized (trypsin, catalog number 25200-056, life technologies) and loaded on the 3D NEP chip for 4 hours. Within this culture time, living cells attach to the chip surface. A vacuum was also applied during cell loading in order to enhance the contact of cells to the nanopores, according to the protocol in the literature. Different from MEP, the vacuum effect on a NEP chip is extremely low due to the high flow-resistivity through the nanochannel, and therefore it is quite safe to cells. After NEP and MEP, cells were trypsinized and transferred back to petri-dishes for culture. Cell viability in the MEF experiments was determined by comparing the portion of cells showing typical fibroblast morphologies to the total cell numbers. Dead cells usually detached from the dishes. Two-sided student's t-test was used to determine the significance for data with Gaussian distribution and equal variances. The groups with p values <0.05 were considered statistically significant.Results and discussion
3D nanochannel array chip
The cross-section of the 3D NEP chip illustrating microchannel–nanochannel connections is shown in Fig. 1a. Using projection photolithography and DRIE, a uniform array of nanochannels (650 nm in diameter, 20 μm in length, with a high aspect ratio of up to 40:1) was developed as shown in Fig. 1b. A microchannel array (50 μm in diameter) was patterned and DRI-etched from the other side until connection with the nanochannel array was established. The nanopore array on the top surface (Fig. 1c) is used for loading and electroporating cells. The method described demonstrates the feasibility of fabricating a uniform nanochannel array with controllable density and micro-/nano-channel length. In our experiments, the NEP chip we used has an average nanochannel length of 13 μm, and a nanochannel to nanochannel spacing of 50 μm, which determines the throughput capability, i.e. 40000 cells with a 1 cm2 chip. The throughput could be easily scaled up to a million cells per wafer for applications requiring such a population of cells.
 | ||
| Fig. 1 Schematic and SEM micrographs of the silicon 3D NEP chip. (a) The cross-section configuration of the microchannel–nanochannel array chip. For NEP, the flat side is used as the top surface for seeding cells. (b) The cross-sectional cut of the chip shows the array of nanochannels (650 nm in diameter, ∼20 μm in length) connecting the microchannel (50 μm in diameter). Scale bar (from left to right): 20 μm, 3 μm. (c) The nanopore array pattern on the top surface. Scale bar (from left to right): 15 μm, 2 μm, 500 nm. | ||
3D NEP system setup
The 3D NEP system consists of four parts, including the 3D NEP chip, a support platform, PDMS (polydimethylsiloxane) spacers, and a pair of electrodes (Fig. 2a). A top chamber was confined by a PDMS spacer for cell loading and NEP. A bottom chamber is compartmentalized by the chip on the support platform made of PMMA (poly(methyl methacrylate)), which is mounted on gold-coated glass serving as the bottom electrode. The second PDMS spacer is sealed between the chip and the platform to avoid leakage. All components are stacked on a PMMA substrate with two clamps (Fig. 2b). The photograph of the 3D NEP system is shown in Fig. S2.† Cargos (such as DNA, RNA) to be injected are filled into the bottom chamber. In NEP, high voltage pulses are applied between the top electrode and the bottom electrode. NEP offers the advantage of being capable of accelerating the charged molecules to transport them across cell membrane, a feature which cannot be realized by MEP and BEP. The connection of electrodes is dependent on the charges of the cargos, for example, the delivery of DNA and RNA (negatively charged) requires that the top electrode is connected to the positive port of the power supply while the bottom electrode is connected to the negative port.19 Different from 2D NEP which is a planar device, 3D NEP electro-injects cargos into cells through independent channels in the Z-direction.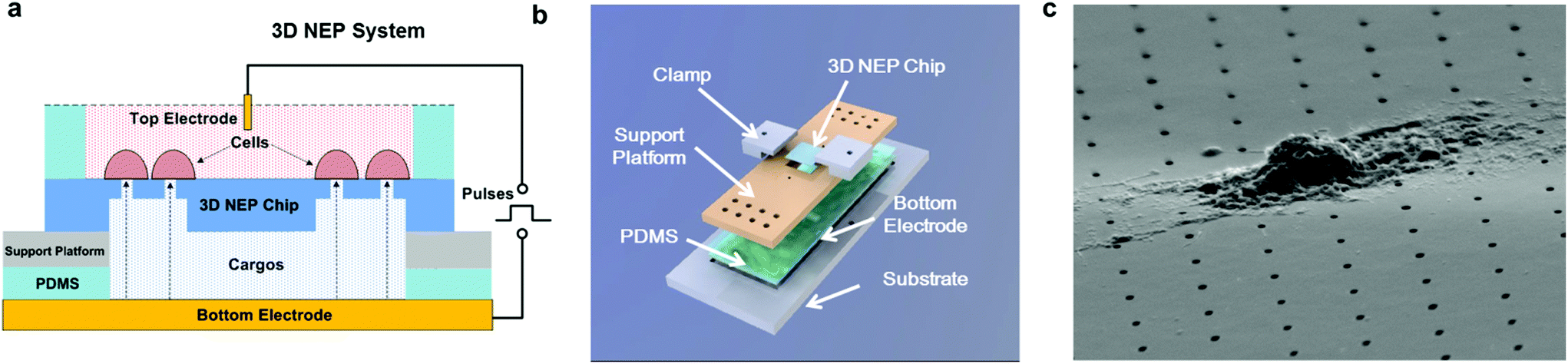 | ||
| Fig. 2 The system setup of 3D NEP. (a) The cross-sectional schematic of the 3D NEP system, which consists of a 3D NEP chip, a support platform, two PDMS spacers, and a bottom electrode. Cells are loaded in the top chamber and the bottom chamber is filled with to-be-injected cargos. The electric field (black dot arrows) is generated by the voltage pulses applied between the top planar electrode and the bottom electrode, by which the cells are electroporated while the cargos are injected into the cells aligned on the nano-pore array. (b) The 3D model showing all components are stacked on a substrate and boned with two clamps. (c) SEM micrograph of a cell loaded onto the nano-channel side of the 3D NEP chip ready for electroporation. | ||
Working conditions
In NEP, only a cell placed in close proximity of a nanopore can be locally electroporated. The applied voltage and the cell-to-channel distance was analyzed with a 3D NEP model using COMSOL, in order to identify the minimum applied bias (VA) that leads to a sufficient electroporation. The schematic with relevant parameters is seen in Fig. 3a where D is the cell diameter, d is the spacing between the cell and the nanochannel entrance, l is the nanochannel length, w is the nanochannel diameter and R is the micro-channel diameter. The system parameters in the simulation are set in accordance with the chip used in NEP experiments: D is set to 10 μm (an acceptable cell size), l to 13 μm (average nanochannel length), R to 50 μm and w to 650 nm. Other parameters including extracellular fluid (phosphate buffered saline, PBS) and cytosol conductivities are 0.8 S m−1 and 0.2 S m−1, respectively. The cellular membrane is estimated to be 5 nm thick with a conductivity of 5 × 10−7 S m−1.32 The governing equation for the system (static electric field) used is: ∇(σ∇V) = 0, where σ is the conductivity. The 3D model in Fig. 3b shows the overview of potential drop in the microchannel–nanochannel-cell system when the applied voltage is VA = 100 V, while Fig. 3c gives the 2-D slice of the potential distribution, where the vast majority (∼97%) of the potential drop is observed over the nanochannel. Fig. 3d zooms in the close proximity of nanochannel to the nearest point of the cell. A low potential of 3–3.5 V is distributed across the single-cell, which explains the reason for promising cell safety offered by NEP. Fig. 3e evaluates the effective potential imposed upon the cell at different conditions for VA. We observe that the voltage imposed upon by the cell is 3.4 V under 100 V, which is visualized by Fig. 3d. In this model, Vs is defined as the threshold voltage for membrane permeabilization, while Vm is the resting transmembrane potential.33 A successful cell electroporation must meet the condition of Vs > Vm. Based on previous research,34 1 V is assumed safe as a conservative estimation for Vm, according to which Fig. 3e indicates that the minimum voltage bias would need to be at least 50 V for successful electroporation within the system, yet the value of Vs = 0.5 V at VA = 15 V would likely be insufficient. Within this estimation, “d” is set to 200 nm, a considered proximate distance from cell to nanochannel opening. However, d is another factor having a significant impact on threshold voltage, as demonstrated in Fig. 3f. For instance, when d = 100 nm, the VA = 50 V can reach the Vs = 1 V mark. Nevertheless, this potential may be insufficient when d > 400 nm. This confirms the importance of localized electroporation in the NEP system. In this work, cells are loaded on the chip while a vacuum is applied underneath the NEP chip to drive the cell towards the nanopore for alignment and contact against the nanopore. In addition, both types of cells we used in this work are adherent cells which, naturally, attach on the chip surface and spread, as shown in Fig. 2c. Therefore, though it is difficult to realize “precise” gap control, a close-contact with d < 100 nm is always guaranteed for cells studied in this work. To handle suspension cells, other techniques, such as dielectrophoresis and magnetic tweezers, are under investigation to achieve efficient cell loading and immobilization on nanochannel arrays. To confirm the identified threshold voltage, H9C2 cells (mouse cardiomyoblasts) were delivered with FAM labelled oligonucleotides (FAM-ODN) by 3D NEP at both VA = 15 V and VA = 50 V. The immediate fluorescence images for both cases are seen in the inset of Fig. 3e. The VA = 15 V case shows little if any observable fluorescence, while using VA = 50 V, the internalized fluorescence intensity is significantly increased. These results imply: (1) 15 V is an insufficient VA even with a close proximity between a cell and a nanochannel; (2) transfection results of 50 V could vary, dependent on the condition of the cell close to the nanochannel.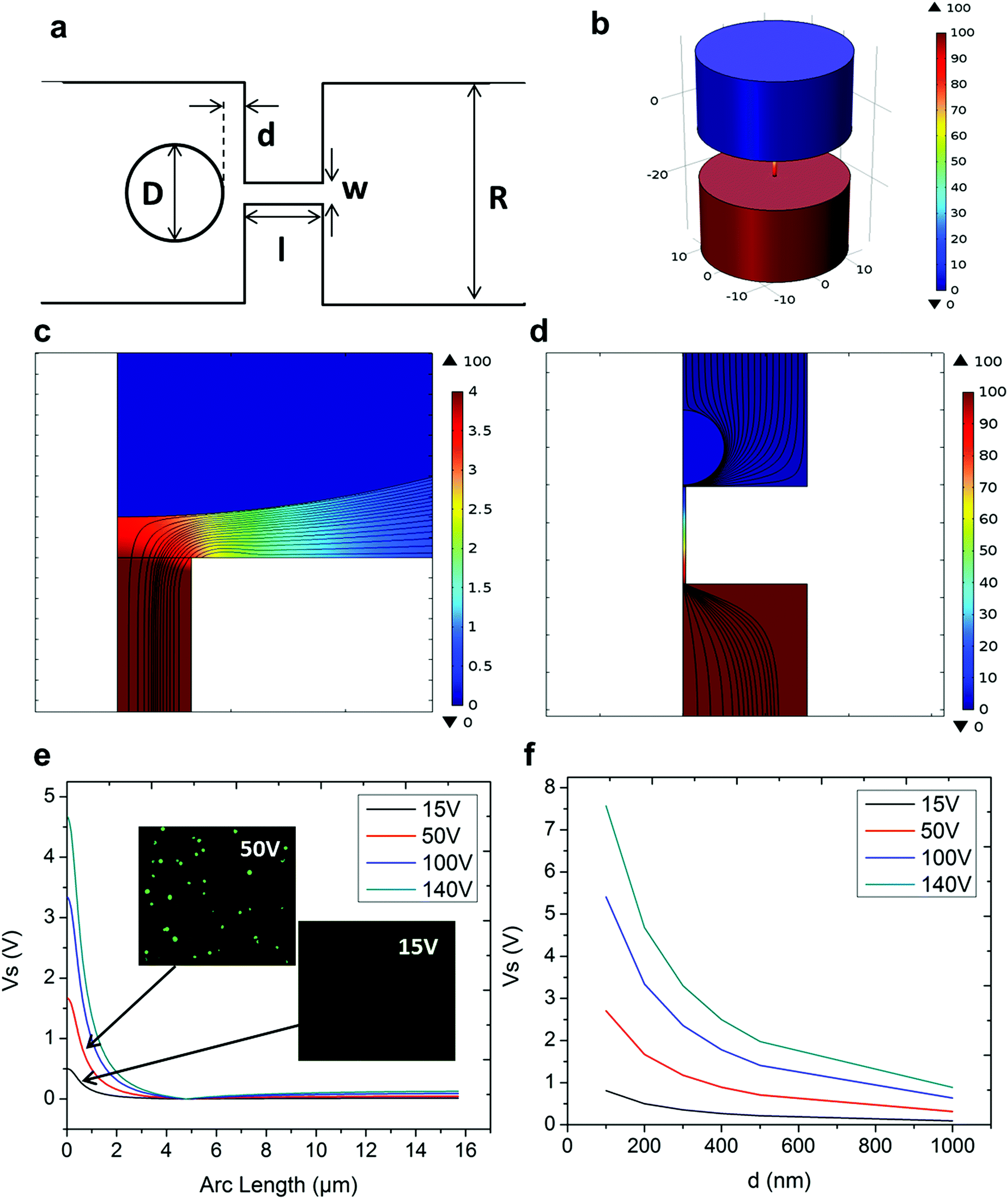 | ||
| Fig. 3 Working conditions of a 3D NEP device. (a) Illustration of a single nano-pore setup and relevant parameters for simulation. (b) 3D layout of a single nanochannel on the device under a biased voltage of 100 V. (c) Electrical field lines from an electroporation simulation at VA = 100 V. (d) Zoom-in of (c) showing field lines at the cell–nanochannel interface. (e) Effective voltage Vs as a function of arc length, where d = 200 nm. Simulation values for VA are 15, 50, 100, and 140 V. (f) Vs as a function of d at varied applied biases (VA), showing required VA for successful electroporation at various d values. | ||
We tested the delivery performance of 3D NEP under different voltages (pulse duration/number/interval were fixed at 10 ms/5 pulses/1 s). H9C2 cells were loaded onto the chip, and FAM-labelled oligonucleotides (FAM-ODN) were then injected into cells seated on the nanopores. Fig. 4a statistically compares the fluorescence intensities of transfected cells with different voltages. No significant difference is observed in the average fluorescence intensity of FAM under 15 V as compared with the cells in the control group (p = 0.188), which further confirms the conclusions given by the simulation. The intensities significantly increase under 50 V (p < 0.005), which gives a clue about the general threshold for sufficient electroporation in the 3D NEP system. When the voltage increases from 50 V to 100 V, the intensities are significantly increased, indicating that larger amounts of FAM-ODN were delivered into individual cells by NEP. Interestingly, increasing the voltage to 140 V does not further enhance the delivery outcomes. One explanation is that in the NEP environment, the electric field can only affect the membrane in contact with the nanochannel. Within the low-voltage range (e.g. from 0 V to 100 V), increasing the voltage could enlarge the size of the single nanopore, or increase the number of nanopores, generated on the cell membrane.35 However, the maximum size of the nanopore or the number of nanopores, occurs when the voltage reaches a saturated value (e.g. above 100 V). Beyond this threshold the high voltage (e.g. 200 V) does not facilitate delivery, but may cause excessive heat, damaging cells, if increased without prudence. Therefore, we used 100 V–140 V as a safe and efficient range, since no obvious joule heating and cell damage phenomena were observed at this biasing range.
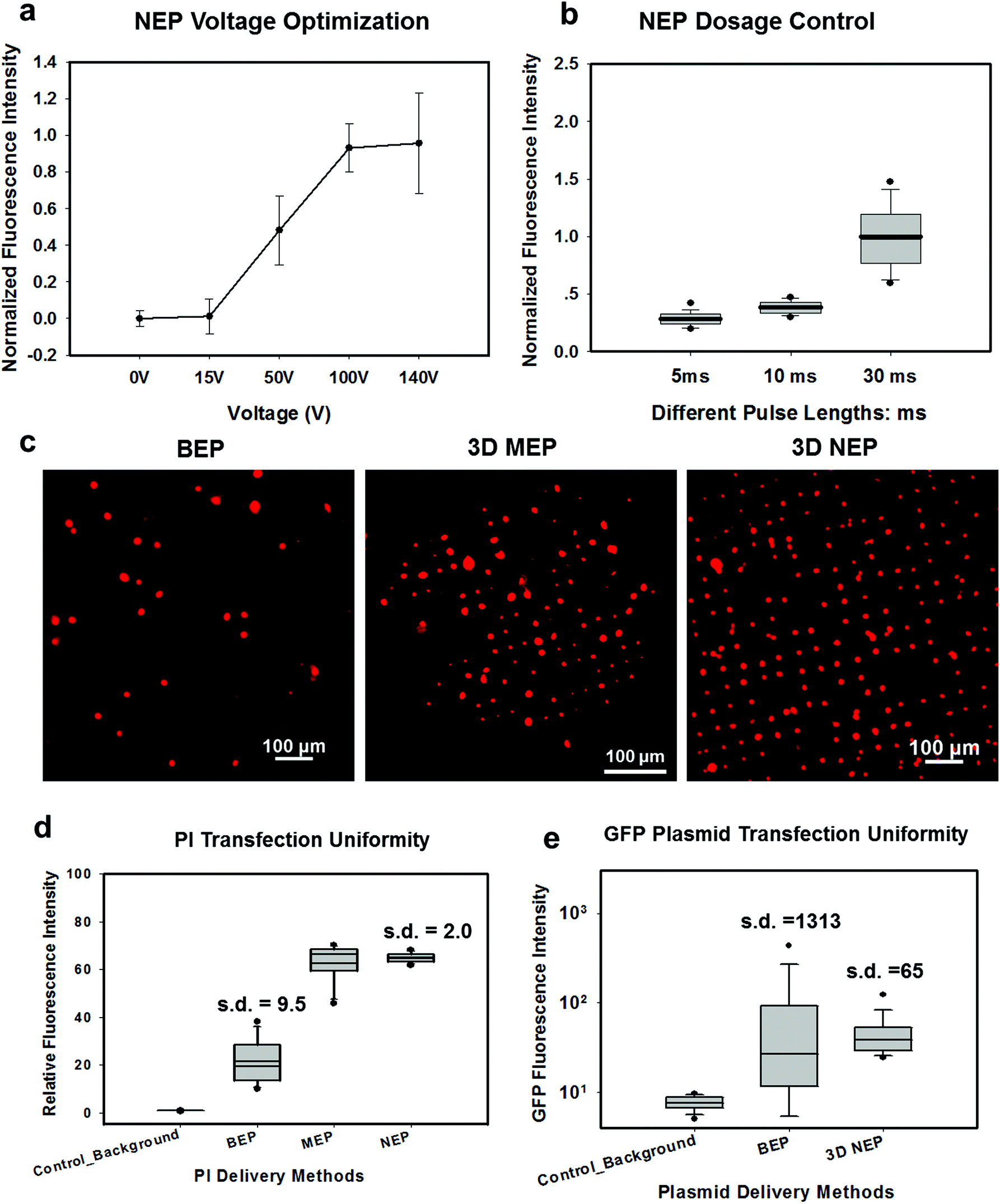 | ||
| Fig. 4 Dosage controllability and transfection uniformity of the 3D NEP for high-throughput cell transfection. (a) The normalized fluorescence intensities of transfected cells under different voltages in delivery of FAM-ODN, compared to the control group (counted cell number in each group, n = 100). (c) The fluorescence intensity of FAM-ODN delivered by NEP is directly proportional to the pulses lengths (n = 50); (C) a uniform transfection, with formation of a fluorescence spot array on a chip, is achieved within 1 min after electroporation by 3D NEP. In comparison, a stochastic and slow fluoresce induced by BEP took more than 30 min, while MEP required 10–30 minutes. (d) The comparison of BEP, 3D MEP, and 3D NEP on the delivery uniformity in the process of PI delivery (n = 400). (e) The comparison of BEP and 3D NEP on the transfection uniformity for GFP plasmid delivery (n = 1000). | ||
Dosage controllability in high throughput cell transfection
In Fig. 4b, we demonstrate the signature feature of 3D NEP, showing dosage control in conjunction with a high-throughput cell transfection. FAM-ODN was delivered into H9C2 cells under an applied bias of 140 V with three different pulse durations (5 ms, 10 ms and 30 ms). The internal fluorescence intensities are directly proportional to the pulse durations. The dosage controllability of NEP is due to the electrophoresis of biomolecules within the nanochannel under an electric field, which is independent of the diffusion when the applied pulse duration is within a small range. However, interestingly, we observed the variation of fluorescence intensity among individual cells increased as the pulse duration increased as shown in Fig. 4a. When the pulse is turned-off, the nanopores in the cell membrane may still last for a while (several ms to several seconds). The accumulation of the biomolecules in the vicinity of the cell would cause diffusion-based delivery in the final stage.36 So the dosage control is still realized by electrophoresis but the variation on dosage gets larger as the pulse duration increases. Depending on the pulse duration, the membrane recovery time could be significantly increased, providing an additional opportunity for molecules to diffuse into the cells.37,38Transfection uniformity
We investigated the transfection uniformity of the 3D NEP system compared to its reported high throughput counterparts, including both BEP and 3D MEP. The PI dye was injected into the loaded H9C2 cells with the given NEP conditions: 140 V/10 ms/2 pulses. In NEP, only cells localized on the array of nanopores were injected with the PI dye, forming a fluorescence spot-array after electroporation (Fig. 4c). Counting the time for image capture, the delivery process was completed in much lesser time than 1 minute, consistent with the results in 2D NEP.19 In contrast, commercial BEP (Neon Transfection System, Life technology) delivery results in a slow (lasting for 1 hour) but random fluorescing (with ∼10% transfection efficiency) among a large number of cells (Fig. 4c). We also tested 3D MEP for the delivery of the PI dye. Cells were trapped on the chip with a microchannel array (5 μm in diameter) using a vacuum and were electroporated with the protocol in previous work. 3D MEP also offers a high efficiency, as shown in Fig. 4c. Fig. 4d shows the performance analysis of 3D NEP, BEP and 3D MEP with respect to transfection uniformity. In each group, the fluorescence intensities of 400 randomly-picked individual cells are counted. 3D NEP shows significantly higher transfection uniformity (s.d. = 2) than that of BEP (s.d. = 9.5) and 3D MEP (s.d. = 8.1). Note that at low mean intensity, 3D MEP works well for intracellular delivery of small molecules too. We further investigated the efficacy of BEP and MEP for the delivery of large molecular weight plasmids (pmaxGFP, 3.5 kbp) compared to NEP. Fig. 4e analyzes the GFP fluorescence intensities of 1000 cells in BEP and NEP groups. 3D NEP has 20-fold better uniformity than BEP on plasmid delivery (s.d = 65 vs. s.d. = 1313), implying the superior performance of NEP in regard to transfection uniformity when attempting to transfect cells with large molecules. NEP also showed significantly higher efficiency for plasmid delivery as compared to MEP platforms. GFP fluorescence was not observed in 3D MEP experiments. When the applied voltage was increased to 10 V, however, all cells loaded on the micro-pores were lysed due to joule heating, consistent with the previous work on MEP.39,40Large weight plasmids delivery in primary cells
We further explored the outcomes specific to the delivery of large molecular weight plasmids (OCT-4, SOX2, KCL-1, and MYC transcription factors, OSKM encoded on a pCAG plasmid backbone, with 13 kbp) into the primary cells (mouse embryonic fibroblasts) on the 3D NEP platform. The plasmid labelled with YOYO-1 dye, is capable of improving the fluorescence intensity 1000-fold once it binds with dsDNA in either the cytosol or nucleus.36 We observed a strong YOYO-1 fluorescence signal co-localized with the cells 1 min after NEP (Fig. 5a), indicating the internalization of the plasmid. Fig. 5a also demonstrates that NEP provides significantly higher efficiency than BEP when delivering such large molecular weight cargos (∼10% vs. <1%). For NEP, it should be noted that as mentioned earlier, only cells seated on nanopores can be transfected, which is why the seemingly low transfection efficiency of 10%, as all cells loaded on the chips were counted rather than only those seated on nanopores. A cell manipulation technique for high-throughput cell trapping on a 3D NEP chip is under investigation now, which would realize efficient cell-array trapping, allowing a better alignment with the nanochannel array. If we count only cells localized on the nanopores, the transfection efficiency would be at ∼90%. The result of extremely low-efficiency obtained by BEP was consistent with previous work for OSKM plasmid delivery.41 We also evaluated the cell viability after electroporation. In the NEP group, 90% of transfected cells are still alive (Fig. 5b), attached and showed typical fibroblast morphologies 24 hours after electroporation (Fig. 5a). In comparison, in the BEP group, only 9% cells are still considered surviving while 91% cells display shrunken, spherical features and have detached from the cell culture dish, a typical behaviour of dead or dying cells (Fig. 5a). The reason for negligible cell damage with NEP has been discussed above and in previous work.19 The unique cell-nanochannel configuration shields the majority of potential drops, allowing a safe but sufficient voltage potential to porate cells. The extremely low efficiency of BEP supports the fact that BEP is a diffusion based process where without additional propulsion, the difficulty of cross-membrane transport is too great. This also suggests that for a successful BEP, an increase in the applied voltage would be required in order to improve delivery efficiency. However, this would cause irreversible electroporation to occur, leading to permanent cell damage.38 We also compared the fluorescence intensities between two groups, and NEP consistently shows significantly better uniformity (s.d. = 4306) than BEP (s.d. = 12347) (Fig. 5c). The OSKM plasmid was also cloned with a reporter gene (RFP) to verify if the plasmids were transcribed and translated in living cells. Fig. 5d shows RFP (red fluorescence) observed 24 hours after NEP, indicating the successful transcription of the OSKM factors. All cells showing RFP can be co-localized with those emitting YOYO-1 fluorescence, demonstrating that the RFP signal is real and reliable. However, only a portion of delivered cells (based on YOYO-1) showed the RFP signal, implying OSKM factors were not suitably expressed in some cells. Our hypothesis is that the current OSKM plasmid is an integrative backbone cloned with five functional genes (OCT-4, SOX2, KCL-1, MYC, and RFP), which increases the difficulties of correct expression in the nucleus.
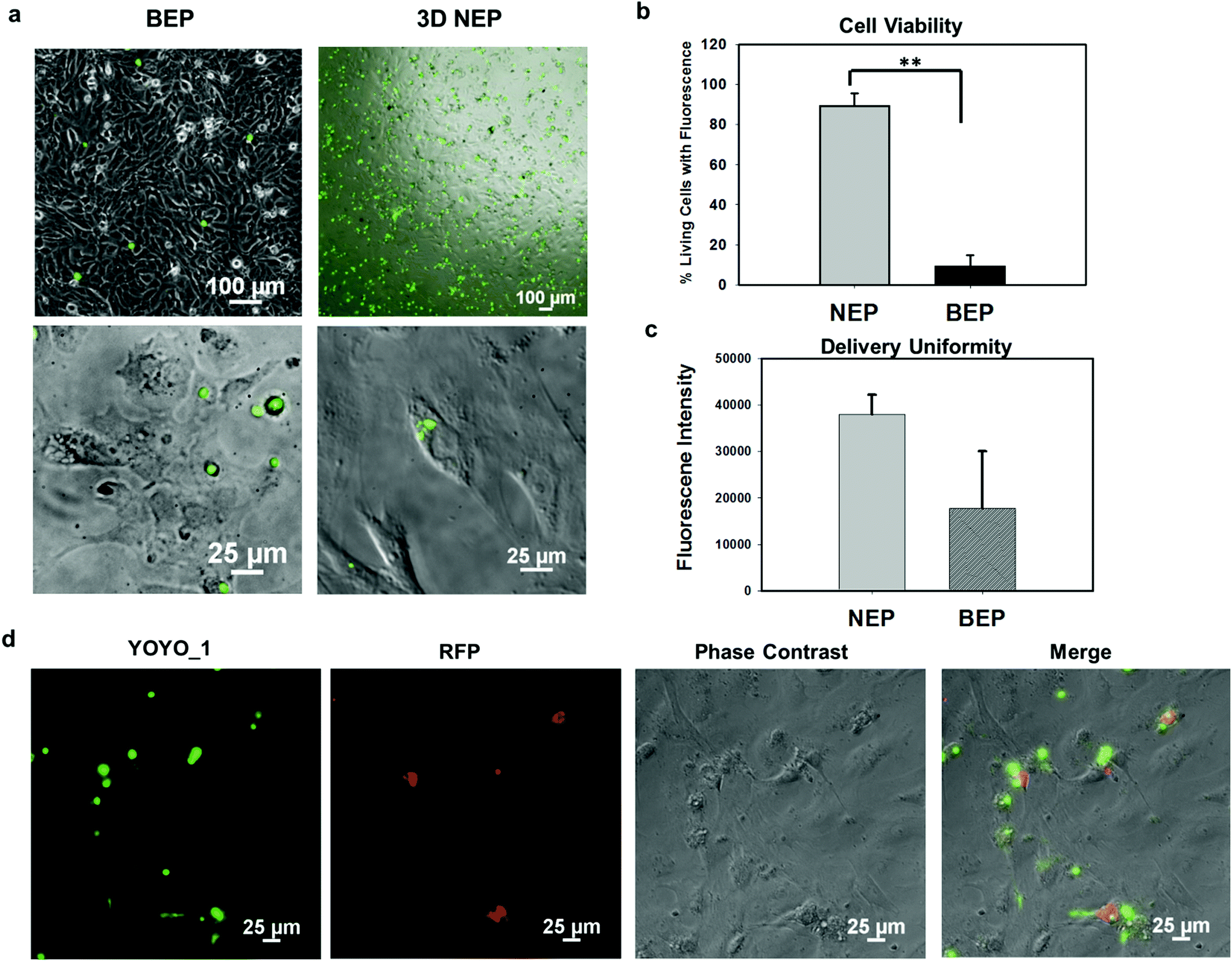 | ||
| Fig. 5 Delivery of MEFs with large molecular weight cargos (13 kbp OSKM plasmid labelled with YOYO-1 dye) using a 3D NEP platform and BEP. (a) The images merged with YOYO-1 green fluorescence and phase contrast show that the delivery efficiency of 3D NEP is significantly higher than that of BEP (the first panel). The zoom-in of individual cells demonstrates that cells transfected by NEP are still alive, with typical MEF morphologies, while most cells delivered by BEP show spherical morphologies and detach from dishes, indicating cell death (the second panel). (b) Statistics of cell viability between NEP and BEP (n = 100). (c) The transfection uniformity of NEP as compared to BEP, according to the internalized YOYO-1 fluorescence (n = 100). (d) Successful expression of OSKM transcriptional factors after 3D NEP. The RFP fluorescence (reporter gene encoded on OSKM plasmid) was observed 24 h after NEP, indicating the successful translation and transcription of OSKM factors. **: p < 0.01. | ||
Conclusions
The manipulation of cellular behaviour and programming may prove essential to the future of clinical medicine and therapeutics. The development of a highly reliable, controllable, and low-cost system is ideal to allow for a smooth transition from research to clinical medicine. BEP, while commercially available and easily performed, is extremely detrimental to cell function and longevity. While BEP allows for high-throughput, it is unable to achieve high cell viability and any dosage control. MEP improves upon this system by being able to achieve high-throughput and higher cell viability, but it is still unable to achieve good dosage control and uniform transfection and to deliver large molecular weight cargos. The 3D NEP system demonstrated here allows for high-throughput cell transfection with high cell viability, dosage control, and high uniformity. These features promote the qualities of gene delivery and transfection in gene therapy, adoptive immunotherapy and cell reprogramming. This platform also offers high efficiency and cell viability in delivering cells with very large molecular weight cargos (e.g. 13 kbp OSKM plasmids) which cannot be achieved with comparable outcomes by either MEP or BEP. The 3D NEP system used in this experiment can handle 40000 cells on a single chip with 1 cm2 area. If a larger number of cells are required, the demonstrated approach can be readily scaled up to deal with millions of cells per wafer.
Author contribution statement
W.L. and L.J.L. conceived and designed the experiments. L.C., P.B., D.G.-P., and V.M. conducted 3D NEP chip fabrication. L.C., F.C., T.K. assembled the 3D NEP system and conducted 3D NEP experiment. Z.Y. and C.-L.C. assisted with cell culture and bio-statistics. P.B. performed the simulation. L.C. and P.B. made the data analysis and wrote the paper. W.L. and L.J.L. revised and finalized the paper.Acknowledgements
The authors are grateful to Mark Brenner from ECE Department at OSU for providing technical support of DRIE technique, and Cheng Kang from the Department of Pharmacology for providing cells. The authors thank the National Science Foundation (EEC-0914790) for supporting this study.Notes and references
- M. Costa, M. Dottori, K. Sourris, P. Jamshidi, T. Hatzistavrou, R. Davis, L. Azzola, S. Jackson, S. M. Lim, M. Pera, A. G. Elefanty and E. G. Stanley, Nat. Protoc., 2007, 2, 792–796 CrossRef CAS PubMed.
- K. Kaji, K. Norrby, A. Paca, M. Mileikovsky, P. Mohseni and K. Woltjen, Nature, 2009, 458, 771–U112 CrossRef CAS PubMed.
- L. Naldini, U. Blomer, P. Gallay, D. Ory, R. Mulligan, F. H. Gage, I. M. Verma and D. Trono, Science, 1996, 272, 263–267 CAS.
- R. King, Methods Mol. Biol., 2004, 245, 167–174 CAS.
- H. Chantrenne, Nature, 1977, 269, 202–202 CrossRef CAS PubMed.
- S. Mehier-Humbert and R. H. Guy, Adv. Drug Delivery Rev., 2005, 57, 733–753 CrossRef CAS PubMed.
- M. Zernicka-Goetz, Nature, 2000, 405, 733–733 CrossRef CAS PubMed.
- U. K. Tirlapur and K. Konig, Nature, 2002, 418, 290–291 CrossRef CAS PubMed.
- P. Chakravarty, W. Qian, M. A. El-Sayed and M. R. Prausnitz, Nat. Nanotechnol., 2010, 5, 607–611 CrossRef CAS PubMed.
- E. Kang, J. Ryoo, G. S. Jeong, Y. Y. Choi, S. M. Jeong, J. Ju, S. Chung, S. Takayama and S. H. Lee, Adv. Mater., 2013, 25, 2167–2173 CrossRef CAS PubMed.
- M. R. Prausnitz, V. G. Bose, R. Langer and J. C. Weaver, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 10504–10508 CrossRef CAS.
- F. Liu, S. Heston, L. M. Shollenberger, B. Sun, M. Mickle, M. Lovell and L. Huang, J. Gene. Med., 2006, 8, 353–361 CrossRef PubMed.
- S. Y. Ho and G. S. Mittal, Crit. Rev. Biotechnol., 1996, 16, 349–362 CrossRef CAS PubMed.
- L. H. Li, M. Wood, R. Shivakumar, S. Feller, S. Wang, V. Singh, J. Holaday, J. Fratantoni and L. N. Liu, Blood, 2001, 98, 425a–425a Search PubMed.
- R. P. Joshi and K. H. Schoenbach, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 2002, 66 Search PubMed.
- Y. C. Wu, T. H. Wu, D. L. Clemens, B. Y. Lee, X. Wen, M. A. Horwitz, M. A. Teitell and P. Y. Chiou, Nat. Methods, 2015, 12(4), 439–444 CrossRef CAS PubMed.
- B. Marrero and R. Heller, Biomaterials, 2012, 33, 3036–3046 CrossRef CAS PubMed.
- E. G. Guignet and T. Meyer, Nat. Methods, 2008, 5, 393–395 CrossRef CAS PubMed.
- P. E. Boukany, A. Morss, W. C. Liao, B. Henslee, H. C. Jung, X. L. Zhang, B. Yu, X. M. Wang, Y. Wu, L. Li, K. L. Gao, X. Hu, X. Zhao, O. Hemminger, W. Lu, G. P. Lafyatis and L. J. Lee, Nat. Nanotechnol., 2011, 6, 747–754 CrossRef CAS PubMed.
- W. Kang, J. P. Giraldo-Vela, S. S. P. Nathamgari, T. McGuire, R. L. McNaughton, J. A. Kessler and H. D. Espinosa, Lab Chip, 2014, 14, 4486–4495 RSC.
- T. Geng and C. Lu, Lab Chip, 2013, 13, 3803–3821 RSC.
- T. Geng, Y. Zhan, J. Wang and C. Lu, Nat. Protoc., 2011, 6, 1192–1208 CrossRef CAS PubMed.
- L. Yan, J. Zhang, C. S. Lee and X. Chen, Small, 2014, 10, 4487–4504 CrossRef CAS PubMed.
- K. L. Gao, L. Li, L. N. He, K. Hinkle, Y. Wu, J. Y. Ma, L. Q. Chang, X. Zhao, D. G. Perez, S. Eckardt, J. Mclaughlin, B. Y. Liu, D. F. Farson and L. J. Lee, Small, 2014, 10, 1015–1023 CrossRef CAS PubMed.
- W. M. Kang, F. Yavari, M. Minary-Jolandan, J. P. Giraldo-Vela, A. Safi, R. L. McNaughton, V. Parpoil and H. D. Espinosa, Nano Lett., 2013, 13, 2448–2457 CrossRef CAS PubMed.
- C. Xie, Z. Lin, L. Hanson, Y. Cui and B. Cui, Nat. Nanotechnol., 2012, 7, 185–190 CrossRef CAS PubMed.
- X. Xie, A. M. Xu, S. Leal-Ortiz, Y. H. Cao, C. C. Garner and N. A. Melosh, ACS Nano, 2013, 7, 4351–4358 CrossRef CAS PubMed.
- Z. Z. Fei, S. N. Wang, Y. B. Xie, B. E. Henslee, C. G. Koh and L. J. Lee, Anal. Chem., 2007, 79, 5719–5722 CrossRef CAS PubMed.
- Z. Z. Fei, X. Hu, H. W. Choi, S. N. Wang, D. Farson and L. J. Lee, Anal. Chem., 2010, 82, 353–358 CrossRef CAS PubMed.
- L. Chang, M. Howdyshell, W. C. Liao, C. L. Chiang, D. Gallego-Perez, Z. Yang, W. Lu, J. C. Byrd, N. Muthusamy, L. J. Lee and R. Sooryakumar, Small, 2015, 11, 1818–1828 CrossRef CAS PubMed.
- Y. C. Liu, W. Y. Lin, Y. R. Jhang, S. H. Huang, C. P. Wu and H. T. Wu, Appl. Biochem. Biotechnol., 2011, 164, 1172–1182 CrossRef CAS PubMed.
- T. Kotnik, F. Bobanovic and D. Miklavcic, Bioelectrochem. Bioenerg., 1997, 43, 285–291 CrossRef CAS.
- Y. G. Man, T. Vinh, C. Zhao, A. Walker and R. Barner, Mod. Pathol., 2005, 18, 42a Search PubMed.
- G. Sersa, T. Jarm, T. Kotnik, A. Coer, M. Podkrajsek, M. Sentjurc, D. Miklavcic, M. Kadivec, S. Kranjc, A. Secerov and M. Cemazar, Br. J. Cancer, 2008, 98, 388–398 CrossRef CAS PubMed.
- S. Movahed and D. Q. Li, J. Colloid Interface Sci., 2012, 369, 442–452 CrossRef CAS PubMed.
- C. Gupta, W. C. Liao, D. Gallego-Perez, C. E. Castro and L. J. Lee, Biomicrofluidics, 2014, 8 Search PubMed.
- S. I. E. Lin, Adv. Mater. Res., 2012, 586, 421–429 CrossRef CAS.
- A. Golberg and M. L. Yarmush, IEEE Trans. BioMed. Eng., 2013, 60, 707–714 CrossRef PubMed.
- Y. Huang and B. Rubinsky, Sens. Actuators, A, 2001, 89, 242–249 CrossRef CAS.
- A. Valero, J. N. Post, J. W. van Nieuwkasteele, P. M. Ter Braak, W. Kruijer and A. van den Berg, Lab Chip, 2008, 8, 62–67 RSC.
- M. C. N. Marchetto, G. W. Yeo, O. Kainohana, M. Marsala, F. H. Gage and A. R. Muotri, PLoS One, 2009, 4 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5nr03187g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |