
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew dinuclear hydrido-carbonyl rhenium complexes designed as photosensitizers in dye-sensitized solar cells
Lorenzo
Veronese
a,
Elsa Quartapelle
Procopio
a,
Francesca
De Rossi
 b,
Thomas M.
Brown
b,
Pierluigi
Mercandelli
acd,
Patrizia
Mussini
a,
Giuseppe
D'Alfonso
ac and
Monica
Panigati
*acd
b,
Thomas M.
Brown
b,
Pierluigi
Mercandelli
acd,
Patrizia
Mussini
a,
Giuseppe
D'Alfonso
ac and
Monica
Panigati
*acd
aDipartimento di Chimica, Università degli Studi di Milano, Via Golgi, 19, 20133, Milano, Italy. E-mail: monica.panigati@unimi.it
bCHOSE, Dipartimento di Ingegneria Elettronica, Università degli Studi di Roma Tor Vergata, Via del Politecnico 1, 00133 Roma, Italy
cConsorzio Interuniversitario Nazionale per la Scienza e Tecnologia dei Materiali (INSTM), Via G. Giusti 9, 50121 Firenze, Italy
dIstituto per lo Studio delle Macromolecole, Consiglio Nazionale delle Ricerche (ISMAC-CNR), Via E. Bassini, 15, 20133 Milano, Italy
First published on 25th January 2016
Abstract
The possible use of some dinuclear rhenium complexes as sensitizers for dye sensitized solar cells (DSSCs) has been investigated. They have general formula [Re2(μ-X)(μ-Y)(CO)6(μ-pyridazine-4-COOH)], with X = Y = Cl (1), X = H, Y = benzoato (2), and X = H, Y = 4-diphenylaminobenzoato (3). An original synthetic strategy has been set for preparing the hydrido-carboxylato derivatives 2 and 3. They have been indicated by DFT and TD-DFT computations as the most promising dyes, endowed with good light harvesting capability. The complexes have absorption maxima in the range of 405–443 nm, on TiO2 films, arising from metal-to-ligand-charge transfer transitions. Cyclic voltammetry experiments have been performed on the derivatives containing the methyl ester of the pyridazine-4-COOH acid, showing electrochemical band gaps in the range of 2.25–1.63 eV. The best DSSC results have been obtained using complex 3, with an overall solar-to-electric conversion efficiency of 1.0%. Noteworthy the presence of a hydrido ligand did not show any detrimental effect on the stability of the sensitizers under the operating conditions.
Introduction
A new a family of dinuclear rhenium complexes with general formula [Re2(μ-X)(μ-Y)(CO)6(μ-diazine)], containing two “Re(CO)3” units connected by a bridging 1,2-diazine ligand and two anionic ancillary ligands, such as halides,1 hydrides,2 alcohoxides or sulphides,3 has been recently reported. Upon optical excitation, many of these complexes exhibit broad and featureless photoluminescence, centred between 520 and 660 nm, arising from the lowest-lying triplet-manifold excited state (T1 → S0), with metal-to-ligand charge transfer (3MLCT) nature. Emission quantum yields up to 0.5, both in solution and in the solid state, have been measured for some members of this family,4 which have also found applications in OLED devices as a dopant in the emissive layer.5Moreover very recently the first molecular dyad containing a dinuclear rhenium(I) moiety has been reported.6 In this species a [Re2(μ-Cl)2(CO)6(μ-pyridazine)] complex constitutes the chromophoric subunit, covalently linked to a fullerene unit by a carbocyclic molecular bridge. In this dyad the rhenium complex does not exhibit any emission, since its strongly emissive triplet charge-transfer state is efficiently quenched by the fullerene subunit, via oxidative electron transfer, with a time constant of about 110 ps.
These results prompted us to investigate the possible use of dinuclear rhenium complexes as dyes in dye sensitized solar cells (DSSCs).7–9 Actually sensitizers suitable for such applications must be able to transfer electrons from an excited state to the conductive band of an oxide (most often TiO2) acting as the anode of the solar cell. The most commonly used dyes are ruthenium complexes containing poly-pyridine ligands functionalized with carboxylic substituents for anchoring to TiO2.10 These complexes exhibit broad absorptions, due to MLCT transitions, long excited state lifetime, and high chemical stability.11 The highest efficiency reported for small area DSSCs based on the ruthenium dye and iodide/triiodide electrolyte is 12.1%,12,13 while an efficiency as high as 13% was achieved using a molecularly engineered porphyrin dye in combination with a cobalt-based electrolyte.14
Alternative dyes based on different metal complexes, such as iron,15 copper,16 and platinum,17 mainly containing bipyridine ligands, have been investigated so far.18 In this framework, the use of rhenium polypyridine complexes has not been largely investigated. Indeed, beside some preliminary studies concerning the electron transfer from Re polypyridyl complexes to nanocrystalline TiO2,19,20 only one study briefly exploring the photovoltaic performances of tricarbonyl rhenium complexes can be found.21
The dirhenium complexes here presented (Chart 1) have been designed by the use of combined density functional (DFT) and time-dependent density functional (TD-DFT) studies. All of them show 1MLCT transitions, in which the excited electron is transferred to the diazine ligand, where the LUMO is mainly localized.1–4 The diazine has been functionalized by a carboxylic group, to allow the anchoring of the dye to the TiO2 surface and the fast injection of the electron from the diazine to the TiO2 conduction band. The ancillary ligands have been selected in order to maximize the light harvesting capability of the dye. The computational studies showed that complexes containing one hydrido and one benzoate bridging ligands were the most promising dyes. The synthesis of hydrido-carboxylato derivatives required the setting of a new general synthetic procedure, which has been exploited for the preparation of complexes 2 and 3 of Chart 1. Their electrochemical and spectroscopical properties have been measured and compared with those of the previously known complex 1, containing two bridging chloride anions. Finally, solar cells with each of the three complexes have been prepared and their performances are discussed herein, in relation to the molecular structure of the dyes and to the nature of the ancillary ligands.
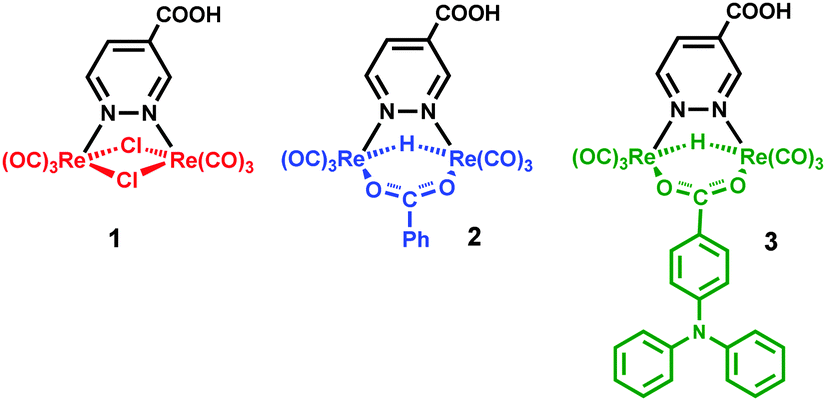 | ||
| Chart 1 Molecular structure of the three dyes. | ||
Experimental section
Synthesis
All reactions were performed under a nitrogen atmosphere unless specified otherwise. All solvents were deoxygenated and dried by standard methods before use, while toluene has been distilled on Na(s)-benzophenone and dichloromethane on P2O5, both under a N2 atmosphere. The reagents were purchased from Aldrich, Fluka and Lancaster and used as received. Commercial deuterated solvents were used as received. Column chromatography was performed using Alfa Aesar silica gel 60 (0.032–0.063 mm). [Re4(μ3-H)4(CO)12](C6H6)2,22 [Re2(μ-H)2(CO)6(μ-ppd)] (4, ppd = 2,5-diphenyl-1,3,4-oxadiazole)23 and [Re2(μ-Cl)2(CO)6(μ-pydz-4-COOH)] (1, pydz-4-COOH = pyridazine-4-carboxylic acid)24 were synthesized according to the literature procedures. 1H NMR spectra were recorded on Bruker DRX-300 and Bruker DRX-400 NMR spectrometers. Solution and thin-film UV/Vis absorption spectra were obtained using an Agilent 8453 spectrophotometer, fused quartz cuvettes (10 mm optical path), and CHROMASOL V grade solvents. IR spectra were acquired on a Bruker Vector 22 FT spectrophotometer. MS spectrum of 1 was carried out on a Finnigan™ LCQ™ Advantage MAX ion trap instrument using methanol as the solvent (ESI-MS: m/z 736). FAB spectrum of 3 was carried out on a mass spectrometer VG Autospec M246 using nitrobenzyl alcohol as the matrix.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), then dried under vacuum, yielding 30 mg (0.034 mmol) of white powder (isolated yield 65%). IR (CH2Cl2) ν(CO): 2041 (m), 2022 (vs), 1935 (vs), 1914 (s) cm−1. 1H NMR (CD2Cl2, 300K, 400 MHz) δH (ppm) 8.15 (d, 4H, Hortho), 7.87 (t, 2H, Hpara), 7.77 (t, 4H, Hmeta), 8.03 (d, 2H, Hortho), 7.51 (t, 1H, Hpara), 7.41 (dt, 2H, Hmeta), −7.08 (s, 1H, hydride). Elemental anal. calcd for C27H16N2O9Re2: C 36.65, H 1.82, N 3.17. Found: C 36.70, H 1.96, N 3.14.
:1, a pale yellow precipitate appears, then stirred for 30 minutes in an ice bath, and filtered again. The resulting solution was dried under vacuum affording 270 mg (0.93 mmol) of pure microcrystalline product (isolated yield 50%). 1H NMR (CD2Cl2, 300 K, 400 MHz) δH (ppm) 7.91 (d, 2H), 7.35 (t, 4H), 7.17 (m, 6H), 7.01 (d, 2H).
:1), affording the desired product as white powder (32 mg, 0.034 mmol, isolated yield 55%). IR (CH2Cl2) ν(CO): 2041 (m), 2022 (vs), 1936 (vs), 1915 (s) cm−1. 1H NMR (CD2Cl2, 300 K, 400 MHz) δH (ppm) 8.16 (d, 4H), 7.77 (m, 4H), 7.89 (t, 2H), 7.84 (dd, 2H), 7.32 (t, 4H), 7.14 (m, 6H), 6.95 (dd, 2H), −7.09 (s, 1H, hydride). Elemental anal. calcd for C39H25N3O9Re2: C 44.52, H 2.40, N 3.99. Found: C 44.93, H 2.45, N 3.96.
:1), then dried under vacuum, yielding 30 mg (0.034 mmol) of white powder (isolated yield 65%). IR (CH2Cl2) ν(CO): 2041 (m), 2022 (vs), 1935 (vs), 1914 (s) cm−1. 1H NMR (CD2Cl2, 300K, 400 MHz) δH (ppm) 8.15 (d, 4H, Hortho), 7.87 (t, 2H, Hpara), 7.77 (t, 4H, Hmeta), 8.03 (d, 2H, Hortho), 7.51 (t, 1H, Hpara), 7.41 (dt, 2H, Hmeta), −7.08 (s, 1H, hydride). Elemental anal. calcd for C27H16N2O9Re2: C 36.65, H 1.82, N 3.17. Found: C 36.70, H 1.96, N 3.14.
:1, a pale yellow precipitate appears, then stirred for 30 minutes in an ice bath, and filtered again. The resulting solution was dried under vacuum affording 270 mg (0.93 mmol) of pure microcrystalline product (isolated yield 50%). 1H NMR (CD2Cl2, 300 K, 400 MHz) δH (ppm) 7.91 (d, 2H), 7.35 (t, 4H), 7.17 (m, 6H), 7.01 (d, 2H).
:1), affording the desired product as white powder (32 mg, 0.034 mmol, isolated yield 55%). IR (CH2Cl2) ν(CO): 2041 (m), 2022 (vs), 1936 (vs), 1915 (s) cm−1. 1H NMR (CD2Cl2, 300 K, 400 MHz) δH (ppm) 8.16 (d, 4H), 7.77 (m, 4H), 7.89 (t, 2H), 7.84 (dd, 2H), 7.32 (t, 4H), 7.14 (m, 6H), 6.95 (dd, 2H), −7.09 (s, 1H, hydride). Elemental anal. calcd for C39H25N3O9Re2: C 44.52, H 2.40, N 3.99. Found: C 44.93, H 2.45, N 3.96.
Electrochemical measurements
The cyclovoltammetric study of the complexes has been performed at scan rates typically ranging from 0.02 to 10 V s−1 in HPLC-grade acetonitrile (MeCN) solutions at 2.5 × 10−4 M concentration in each substrate, deaerated by N2 bubbling, with 0.1 M TBAPF6 (TBA = NBu4, Aldrich) as the supporting electrolyte, at 298 K. The ohmic drop has been compensated by the positive feedback technique. The experiments were carried out using an AUTOLAB PGSTAT potentiostat (EcoChemie, The Netherlands) run by a PC with GPES software. The working electrode was a glassy carbon one (AMEL, diameter 1.5 mm) cleaned by diamond powder (Aldrich, diameter 1 μm) on a wet cloth (STRUERS DP-NAP); the counter electrode was a platinum wire; the reference electrode was an aqueous saturated calomel electrode, having in our working medium a difference of −0.385 V vs. the Fc+|Fc couple (the intersolvent redox potential reference currently recommended by IUPAC).Computational details
Ground state geometries were optimized by means of density functional calculations. The parameter-free hybrid functional PBE025,26 was employed along with the standard valence double-ζ polarized basis set 6-31G(d,p) for C, H, Cl, N, O and S. For Re the Stuttgart–Dresden effective core potentials were employed along with the corresponding valence triple-ζ basis set. Preliminary calculations were done without imposing any symmetry. However, almost all the optimized structures are symmetric (apart from 2, 3 and TPA-COOH), as reported in Table 1. The nature of all the stationary points was checked by computing vibrational frequencies and all the geometries were found to be true minima. In order to simulate the absorption electronic spectrum down to 300 nm the lowest 20 singlet excitation energies were computed by means of time-dependent density functional calculations. For complexes 1–3 calculations were done also in the presence of solvent (toluene, used in the photophysical characterizations) described by the conductor-like polarizable continuum model (CPCM). All the calculations were done with Gaussian 09.27| Compound | E HOMO [eV] | E LUMO [eV] | ΔE [eV] | λ [nm, eV] (f) | Transitiona |
|---|---|---|---|---|---|
| a A description of the electronic transition in terms of one-electron excitation between the pair of ground-state orbitals mainly involved (H = HOMO, L = LUMO). b π–π* transition. | |||||
| C s-[Re2Cl2(CO)6(pydz-4-COOH)] (1) | −6.91 | −3.99 | 2.92 | 428, 2.90 (0.178) | H−3 → L |
| C 1-[Re2H(OOCPh)(CO)6(pydz-4-COOH)] (2) | −6.53 | −3.78 | 2.75 | 526, 2.36 (0.118) | H−1 → L |
| C 1-[Re2H(OOC-TPA)(CO)6(pydz-4-COOH)] (3) | −5.48 | −3.71 | 1.77 | 530, 2.34 (0.079) | H−3 → L |
| 344, 3.60 (0.672)b | H → L+2 | ||||
| C 2v-[Re2Cl2(CO)6(pydz)] | −6.84 | −3.52 | 3.32 | 385, 3.22 (0.136) | H−3 → L |
| C 2v-[Re2(OPh)2(CO)6(pydz)] | −6.31 | −3.36 | 2.95 | 387, 3.20 (0.130) | H−9 → L |
| C 2-[Re2(SPh)2(CO)6(pydz)] | −6.27 | −3.22 | 3.05 | 415, 2.98 (0.059) | H−5 → L |
| C 2-[Re2(OOCPh)2(CO)6(pydz)] | −6.50 | −3.19 | 3.32 | 446, 2.78 (0.091) | H−2 → L |
| 411, 3.01 (0.090) | H−4 → L | ||||
| C s-[Re2HCl(CO)6(pydz)] | −6.69 | −3.45 | 3.24 | 406, 3.06 (0.113) | H−3 → L |
| C s-[Re2H(OPh)(CO)6(pydz)] | −6.39 | −3.38 | 3.00 | 397, 3.12 (0.096) | H−6 → L |
| C s-[Re2H(SPh)(CO)6(pydz)] | −6.27 | −3.29 | 2.97 | 386, 3.21 (0.128) | H−1 → L+1 |
| C s-[Re2H(OOCPh)(CO)6(pydz)] | −6.44 | −3.31 | 3.13 | 469, 2.64 (0.117) | H−1 → L |
| TPA-COOH | −5.48 | −1.04 | 4.44 | 327, 3.79 (0.473)b | H → L |
Fabrication and characterization of DSSCs
FTO glass substrates (Pilkington, 8 U sq−1) were ultrasonically cleaned sequentially in acetone and in isopropyl alcohol for 10 min and then immersed in a 40 mM aqueous solution of TiCl4 at 70 °C for 30 min. The as treated substrates were rinsed with deionized water and ethanol and dried at room temperature. An opaque TiO2 single-layer was screen-printed on clean FTO glass substrates using a commercial TiO2 paste (18 NR-AO paste, Dyesol). Over each 4 × 2 cm2 glass sample, three 8 μm-thick TiO2 active areas of 0.5 × 0.5 cm2 were deposited and electrically isolated one from another by laser scribing the FTO. The electrodes were then sintered in an oven with a final step at 525 °C for 30 min. The sintered photoanodes were left in a 40 mM aqueous solution of TiCl4 at 70 °C for 30 min. After rinsing with deionized water and ethanol, the samples were treated at 500 °C for 30 min. For each dye, two 0.3 mM ethanol solutions were prepared, respectively, with and without adding 3 mM of chenodeoxycholic acid (CDCA from Sigma Aldrich). After 2 h of immersion in the dye solution, photoelectrodes were assembled with platinized counter electrodes, prepared by pyrogenic decomposition at 420 °C for 15 min of a screen-printed platinum-based paste (Pt10 004F-05, Chimet), using a thermoplastic gasket (Surlyn 25, Solaronix). For comparison, a reference sample was sensitized by a commercial N719 dye (Dynamo AB). Finally, either a commercial electrolyte (Dyesol's EL-HSE High Stability Electrolyte) or the EL14 electrolyte, containing smaller amounts of the redox couple I−/I3−, was injected by vacuum back-filling. All devices were assembled in air and no secondary encapsulants were utilized. Photovoltaic performance of the cells was measured using a computer-controlled Keithley 2420 source meter, under a solar simulator (SolarTest 1200 KHS Class B) at 1000 W m−2, AM 1.5. The incident power was measured using a Skye SKS 1110 sensor; before measurement, a black 0.7 × 0.7 cm2 adhesive mask was applied over each cell to avoid overestimations by light collection from areas surrounding the active area of the cell. Incident photon-to-current efficiency (IPCE) measurements were carried out using a set-up consisting of a 150 W xenon lamp (Newport Model 70612) coupled with a monochromator (Cornerstone 130) and a Keithley 2420 source meter.Results and discussion
Computational study
DFT and TD-DFT computations have been previously used to investigate in detail the frontier orbitals and the electronic transitions of many [Re2(μ-X)(μ-Y)(CO)6(μ-diazine)] complexes.1–4 None of them contained a carboxylic group on the bridging diazine ligand, therefore the first aim of the present study was to understand the effects caused by the presence of a COOH substituent on the diazine. The β position was chosen, since, as previously reported,1 the presence of substituents in the α position has detrimental effects on the stability of the complex, due to the steric hindrance with the equatorial carbonyl ligands.To enlighten the effects of the COOH substituent on the molecular energy levels, we have investigated the [Re2(μ-Cl)2(CO)6(μ-pydz-4-COOH)] complex (1) and compared its data with those of the progenitor [Re2(μ-Cl)2(CO)6(μ-pydz)] complex.1
In agreement with the results previously reported for some other members of this family of complexes,1,4b DFT calculations on 1 show that the six highest occupied molecular orbitals correspond to the “t2g” set of the two rhenium atoms in a pseudo-octahedral environment (three of them are combination of non-bonding d orbitals and the remaining three are Re-(μ-Cl) π* orbitals) while the two lowest unoccupied molecular orbitals are the lowest-lying π* orbitals of the diazine. These orbitals lie at much lower energy than in the parent compound containing unsubstituted pyridazine (−0.47 eV, Table 1), in accordance with the electron-withdrawing nature of the carboxylic group. In agreement with the nature of the frontier orbitals, the HOMO level is poorly affected (−0.07 eV).
The orbitals involved in the strongest MLCT electronic transition computed for 1 (see Table 1) are represented in Fig. 1. This transition is red-shifted with respect to the corresponding transition computed for the pyridazine derivative, due to the stabilization of the LUMO. In addition, Fig. 1 clearly shows that the LUMO in 1 is delocalized over the carboxylic substituent, used to anchor the sensitizer to the nanocrystalline TiO2, possibly allowing a fast charge-injection process into the surface. We have then investigated the possibility to further shift to longer wavelengths the absorption band, in order to increase the light harvesting ability of the dye.
 | ||
| Fig. 1 Isodensity surface plots of some relevant molecular orbitals of [Re2(μ-Cl)2(CO)6(μ-pydz-4-COOH)] (1): HOMO−3 (left) and LUMO (right). | ||
Taking into account the possibility to tune the energy-gap for these species not only modifying the substituents on the diazine (i.e. shifting the LUMOs) but also changing the nature of the ancillary ligands (i.e. shifting the HOMOs), we have systematically computed the energies of the frontier molecular orbitals and the electronic transition energies for two series of complexes containing different ancillary ligands and an unsubstituted pyridazine: [Re2(μ-X)2(CO)6(μ-pydz)] and [Re2(μ-H)(μ-X)(CO)6(μ-pydz)] (X = Cl, OPh, SPh, OOCPh). It is important to compute both the energy gap and the strongest MLCT electronic transition energy because in these species the HOMO → LUMO transition is either symmetry forbidden or shows a very low oscillator strength. Therefore no clear correlation between the energy gap (as measured for instance from electrochemical data) and the maximum of the absorption spectra can be found. Moreover, the “t2g” set of orbitals of the two rhenium atoms (involved in the MLCT transition) are not necessarily the six HOMOs for these species, since the presence of aromatic moieties in the ancillary ligands (OPh, SPh and OOCPh) determines the intercalation of some π orbitals between them. The data reported in Table 1 show that a more red-shifted absorption can be attained by using the hydrido derivative containing a μ-benzoato bridging ligand.
On this ground, we focused our attention on complex 2, in which the pyridazine-4-carboxylic acid ligand is combined with one hydrido and one benzoato ligand, and on the analogous derivative containing the 4-diphenylamino-benzoate anion (compound 3), due to the recognized ability28 of the triarylamine moiety to promote charge separation after UV excitation. Fig. 2 shows the electronic density difference maps for the two most intense electronic transitions computed for 3. In the MLCT transition it is clearly recognizable the electronic transfer from the Re2(μ-H)(μ-X) moiety to the pyridazine ligand (and its carboxylic substituent). Apparently the diphenylamino substituent is not directly involved in this transition, however it drastically changes the energy of the HOMO (not a rhenium centred orbital anymore). Its presence gives rise to an additional π–π* transition, that can be described as a charge transfer from the diphenylamino moiety to the benzoato bridging ligand, and is very similar to the corresponding transition computed for the free ligand, even if their energies are quite different, as a consequence of their different charge distribution.
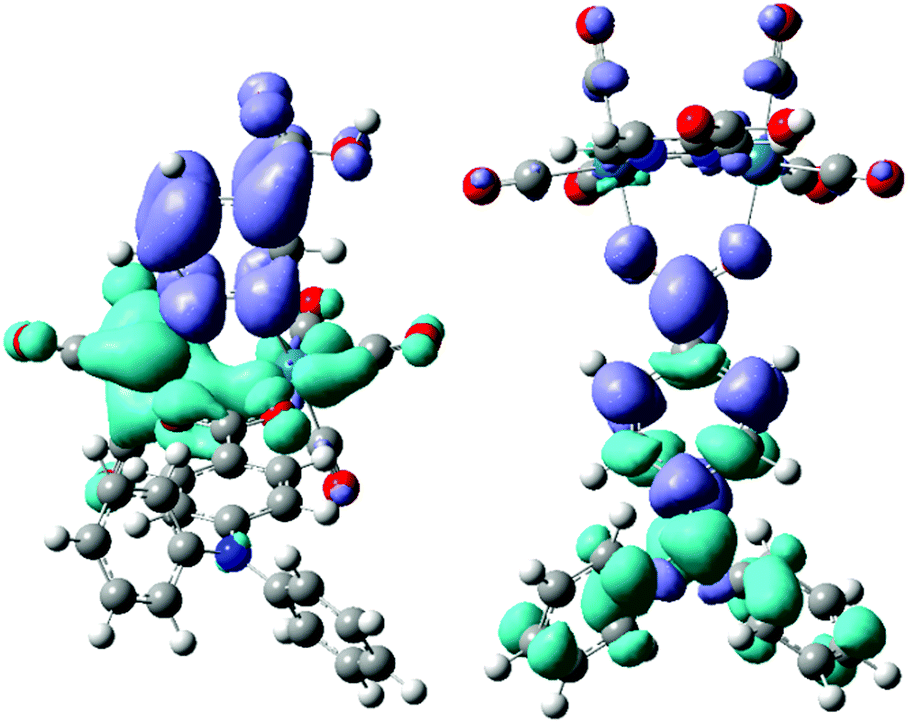 | ||
| Fig. 2 Electronic density difference maps for the two most intense electronic transitions computed for the rhenium complex [Re2H(OOC-TPA)(CO)6(pydz-4-COOH)] (3): the MLCT at 530 nm (left) and the π–π* at 344 nm (right). Light blue indicates a decrease in electron density, blue indicates an increase. | ||
Synthesis of the designed molecules
A new synthetic strategy was designed for obtaining the hydrido-carboxylato derivatives 2 and 3 (whereas the dichloride complex 1 was prepared according to a literature procedure, involving the high temperature reaction of ReCl(CO)5 with 0.5 equivalents of pyridazine-4-carboxylic acid).1,24The three step procedure shown in Scheme 1 was followed, which is based on the peculiar reactivity of the tetrahedral hydrido-carbonyl cluster [Re4(μ3-H)4(CO)12] with bridging donor ligands.2 Indeed this tetrahedral cluster, which possesses four electrons less than that required by the effective atomic number rule (56 instead of 60 valence electrons, v.e.s),29 easily reacts with any (even weakly) donor species L.30 By using pyridazine as a donor ligand, the main reaction products are the tetrametallic derivatives [Re4(μ-H)4(CO)12(μ-pydz)2], with a square geometry of the Re(μ-H)Re skeleton and two pyridazine molecules bridging on opposite edges of the squares, in trans or cis positions with respect to the plane of the cluster.2 The reaction produces also the dinuclear unsaturated complex [Re2(μ-H)2(CO)6(μ-pydz)], which possesses 32 v.e.s, instead of the 34 v.e.s required by the effective atomic number rule.2 Such dinuclear species is not formed in high yields in this reaction. In contrast dinuclear species of this kind are the only reaction products using 3,6-disubstitued pyridazine ligands, indicating the occurrence of a [2+2] fragmentation pathway of the parent tetranuclear cluster, without any spectroscopically recognizable intermediate. This is probably due to steric hindrance, which prevents the formation of the square planar species by destabilizing some key intermediate of the addition reaction. Analogously the selective formation of a dinuclear product is observed by reacting [Re4(μ3-H)4(CO)12] with 2,5-diphenyl-1,3,4-oxadiazole (ppd), which contains two aromatic substituents in the α position of the oxadiazole ring.23 The labile ppd ligand can be easily replaced by a diazine molecule, and this provides a general route to [Re2(μ-H)2(CO)6(μ-diazine)] complexes.
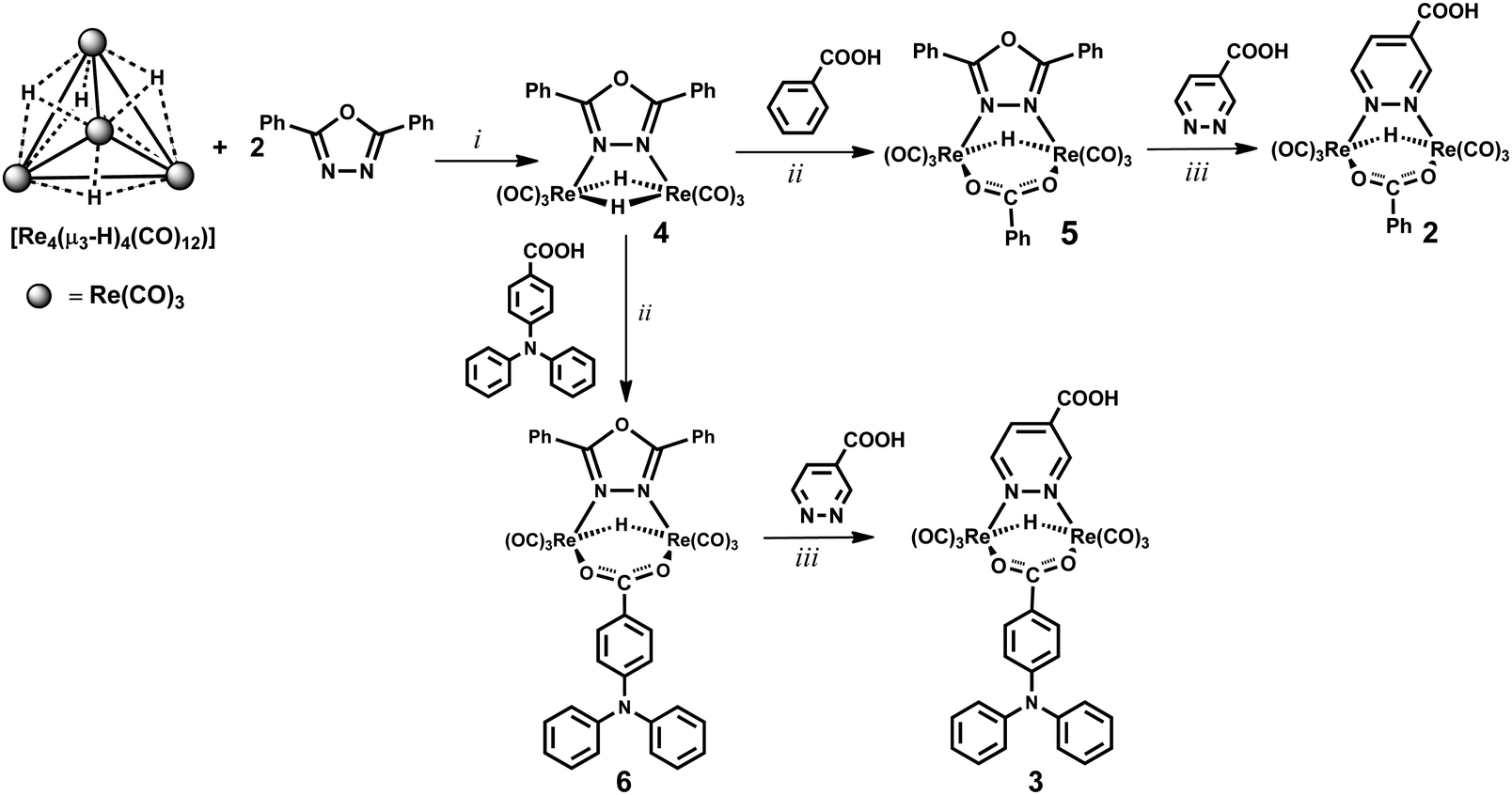 | ||
| Scheme 1 Synthetic pathways for the preparation of rhenium complexes 2 and 3. Reaction conditions: (i) CH2Cl2, room temperature, 24 h; (ii) toluene reflux, 2 h; (iii) THF reflux, 12 h. | ||
Therefore, at first the complex [Re2(μ-H)2(CO)6(μ-ppd)] (4) was prepared in high yields by reacting [Re4(μ3-H)4(CO)12] with 2 equiv. of ppd, at room temperature in dichloromethane solution.23 Then the mixed hydrido-carboxylato complex [Re2(μ-H)(μ-OOCPh)(CO)6(μ-ppd)] (5, Scheme 1) was obtained by reaction 1, which exploits the reactivity of the hydride ligands of 4 towards the acidic proton of the carboxylic acid.
| [Re2(μ-H)2(CO)6(ppd)] + PhCOOH → [Re2(μ-H) (OOCPh)(CO)6(ppd)] + H2 | (1) |
Finally, substitution of the oxadiazole by the pyridazine-4-carboxylic acid afforded the desired product 2. It is important to note that the introduction of the diazine ligand must be the last step of the synthesis, since both [Re4(μ3-H)4(CO)12] and [Re2(μ-H)2(CO)6(μ-ppd)] (4) could react with the carboxylic substituent of the diazine ring, affording undesired byproducts.
The same synthetic procedure has been exploited to carry out the synthesis of complex 3, containing the 4-diphenylamino-benzoate anion as an ancillary ligand.
Electrochemical characterization
The cyclic voltammetry analysis has been performed on the derivatives 1-COOMe, 2-COOMe and 3-COOMe, containing the methyl ester of the pydz-4-COOH ligand, in order to avoid the reduction of the acidic proton, affording H2 evolution. The cyclic voltammetry behaviour in acetonitrile, at 298 K, is reported in Fig. 3 and the peak potentials, together with the related HOMO and LUMO energy values, are collected in Table 2. In the cathodic region all the complexes show one monoelectronic reduction peak at about −0.9 V (vs. Fc+|Fc, see Table 2), which is centred on the diazine ligand and whose chemical reversibility is confirmed by the presence of its anodic counterpart. Moreover, the position of this peak is unaffected by the scan rate, in agreement with the electrochemical reversibility of this reductive process. The peak potential is slightly modulated by the different nature of the ancillary ligand (see Table 2). Comparing the CV curve of the complexes 1–3 with those of analogous complexes of this family containing an unsubstituted pydz ligand, a shift in the positive direction of the reduction potential can be observed (see for instance 1-pydz in Table 2),31 consistently with the presence of the electron-withdrawing substituent on the aromatic ring. These features are in agreement with the localization of the reduction process on the diazine ring, as indicated also by the description of the LUMO provided by the DFT computations.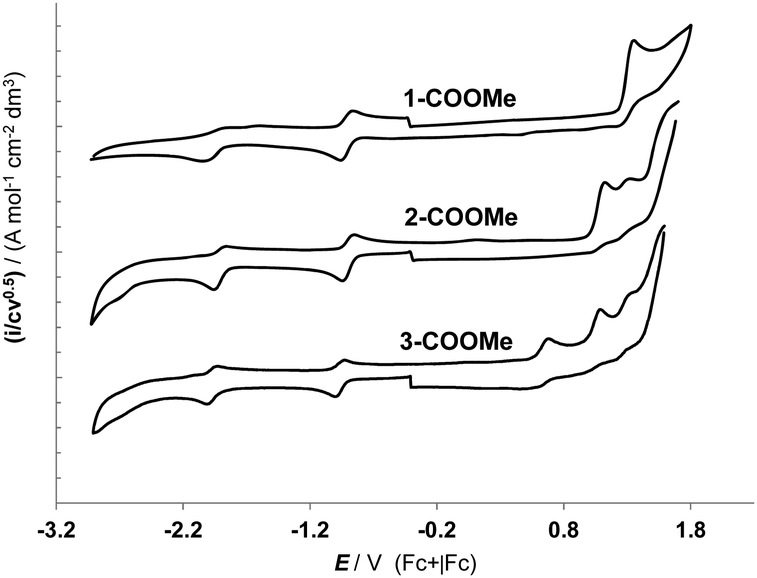 | ||
| Fig. 3 Normalized CV features of the methyl-ester derivatives of the three dyes on GC electrodes, in MeCN +0.1 M (NBu4)PF6 solution, at 0.2 V s−1 with ohmic drop compensation. | ||
| Compound | E p,c [V] | E p,a [V] | ΔEe [eV] | ΔEs [eV] | E LUMO [eV] | E HOMO [eV] |
|---|---|---|---|---|---|---|
| a The spectroscopic (ΔEs) energy gaps is the energy associated with the electronic transition determined from the maximum of the 1MLCT absorption band. b Fc+|Fc potential is 0.385 V vs. SCE in acetonitrile solution. c First oxidation peak potential not located on the metal core. | ||||||
| 1-pydz | −1.34 | 1.31 | 2.66 | 3.31 | −3.46 | −6.11 |
| 1-COOMe | −0.91 | 1.34 | 2.25 | 2.98 | −3.88 | −6.14 |
| 2-COOMe | −0.89 | 1.11, 1.32 | 2.00 | 2.56 | −3.90 | −5.91 |
| 3-COOMe | −0.97 | 0.66,c 1.07, 1.32 | 1.63 | 2.56 | −3.82 | −5.46 |
In all cases, the first reduction peak is followed by another reversible reduction peak, which is probably centred on the diazine ligand, as suggested by the invariance of the potential on varying the ancillary ligands. This agrees with DFT computations, that indicated that the two lowest LUMOs are both π* orbitals of the diazine.
In the anodic region significant differences have been observed for the three complexes. 1-COOMe exhibits an oxidation potential at 1.34 V (vs. Fc+|Fc), which corresponds to a metal-centred bi-electronic oxidation and appears to be irreversible.1 However, it becomes partially reversible at a higher scan rate, as demonstrated by the presence of the return peak.1 This clearly points to an electrochemically quasi-reversible electron transfer step and a subsequent chemical step. The resulting electrochemical HOMO–LUMO gap was 2.25 eV and the HOMO and the LUMO energy levels of 1 are calculated to be −6.14 eV and −3.88 eV respectively.
Interesting differences appear in the anodic behaviour of the complexes 2-COOMe and 3-COOMe. As already observed in the derivatives containing alcoholate or phenolate anions as ancillary ligands,3 also in these complexes a close sequence of two monoelectronic oxidation peaks is observed, instead of the bi-electronic one. These two oxidation processes are clearly localized on the metal core (as confirmed also by the computational analysis)3 and strongly indicate that the oxidation process is markedly different in the case of the oxygen-bridged derivatives with respect to the dichloride ones, also when only one oxygenated ancillary ligand is present. This might be related to the closer Re(μ-X)2Re scaffold and to the harder nature of the bridging ligands in the case of the derivatives containing oxygen ligands. This feature could afford a less-efficient stabilization of the cationic products, with respect to the softer anions, such as halides.
The first oxidation peak for 2-COOMe is observed at +1.11 V (vs. Fc+|Fc), and it appears chemically irreversible. It is followed by a second peak, at 1.32 V, which is reversible in the whole scan rate range explored. This lower oxidation potential affords a higher HOMO level, in agreement with the DFT calculations, and the resulting electrochemical energy gap is reduced to 2.00 eV.
On the other hand, for complex 3-COOMe, beside the two oxidation peaks centred on the metal core (at 1.07 V and at 1.32 V), another chemically reversible oxidation peak at +0.66 V is observed. This peak is clearly attributed to the formation of a radical cation on the triarylamine (TPA) moiety, as indicated by the comparison with the electrochemical behaviour of the free 4-diphenylamino-benzoic acid (Ep,a = 0.70 V). This confirms the lack of communication between the pyridazine ring and the triarylamine moiety, which is verified also in the UV-Vis absorption data (see the following section).32 In spite of the lack of electronic communication, the presence of the TPA moiety affects (even if slightly) the position of the reduction of the pyridazine ring, which results at more negative potential values than in 2. The slight destabilization of the LUMO level is also confirmed by the computational data (see Table 1).
Even so, the introduction of the TPA moiety in 3 affords a strong reduction of the electrochemical energy gap, which does not parallel the spectroscopic (absorption) gap, as in the case of 1 and 2. This is consistent with the fact that the electronic absorption arises from a 1MLCT transition that in 3 does not involve the HOMO level, which is centred on the TPA moiety. In the case of dyes 1 and 2, in contrast, the electrochemical and spectroscopical energy gaps does parallel each other, although the spectroscopical gaps are always higher than the electrochemical ones, since the electronic transition responsible for the 1MLCT absorption band is not a pure HOMO–LUMO transition.
Photophysical characterization
The UV-Vis absorption spectra of the three dyes in diluted toluene and EtOH solution are reported in Table 3, while Fig. 4 shows the spectra in toluene solution. In toluene solution all the complexes exhibit a broad and featureless absorption band that covers a large part of the visible spectrum between 350 nm and 550–600 nm. The predicted red-shift in the absorption maximum (see the Computational study section) for 2 and 3, with respect to complex 1, is observed. This feature indicates that this band is ascribable to the metal-to-ligand dπ(Re) → π*(diazine) charge transfer transition (1MLCT). This attribution is also supported by the weak intensity of these bands and by the observed solvatochromic behaviour. Indeed a significant blue-shift of the absorption maximum has been observed in a more polar solvent as EtOH (see Table 3). This broad band arises from the convolution of multiple transitions, as testified by the more or less pronounced shoulders observed at longer wavelengths. As evidenced by TD-DFT computations, the electronic transitions responsible for the MLCT absorption band involve as starting orbitals the metal-centred HOMO−n orbitals and, as final orbitals, the two low lying π* orbitals of the diazine, LUMO and LUMO+1.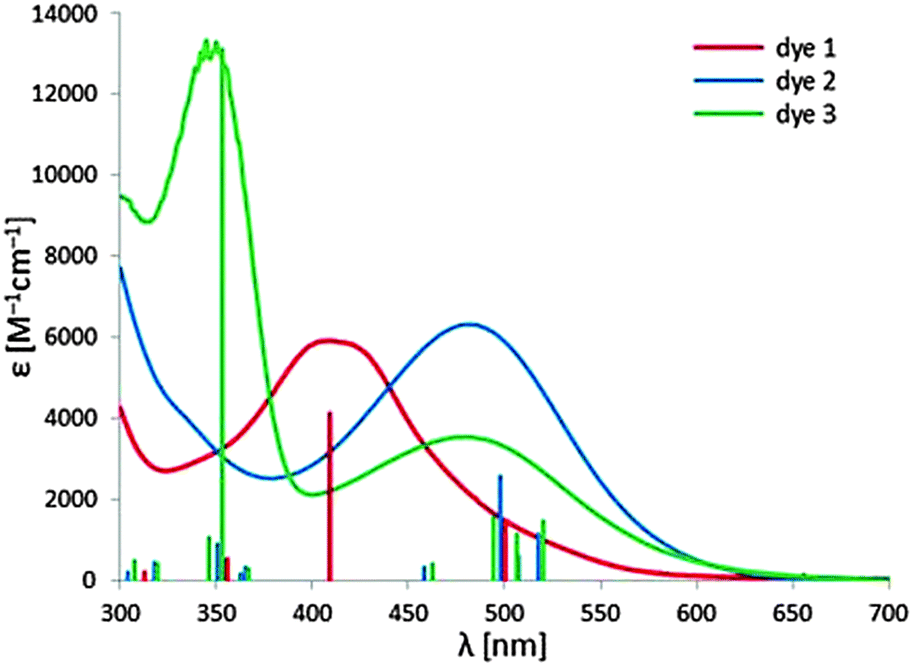 | ||
| Fig. 4 UV-Vis absorption spectra of the three dyes in toluene solution. A comparison with excitation energies and oscillation strengths computed in toluene is reported (vertical lines). | ||
Complex 3 displays another high energy absorption band, at 350 nm, whose position is independent of the polarity of the solvent. In agreement with the electrochemical and computational data, this band is attributed to the π → π* transition involving the triarylamine moiety.
DSSC performances
Photovoltaic measurements have been carried out to evaluate the potential of the three new rhenium complexes as dyes in DSSC devices. The main photovoltaic parameter performances of the solar cells under AM 1.5 G at 1 Sun (1000 W m−2) illumination are presented in Table 4 and the current–voltage (J–V) curves are reported in Fig. 5. Two different electrolytes were used, both based on the iodide/triiodide redox couple. The first one (HSE, Dyesol) is commercially available, while the latter one (labeled El14) was developed in-house, reducing the concentration of iodine (and thus of iodide), which strongly absorbs below 450 nm, competing with the rhenium complexes absorption. Actually, an increase in the photogenerated currents can be observed (see Table 4 and Fig. 5) for all the rhenium-based dyes when El14 substitutes HSE and it is confirmed by the incident photon-to-current conversion efficiency (IPCE) plots for the DSSCs sensitized by 1, 2 and 3, in the presence of the two different electrolytes shown in Fig. 6.| Compound | J SC (mA cm−2) | V OC (mV) | Fill factor | η (%) |
|---|---|---|---|---|
| a Photovoltaic performances based on the commercial HSE electrolyte (Dyesol). | ||||
| N719 | 12 | 0.78 | 0.32 | 3.0 |
| 13.6a | 0.73a | 0.63a | 6.3a | |
| 1 | 2.2 | 0.54 | 0.61 | 0.8 |
| 1.8a | 0.53a | 0.70a | 0.6a | |
| 2 | 1.7 | 0.48 | 0.6 | 0.45 |
| 1.2a | 0.46a | 0.67a | 0.38a | |
| 3 | 3.3 | 0.51 | 0.57 | 0.97 |
| 2.9a | 0.51a | 0.70a | 1.0a | |
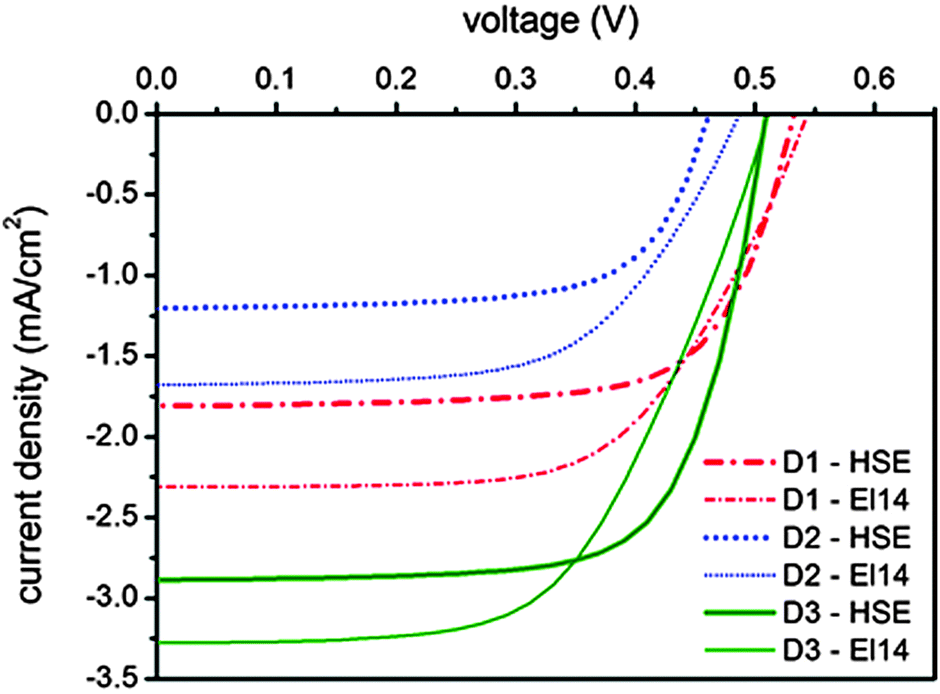 | ||
| Fig. 5 Current–voltage curves for the DSSCs sensitized by the rhenium-based dyes under 1 Sun illumination, AM 1.5. Two different iodide/triiodide electrolytes were used, a commercially available one (HSE, Dyesol) and another developed in-house (labeled El14). | ||
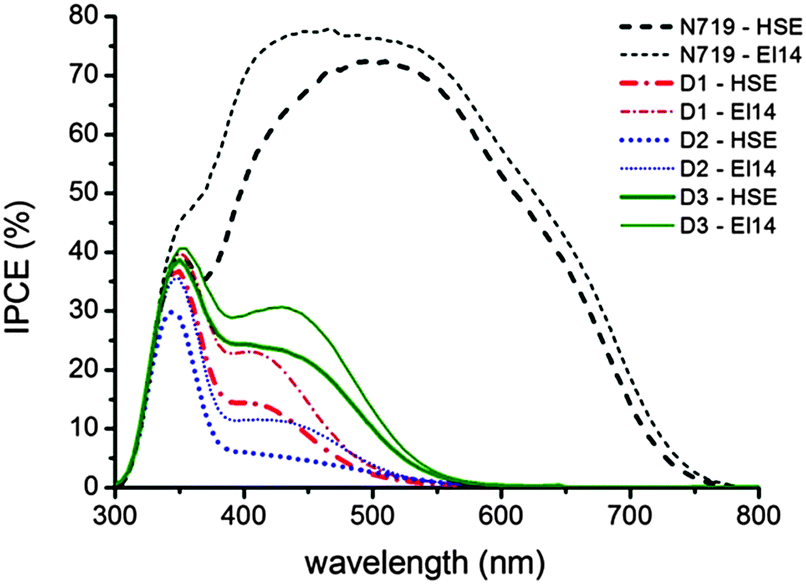 | ||
| Fig. 6 IPCE spectra for DSSC devices sensitized by different dyes: 1 with a HSE electrolyte (thick red dash-dotted line) and with an El14 electrolyte (thin red dash-dotted line); 2 (dotted blue lines); 3 (solid green lines); N719 as reference dye (dashed black lines). Two different iodide/triiodide electrolytes were used, a commercially available one (HSE, Dyesol) and another developed in-house (labeled El14). | ||
The best performing cell, sensitized by complex 3, gives an overall power conversion efficiency of 1% (with short-circuit photocurrent density JSC = 2.9 mA cm−2, open-circuit photovoltage VOC = 0.51 mV, fill factor ff = 0.70). These values are far from those obtained under similar conditions by cells assembled using the conventional ruthenium N719 dye. Indeed these dyes absorb in a narrow spectral range compared to N719 and in addition, below 450 nm, they compete with iodide/triiodide-based electrolytes in the absorption of light. However, the maximum of the absorption spectrum is not the only factor which determines the overall characteristics of the cell. Indeed, despite the red-shift observed in the absorption spectrum, mainly due to the raise of the HOMO level, complex 2 displays the worst performances. This result is probably due to the particularly low LUMO level (see Table 2) which, as in the case of complex 1, hampers the electron injection into the TiO2 conduction band (−3.9 eV).
The influence of the ancillary ligand is confirmed by the results obtained with complex 3. The replacement of the simple benzoate anion with the carboxylate derivative of the triarylamine passing from 2 to 3 has a double role. The LUMO level of the complex was slightly raised, then allowing the injection on the TiO2. Most importantly, the hole transport unit, now localized on the TPA moiety, is placed far away from TiO2 and can better interact with the electrolyte. Moreover the higher steric hindrance of the TPA unit, with respect to the simple benzoate anion, reduces the recombination of the electrons, with the redox species, on the surface of TiO2. This insulating effect is in agreement with the slightly higher VOC value obtained for complex 3.
Conclusions
Dirhenium complexes designed as potential sensitizers in DSSCs have been prepared, characterized and tested in operating solar cells. Light-to-current conversion has been really observed, although with a low efficiency. The best results have been obtained for the hydrido-carboxylato complex 3, in which the introduction of the triarylamine moiety further suppresses the recombination of the injected electron with the oxidized state of the complex, thus improving the charge separation on TiO2. Moreover the bulky triarylamine moiety also reduces the back reaction of the injected electron with the electrolyte.Compared to the standard ruthenium-based N719 dye, the maximum power conversion efficiency of 1% obtained for 3 is obviously too low. However, the data here collected have provided a sufficiently clear picture of the critical factors and of the lines to follow for improving the performances. The future molecular design will take advantage from the possibility to independently tune the ground-state energy levels of the dirhenium diazine complexes, since their frontier orbitals are clearly localized on different regions of the molecule.
So a different choice of the diazine ligand will permit to raise the LUMO level, to overcome the most serious problem concerning the electron injection into TiO2, which is certainly represented by the excessive stabilization of the π* orbitals of the diazine in the complexes here investigated.
On the other hand, wider light harvesting and efficient hole-transfer towards the periphery of the molecule could be obtained through the introduction of a more conjugated moiety on the triarylamine ancillary ligand. Moreover metal oxide photoelectrodes other than TiO2, such as SnO2, and different electrolytes, such as those based on cobalt complexes, could be used. Work towards these directions is in progress in our laboratory.
However, it must be pointed out that, due to the novelty of the employed dyes, the present work did not aim to optimize the cell performances, but rather to understand the relationships between the molecular structure of the complexes and their spectral and photovoltaic properties. This result has been attained.
Two final observations, from the point of view of coordination chemistry. The survival of hydrido complexes in the media and under the operating conditions of solar cells is noteworthy. Clearly the bridging coordination of the hydride contributes to this peculiar stability. Moreover, the three-step synthetic procedure here developed has a wider scope than the preparation of the compounds here described and opens the way to the obtainment of a large number of dinuclear complexes of this family, tailored for specific applications.
Acknowledgements
M. P., P. M., and G. D. thank the Italian Ministero dell'Istruzione, Università e Ricerca, for financial support (PRIN-2012A4Z2RY) and F. D. R. and T. M. B. acknowledge “Polo Solare Organico” Regione Lazio for funding.Notes and references
- D. Donghi, G. D'Alfonso, M. Mauro, M. Panigati, P. Mercandelli, A. Sironi, P. Mussini and L. D'Alfonso, Inorg. Chem., 2008, 47, 4243–4255 CrossRef CAS PubMed.
- M. Panigati, D. Donghi, G. D'Alfonso, P. Mercandelli, A. Sironi and L. D'Alfonso, Inorg. Chem., 2006, 45, 10909–10921 CrossRef CAS PubMed.
- A. Raimondi, M. Panigati, D. Maggioni, L. D'Alfonso, P. Mercandelli, P. Mussini and G. D'Alfonso, Inorg. Chem., 2012, 51, 2966–2975 CrossRef CAS PubMed.
- (a) E. Quartapelle Procopio, M. Mauro, M. Panigati, D. Donghi, P. Mercandelli, A. Sironi, G. D'Alfonso and L. De Cola, J. Am. Chem. Soc., 2010, 132, 14397–14399 CrossRef CAS PubMed; (b) M. Panigati, M. Mauro, D. Donghi, P. Mercandelli, P. Mussini, L. De Cola and G. D'Alfonso, Coord. Chem. Rev., 2012, 256, 1621–1643 CrossRef CAS.
- (a) M. Mauro, E. Quartapelle Procopio, Y. Sun, C.-H. Chien, D. Donghi, M. Panigati, P. Mercandelli, P. Mussini, G. D'Alfonso and L. De Cola, Adv. Funct. Mater., 2009, 19, 2607–2614 CrossRef CAS; (b) M. Mauro, C.-H. Yang, C.-Y. Shin, M. Panigati, C.-H. Chang, G. D'Alfonso and L. De Cola, Adv. Mater., 2012, 24, 2054–2058 CrossRef CAS PubMed.
- F. Nastasi, F. Puntoriero, M. Natali, M. Mba, M. Maggini, P. Mussini, M. Panigati and S. Campagna, Photochem. Photobiol. Sci., 2015, 14, 909–918 CAS.
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef.
- T. M. Brown, F. De Rossi, F. Di Giacomo, G. Mincuzzi, V. Zardetto, A. Reale and A. Di Carlo, J. Mater. Chem. A, 2014, 2, 10788–10817 CAS.
- A. Fakharuddin, R. Jose, T. M. Brown, F. Fabregat-Santiago and J. Bisquert, Energy Environ. Sci., 2014, 7, 3952–3981 CAS.
- (a) M. Nazeeruddin, A. Kay, I. Rodicio, R. Humphry-Baker, E. Müller, P. Liska, N. Vlachopoulos and M. Grätzel, J. Am. Chem. Soc., 1993, 115, 6382–6390 CrossRef CAS; (b) K. Kalyanasundaram and M. Grätzel, Coord. Chem. Rev., 1998, 177, 347–414 CrossRef CAS.
- S. Campagna, F. Puntoriero, F. Nastasi, G. Bergamini and V. Balzani, Top. Curr. Chem., 2007, 280, 117–214 CrossRef CAS.
- M. Nazeeruddin, F. De Angelis, S. Fantacci, A. Selloni, G. Viscardi, P. Liska, S. Ito, B. Takeru and M. Grätzel, J. Am. Chem. Soc., 2005, 127, 16835–16847 CrossRef CAS PubMed.
- D. Kuang, S. Ito, B. Wenger, C. Klein, J. Moser, R. Humphry-Baker, S. Zakeeruddin and M. Grätzel, J. Am. Chem. Soc., 2006, 128, 4146–4154 CrossRef CAS PubMed.
- S. Mathew, A. Yella, P. Gao, R. Humphry-Baker, B. F. E. Curchod, N. Ashari-Astani, I. Tavernelli, U. Rothlisberger, M. K. Nazeerruddin and M. Grätzel, Nat. Chem., 2014, 6, 242–247 CrossRef CAS PubMed.
- (a) S. Ferrere and B. A. Gregg, J. Am. Chem. Soc., 1998, 120, 843–844 CrossRef CAS; (b) P. Balraju, M. Kumar, M. S. Roy and G. D. Sharma, Synth. Met., 2009, 159, 1325–1331 CrossRef CAS.
- (a) T. Bessho, E. C. Constable, M. Grätzel, A. H. Redondo, C. E. Housecroft, W. Kylberg, M. K. Nazeeruddin, M. Neuburger and S. Schaffner, Chem. Commun., 2008, 3717–3719 RSC; (b) C. L. Linfoot, P. Richardson, T. E. Hewat, O. Moudam, M. M. Forde, A. Collins, F. White and N. Robertson, Dalton Trans., 2010, 39, 8945–8956 RSC.
- (a) W. Wu, X. Xu, H. Yang, J. Hua, X. Zhang, L. Zhang, Y. Longa and H. Tian, J. Mater. Chem., 2011, 21, 10666–10671 RSC; (b) E. A. M. Geary, L. J. Yellowlees, L. A. Jack, I. D. H. Oswald, S. Parsons, N. Hirata, J. R. Durrant and N. Robertson, Inorg. Chem., 2005, 44, 242–250 CrossRef CAS PubMed.
- B. Bozic-Weber, E. C. Constable and C. E. Housecroft, Coord. Chem. Rev., 2013, 257, 3089–3106 CrossRef CAS.
- (a) J. B. Asbury, E. Hao, Y. Wang and T. Lian, J. Phys. Chem. B, 2000, 104, 11957–11964 CrossRef CAS; (b) N. A. Anderson, X. Ai, D. Chen, D. L. Mohler and T. Lian, J. Phys. Chem. B, 2003, 107, 14231–14239 CrossRef CAS.
- G. M. Hasselmann and G. J. Meyer, J. Phys. Chem. B, 1999, 103, 7671–7675 CrossRef CAS.
- Y. Chen, W. Liu, J.-S. Jin, B. Liu, Z.-G. Zou, J.-L. Zuo and X.-Z. You, J. Organomet. Chem., 2009, 694, 763–770 CrossRef CAS.
- (a) M. A Andrews, S. W. Kirtley and H. D. Kaesz, Inorg. Chem., 1977, 16, 1556–1561 CrossRef; (b) J. R. Johnson and H. D. Kaesz, Inorg. Synth., 1978, 18, 60–62 CrossRef CAS.
- M. Mauro, M. Panigati, D. Donghi, P. Mercandelli, P. Mussini, A. Sironi and G. D'Alfonso, Inorg. Chem., 2008, 47, 11154–11165 CrossRef CAS PubMed.
- E. Ferri, D. Donghi, M. Panigati, G. Prencipe, L. D'Alfonso, I. Zanoni, C. Baldoli, S. Maiorana, G. D'Alfonso and E. Licandro, Chem. Commun., 2010, 46, 6255 RSC.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS.
- Gaussian 09 (revision D.01), Gaussian Inc., Wallingford, CT, 2013 Search PubMed.
- B.-G. Kim, K. Chung and J. Kim, Chem. – Eur. J., 2013, 19, 5220–5230 CrossRef CAS PubMed.
- (a) R. Saillant, G. Barcelo and H. Kaesz, J. Am. Chem. Soc., 1970, 92, 5739–5741 CrossRef CAS; (b) R. D. Wilson and R. Bau, J. Am. Chem. Soc., 1976, 98, 4687–4689 CrossRef CAS; (c) N. Masciocchi, G. D'Alfonso, W. Kockelmann, W. Schäfer and A. Sironi, Chem. Commun., 1997, 1903–1904 RSC.
- (a) T. Beringhelli and G. D'Alfonso, J. Chem. Soc., Chem. Commun., 1994, 2631–2632 RSC; (b) T. Beringhelli, G. D'Alfonso and M. Zarini, J. Chem. Soc., Dalton Trans., 1995, 2407–2415 RSC; (c) T. Beringhelli, G. D'Alfonso and M. G. Garavaglia, J. Chem. Soc., Dalton Trans., 1996, 1771–1773 RSC.
- The reduction potential is −1.34 V for the parent complex [Re2(μ-Cl)2(CO)6(μ-pyridazine)] and −1.58 V for [Re2(μ-Cl)2(CO)6(μ-4,5-Mez-pyridazine)] containing an electron-rich pyridazine ligand.
- The same behaviour has been observed in the complex containing, as an ancillary ligand, the carboxylate derivative of cyclopentadithiophene. E. Quartapelle Procopio, V. Bonometti, M. Panigati, P. Mercandelli, P. Mussini, T. Benincori, G. D'Alfonso and F. Sannicolò, Inorg. Chem., 2014, 53, 11242–11251 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |