
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of ester-substituted dihydroacridine derivatives and their spectroscopic properties†
Ryota
Suzuki
,
Reiki
Tada
,
Takumi
Hosoda
,
Youhei
Miura
and
Naoki
Yoshioka
*
Department of Applied Chemistry, Faculty of Science and Technology, Keio University, Yokohama, 223-8522, Japan. E-mail: yoshioka@applc.keio.ac.jp
First published on 20th January 2016
Abstract
Three dihydroacridine derivatives, 2,7-bis(4-methoxycarbonylphenyl)-9,9-diphenyl-9,10-dihydroacridine (1), 2,8-bis(4-methoxycarbonylphenyl)-10,10-diphenyl-5,10-dihydrophenazasiline (2), and 2,7,9,9-tetraphenyl-9,10-dihydroacridine (3), were prepared and their spectroscopic properties were investigated. These compounds exhibited relatively high quantum yields in a range of solvents. The emission spectra of 1 and 2 displayed large solvatochromic shifts, while the fluorescence solvatochromic behavior was not observed in 3. The intramolecular charge transfer (CT) process from the electron donating moiety at the NH site to the electron withdrawing ester moiety occurs in the excited states of 1 and 2. The increase in the dipole moment induced by the CT process was determined to cause the positive fluorescence solvatochromism. The differences between the excited and ground state dipole moments based on the Lippert–Mataga expression were estimated. The effect of the push–pull substitution in the dihydroacridine π-conjugated system was also discussed using a computational method.
Introduction
Environment-sensitive fluorescent dyes are an interesting class of molecules exhibiting unique spectroscopic properties depending on the properties of their surroundings such as solvent polarity.1 Typical examples of these solvatochromic fluorophores are 4-dimethylamino phthalimide (4-DMAP),2 2-propionyl-6-dimethylaminonaphthalene (PRODAN),3 4-amino-1,8-naphthalimide (4-DMN),4 and 6-N,N-dimethylamino-2,3-naphthalimide (6-DMN)5 having a push–pull substituted π-conjugated system.6 It is also known that bridging an aromatic ring by a Si atom influences the spectroscopic property due to the hyperconjugation and perturbation of the electronic structure that often enhances the fluorescence intensity.7During the course of the study on the synthesis and physicochemical properties of aromatic nitroxide derivatives having rigid 1,2-dihydroquinole8 and 9,10-dihydroacridine9 backbones, we observed the fluorescence of phenyl substituted 9,10-dihydroacridine, which prompted us to investigate the structure–function relationship in 9,10-dihydroacridine derivatives because they have a planar backbone due to the cross-linked diphenylamine whose rigid structure is suitable for fluorescent molecules. In the present paper, we describe the synthesis of ester substituted derivatives, 2,7-bis(4-methoxycarbonylphenyl)-9,9-diphenyl-9,10-dihydroacridine (1) and its silicon containing analogue 2,8-bis(4-methoxycarbonylphenyl)-10,10-diphenyl-5,10-dihydrophenazasiline (2), and their spectroscopic properties in comparison with that of 2,7,9,9-tetraphenyl-9,10-dihydroacridine (3) (Fig. 1).
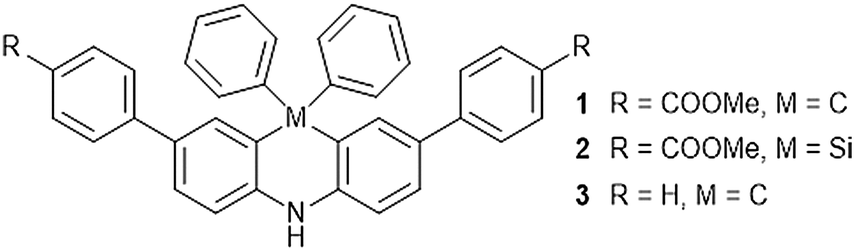 | ||
| Fig. 1 Chemical structures of 1, 2, and 3. | ||
Results
Synthesis
The dihydroacridines, 1 and 3, were prepared in four steps from 9,9-diphenyl-9,10-dihydroacridine (4)10 according to Scheme S1 (ESI†). The aromatic substituent groups were introduced by the Suzuki–Miyaura cross-coupling reaction with good yields between the 2,7-dibromo-9,9-diphenyl-9,10-dihydroacridine-10(9H)-carboxylic acid tert-butyl ester (6) with the 4-methoxycarbonylphenylboronic acid pinacol ester and the phenylboronic acid. 2,7-Dibromo-9,9-diphenyl-9,10-dihydroacridine (5) was protected with the tert-butoxycarbonyl group to enhance the reactivity. The synthetic scheme of the 2,8-diaryldihydrophenazasiline derivative, 2, is outlined in Scheme S2 (ESI†). 2 was prepared in six steps from 2,2′,4,4′-tetrabromodiphenylamine (9).11 The dihydrophenazasiline skeleton was constructed by the reaction between the o-dilithiodiphenylamine derivative12 and SiPh2Cl2, while the p-methoxybenzyl group was used as the protecting group for the NH site. The electron withdrawing group could be introduced by the Suzuki–Miyaura cross-coupling reaction via the same Scheme S1 (ESI†). The deprotection reaction of the BOC group was conducted using sulfuric acid in MeOH because the deprotection reaction using HCl in AcOEt did not provide a good yield.X-ray structural determinations
Block-shaped single crystals of 1 and 2 were obtained by slow evaporation from an acetonitrile solution. Their crystal structures were successfully determined by an X-ray crystallographic analysis (Table S1, ESI†). The ORTEP drawing is depicted in Fig. 2. 2 was crystallized with one molecule of acetonitrile and existed as two crystallographically independent structures (denoted as 2A and 2B). The bond lengths between the carbon atoms adjacent to the bridge atoms (C, Si) were 1: 1.539(3) Å, 1.543(3) Å, 2A: 1.837(5) Å, 1.848(6) Å, and 2B: 1.846(5) Å, 1.856(5) Å. The Si–C bond lengths are almost the same as the reported Si–C distance values which are 1.846 Å and 1.847 Å.13 The angles between the carbon atoms adjacent to the bridge atoms (C, Si) were 1: 108.66(15)°, 2A: 102.6(3)°, and 2B: 102.7(3)°. These angles depend on the atomic radius, and 1 has the larger angle. The long axes of the molecules were 1: 22.101 Å, 2A: 22.844 Å, and 2B: 22.860 Å. The dihedral angles between the dihydroacridine units and the 4-methoxycarbonyl units were 1: 23°, 32°, 2A: 24°, 30°, and 2B: 20°, 27°.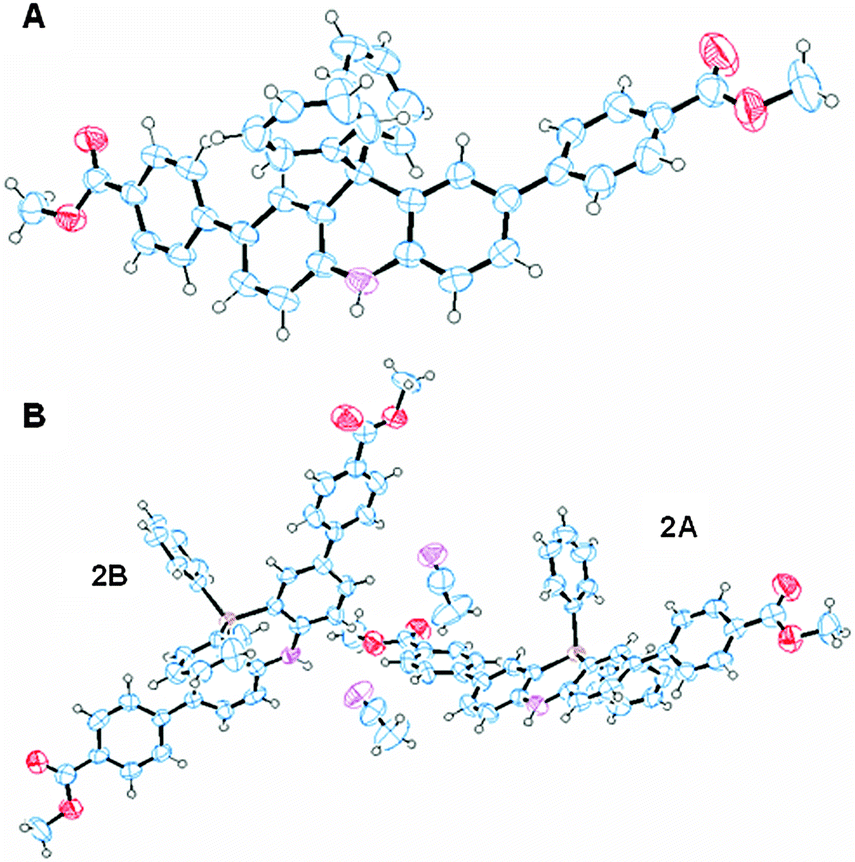 | ||
| Fig. 2 ORTEP diagrams of 1 (A) and 2·CH3CN (B) with thermal ellipsoids at the 50% level. | ||
Spectroscopic properties
The UV-vis and the fluorescence spectra of 1, 2, and 3 in various solvents are summarized in Table 1. The maximum absorption wavelengths of 1, 2, and 3 in MeOH were 370, 376, and 336 nm, respectively. Based on the UV-vis measurements, introduction of the ester group prompted a red-shift in the maximum absorption wavelength (Fig. S1, ESI†). While the wavelengths of the maximum absorptions of 1 and 2 were almost the same, the molar extinction coefficients of 2 were larger than those of 1 in all solvents. 1 and 2 exhibited a fluorescence solvatochromism and the emission spectra were also shifted along with a change in the solvent polarities, while 3 did not exhibit a fluorescence solvatochromism (Fig. 3). Shorter wavelength emission in an apolar solvent and longer wavelength emission in a polar solvent were observed for 1 and 2. The wavelength of the maximum emission of 1 was red-shifted by 98 nm from toluene to MeOH, while it was shifted 77 nm for 2 and 14 nm for 3. The Stokes shifts in toluene were 53 nm for 1, 41 nm for 2 and 48 nm for 3. Those in MeOH were 142 nm for 1, 108 nm for 2 and 58 nm for 3.| Solvent | E T(30) | f(ε) | λ Abs,max [nm] | ε [104 M−1 cm−1] | λ Em,max [nm] | Φ F [%] | ε × ΦF [104 M−1 cm−1] | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 1 | 2 | 3 | 1 | 2 | 3 | 1 | 2 | 3 | 1 | 2 | 3 | |||
| Toluene | 33.9 | 0.24 | 361 | 366 | 332 | 3.9 | 5.5 | 3.2 | 414 | 407 | 380 | 65 | 69 | 63 | 2.5 | 3.8 | 2.0 |
| THF | 37.4 | 0.41 | 371 | 375 | 329 | 4.2 | 5.8 | 3.5 | 441 | 433 | 391 | 68 | 72 | 58 | 2.9 | 4.2 | 2.0 |
| CH2Cl2 | 40.7 | 0.42 | 360 | 367 | 332 | 3.9 | 5.7 | 3.6 | 449 | 439 | 386 | 64 | 67 | 49 | 2.5 | 3.8 | 1.8 |
| DMF | 43.2 | 0.48 | 376 | 381 | 338 | 4.1 | 5.7 | 3.4 | 472 | 462 | 399 | 60 | 62 | 69 | 2.5 | 3.5 | 2.3 |
| MeCN | 45.6 | 0.48 | 363 | 370 | 341 | 4.1 | 4.4 | 3.3 | 466 | 458 | 393 | 54 | 56 | 58 | 2.2 | 2.5 | 1.9 |
| n-BuOH | 49.7 | 0.46 | 378 | 382 | 339 | 3.8 | 5.6 | 3.3 | 472 | 461 | 392 | 53 | 53 | 63 | 2.0 | 3.0 | 2.1 |
| EtOH | 51.9 | 0.47 | 375 | 379 | 338 | 4.1 | 4.8 | 3.5 | 478 | 467 | 391 | 45 | 50 | 62 | 1.8 | 2.4 | 2.2 |
| MeOH | 55.4 | 0.48 | 370 | 376 | 336 | 4.2 | 5.7 | 3.5 | 512 | 484 | 394 | 17 | 25 | 57 | 0.7 | 1.4 | 2.0 |
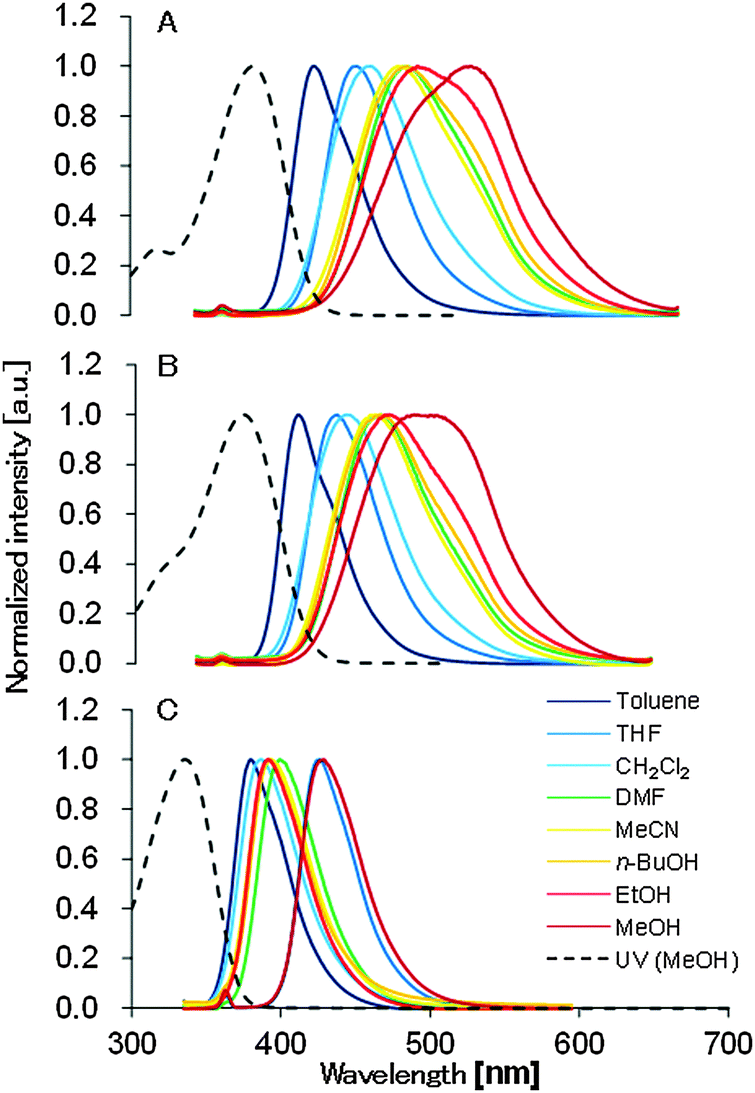 | ||
| Fig. 3 Absorption (dashed line) and emission (solid line) spectra of 1 (A), 2 (B), and 3 (C) in various solvents. Emission spectra were collected under 10−5 mol l−1. | ||
The CV measurements of 1, 2, and 3 were carried out to evaluate their electronic states. They have one reversible oxidation wave, and the half-wave potentials of 1, 2, and 3 were 0.50, 0.53 and 0.43 V (versus Fc/Fc+), respectively (Fig. S2, ESI†). The oxidation potentials of 1 and 2 were higher than that of 3, which is explained by the introduction of the electron withdrawing ester group.
A DFT calculation with B3LYP/6-31G* was conducted to study the electronic structure (Fig. 4). Focusing on the molecular orbitals of the HOMO and LUMO, the HOMO distributes over the electron donating moiety and the LUMO is spread all over the dihydroacridine unit in 3. On the other hand, the LUMO also has distribution over the electron withdrawing ester moiety in 1 and 2. These results indicate the intramolecular CT in the excited states that results in the noticeable solvatochromism. 1 and 2 have almost the same HOMO and LUMO energy levels which are lower than those of 3. The energy gaps of 1, 2, and 3 are 3.78, 3.73 and 4.25 eV, respectively. The energy gaps of 1 and 2 are smaller than that of 3. These calculations agree with the experimental results of the CV, UV-vis, and fluorescence spectra.
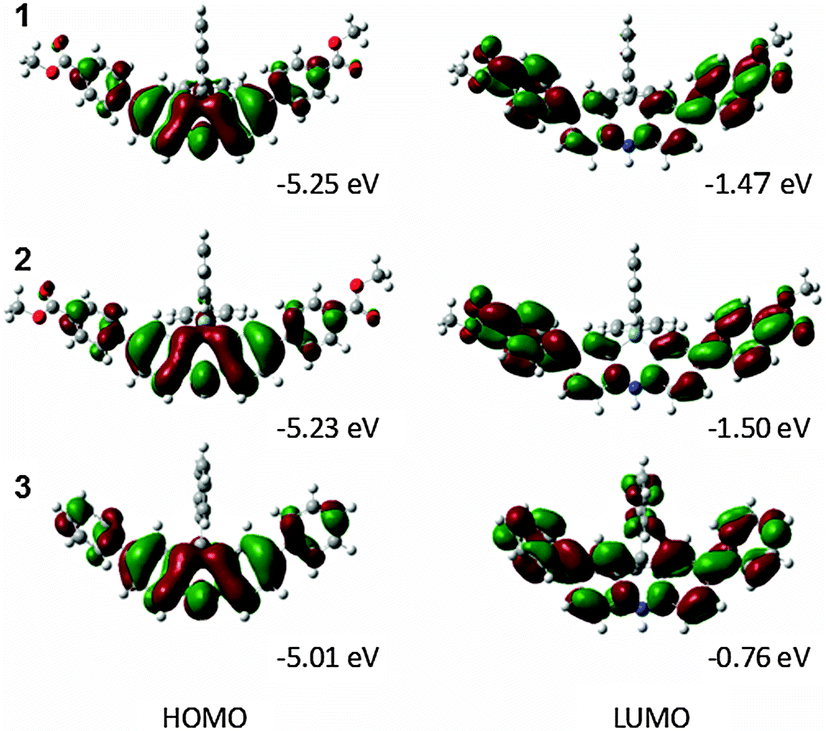 | ||
| Fig. 4 Energy diagrams of 1, 2, and 3 obtained by DFT calculations. | ||
Discussion
We studied the correlation between the emission maxima and the solvent polarity index, ET(30)14 (Fig. 5). Good correlation was found in various solvents and linear fits were used to determine the slopes, i.e., 1: 240 cm−1, 2: 241 cm−1, 3: 78 cm−1. These values of 1 and 2 are comparable to that of PRODAN (220 cm−1)15 and almost the same solvatochromic properties of PRODAN could be achieved using the dihydroacridine-based novel fluorophore.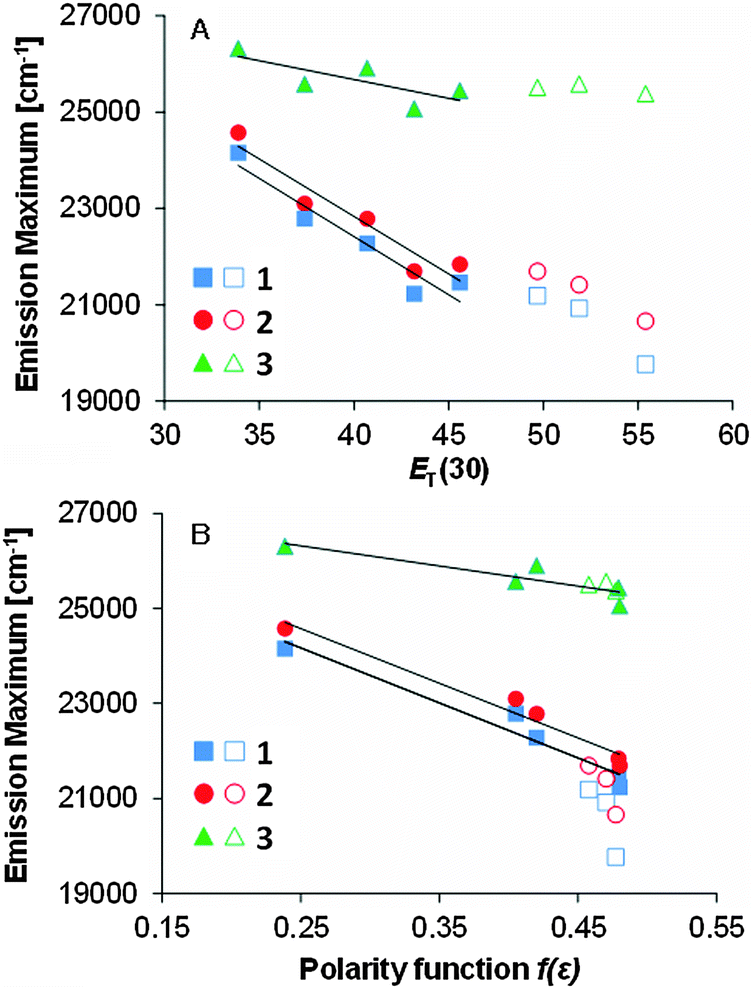 | ||
Fig. 5 The relation between the emission maxima and polarity index ET(30) and dielectric constant function f(ε) of 1, 2, and 3 in aprotic (solid symbols) and protic (open symbols) solvents. Linear fits were obtained only in the aprotic solvents. The slopes for graph A: 240 (R2 = 0.897), 241 (R2 = 0.906), and 78 cm−1 (R2 = 0.584); graph B: 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 (R2 = 0.960), 11600 (R2 = 0.958), and 4200 cm−1 (R2 = 0.784) for 1, 2, and 3, respectively. 600 (R2 = 0.960), 11600 (R2 = 0.958), and 4200 cm−1 (R2 = 0.784) for 1, 2, and 3, respectively. | ||
The data plots in the protic solvents deviate up. For a further understanding, we studied the correlation as a function of the dielectric constant f(ε) which only accounts for the general dipolar interaction.14 A high linearity was also observed in the aprotic solvents. On the other hand, the data plots in the protic solvents deviate low in 1 and 2 though a high linearity was observed in all solvents in 3. Though similar phenomena were reported in 3-hydroxyflavone16 and 3-methoxychromone,17 they could be assigned to the hydrogen bonding effects between the carbonyl group and solvents. To further study the fluorescence solvatochromic behaviors of 1 and 2, the difference between the ground and excited state dipole moments (μE − μG) was estimated by the Lippert–Mataga equation (eqn (1)):18
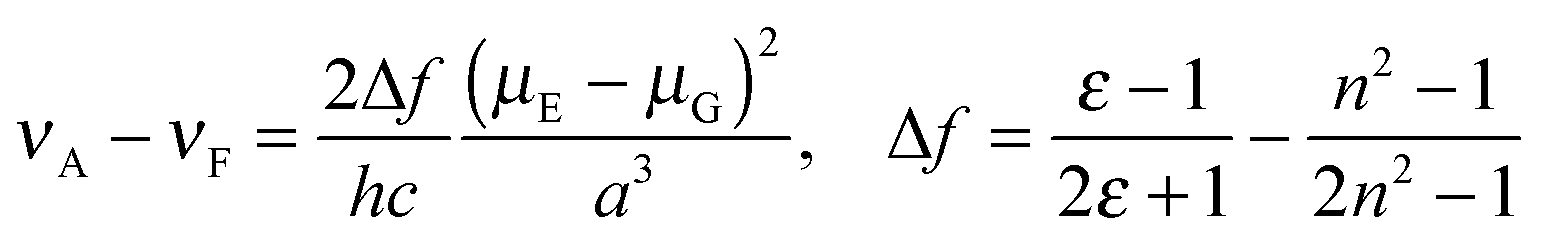 | (1) |
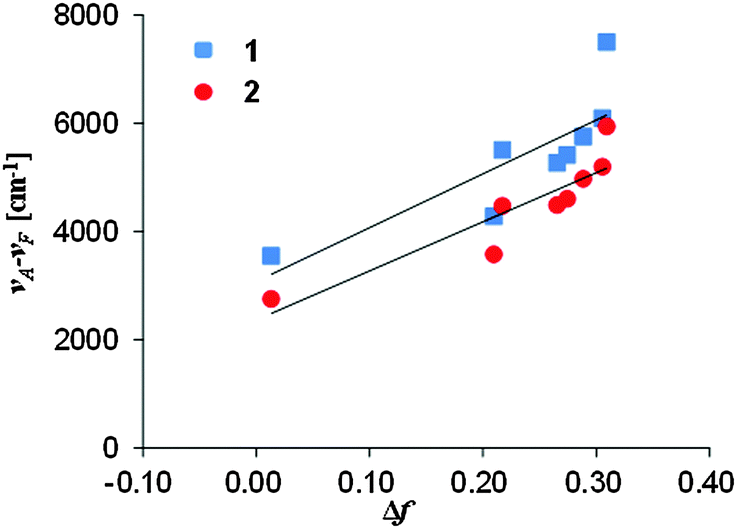 | ||
| Fig. 6 The relation between the Stokes shift and the orientation polarizability of 1 and 2. | ||
The change in the dipole moments μE − μG was estimated as 11.0 D for 1 and 10.5 D for 2 when 50% of the donor–acceptor distance was used for the cavity radius17 and as 26.1 D for 1 and 26.2 D for 2 when 40% of the long axes of the molecular structure19 obtained by X-ray analysis was used for the cavity radius. These values of 1 and 2 are higher than the change in the dipole moments μE − μG of the representative fluorescence solvatochromic dyes, Nile Red (6.8 D)20 and PRODAN (7.9 D).15
Although the quantum yields of 1 and 2 in MeOH were slightly low, the quantum yields of 1 and 2 were almost over 40% in other solvents. The quantum yields of 2 were higher than those of 1 in all solvents except for n-BuOH though their energy gaps between the HOMO and LUMO are almost the same. We assume that this observation is due to the introduction of the Si atom. The quantum yield is affected by radiation and non-radiation processes. The radiation process was accelerated in the Si atom substituted naphthalene derivatives.21 The Si atom might influence radiation and non-radiation processes. The higher molar extinction coefficients of 2 compared to those of 1 might be attributed to the acceleration of the radiation process. It is known that the quantum yield depends on the type of solvent and there are a few dyes which fulfill the noticeable solvatochromism and the high quantum yield in both polar and apolar media at the same time. The quantum yield often decreases in a polar or an apolar solvent although exhibiting the properties of fluorescence solvatochromism with a high quantum yield in both polar and apolar solvents is important for use as a fluorescent probe. The fluorescence spectra of 1 and 2 showed a broad peak in MeOH whereas that of 3 was sharp. The experimental results might be due to the reorganization of the dihydroacridine and ester-substituted phenyl units into a twisted charge transfer excited state conformation22 or aggregation induced in polar solvents.15 It might cause the decrease of the quantum yields in MeOH. On the other hand, the quantum yields of 3 were high in all solvents including MeOH. A TD-DFT calculation (B3LYP/6-31G*) for 1 was performed to estimate the optimized geometry at the excited state in toluene and MeOH (Fig. S3, ESI†). However, obvious difference was not observed between them. The fluorescence spectra of 3 showed a sharp peak in all solvents. The conformation change at the excited state might not occur in 3. 3,8-Dibutyl-6-(piperidin-1-yl)pyrene-1-carbaldehyde (PA) and 1-(3,8-dibutyl-6-(piperidin-1-yl)pyren-1-yl)butan-1-one (PK) are known as the dyes which combine these two properties.15 PRODAN, 7-diethylamino-9,9-dimethyl-9H-fluorene-2-carbaldehyde (FR0),23 1-(7-diethylamino-9,9-dimethyl-9H-fluoren-2-yl)-nonan-1-one (FR8),23 7-dimethylamino-9,9-dimethyl-9H-fluorene-2-carbaldehyde (9FR),24 7-dimethylamino-9,10-dihydrophenanthrene-2-carbaldehyde (9Phen),24 and 1,3-diphenyl-2-[4-(N,N-diphenylamino)phenyl]-benzo[b]phosphole-P-oxide25 have been reported to show these two properties. Our spectroscopic data indicate that 1 and 2 also exhibit a fluorescence solvatochromism with high quantum yields in both polar and apolar solvents.
Conclusions
Spectroscopic properties of three dihydroacridine derivatives, 2,7-bis(4-methoxycarbonylphenyl)-9,9-diphenyl-9,10-dihydroacridine (1), 2,8-bis(4-methoxycarbonylphenyl)-10,10-diphenyl-5,10-dihydrophenazasiline (2), and 2,7,9,9-tetraphenyl-9,10-dihydroacridine (3), were investigated. These compounds exhibited relatively high quantum yields in a range of solvents (1: 17–68%, 2: 25–72%, 3: 49–69%). To evaluate their fluorescence behavior, the correlations between the fluorescence maxima and the empirical polarity parameter, ET(30), were studied. Linear correlations were obtained and the slopes for 1, 2, and 3 were 240, 241, and 78 cm−1, respectively. The differences between the excited and the ground state dipole moments based on the Lippert–Mataga expression were higher than those of the representative fluorescence solvatochromic dyes, Nile Red and PRODAN. The effect of the push–pull substitution in the dihydroacridine π-conjugated system was discussed. Based on the MO calculation, the CT processes from the electron donating NH moiety to the electron withdrawing ester moiety are ascribed to causing the positive fluorescence solvatochromism in 1 and 2.Experimental section
The reactions were performed under N2 with the exception of the hydrogenation reaction. UV-vis and fluorescence spectra were recorded using JASCO V-650 and JASCO FP-777W, respectively. The quantum yields were determined by the absolute measurement using a Shimadzu RF-6000 attached integrating sphere unit. The CV measurements were performed in a CH2Cl2 solution with tetrabutylammonium hexafluorophosphate (0.1 M) as the supporting electrolyte (298 K, 100 mV s−1 scan rate). The observed redox potentials were corrected against the ferrocene/ferrocenium (Fc/Fc+) couple. The X-ray diffraction data were collected by a Bruker D8 VENTURE diffractometer and refined by using SHELX-2014. Gaussian 03 programs were applied for the DFT calculations and Gaussian 09 programs were for the TD-DFT calculations.2,7-Dibromo-9,9-diphenyl-9,10-dihydroacridine (5)
NBS (7.8 g, 4.4 eq.) dissolved in 20 ml of DMF was added to a solution of 9,9-diphenyl-9,10-dihydroacridine (1.7 g) in 10 ml of DMF. The reaction mixture was stirred at room temperature for 10 h. An aqueous solution of K2CO3 was then added to the mixture. The organic phase was extracted with AcOEt and dried over Na2SO4. After removal of the solvent under reduced pressure, the residue was purified by silica gel column chromatography (CH2Cl2/n-hexane = 1/2). The collected material was further purified by recrystallization from CH2Cl2/n-hexane to give a white solid (2.4 g, 95%). Mp 280–283 °C; 1H NMR (300 MHz, acetone-d6): δ = 8.63 (s, 1H), 7.35–7.27 (m, 8H), 6.96–6.92 (m, 6H), 6.86 (d, J = 2.4 Hz, 2H) ppm; 13C NMR (75 MHz, acetone-d6): δ = 146.0, 140.2, 133.1, 131.1, 130.8, 129.9, 128.9, 127.7, 116.7, 112.5, 57.5 ppm; IR (KBr): 3389, 2360, 1665, 1598, 1472 cm−1; MS (MALDI, matrix: SA): 490 [M + H]+; E.A. (%): found: C 61.01, H 3.33, N 2.87, calcd for C25H15Br2N: C 61.13, H 3.49, N 2.85.2,7-Dibromo-9,9-diphenyl-9,10-dihydroacridine-10(9H)-carboxylic acid tert-butyl ester (6)
A solution of 2,7-dibromo-9,9-diphenyl-9,10-dihydroacridine (1.67 g, 3.4 mmol) and 4-dimethylaminopyridine (145 mg, 1.2 mmol, 0.35 eq.) in THF was added to a solution of di-tert-butyl dicarbonate (1.13 g, 5.1 mmol, 1.5 eq.) in THF, and the mixture was stirred for 2 h. After the addition of water, the resulting mixture was extracted with ethyl acetate. The organic extracts were washed with brine and dried (Na2SO4), concentrated, loaded on silica gel and purified by silica gel column chromatography (CH2Cl2/n-hexane = 1/1). The collected material was further purified by recrystallization from CH2Cl2/n-hexane to give 6 as a white solid (1.86 g, 92%). Mp 204–209 °C; 1H NMR (300 MHz, acetone-d6): δ = 7.60 (d, J = 8.4 Hz, 2H), 7.54 (dd, J = 8.9 Hz, 2.4 Hz, 2H), 7.36–7.34 (m, 6H), 6.96 (d, J = 2.7 Hz, 2H), 6.85–6.83 (m, 4H), 1.18 (s, 9H) ppm; 13C NMR (75 MHz, CDCl3): δ = 151.1, 143.2, 142.6, 137.8, 131.1, 130.0, 129.6, 128.1, 127.3, 127.0, 118.3, 82.0, 58.3, 27.7 ppm; IR (KBr): 3049, 2983, 1720, 1464, 1323, 1159 cm−1; MS (MALDI, matrix: SA): 588 [M − H]+; E.A. (%): found: C 60.70, H 4.35, N 2.16, calcd for C30H25Br2NO2: C 60.93, H 4.26, N 2.37.2,7-Bis(4-methoxycarbonylphenyl)-9,9-diphenyl-9,10-dihydroacridine-10(9H)-carboxylic acid tert-butyl ester (7)
To a solution of 6 (59 mg, 0.10 mmol) in dry THF, 2-(4-methoxycarbonylphenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (69 mg, 0.25 mmol, 2.5 eq.), potassium carbonate (276 mg, 2.0 mmol, 20 eq.) and Pd(PPh3)4 (7 mg, 6.0 μmol, 0.06 eq.) were added and the reaction mixture was refluxed for 24 h under nitrogen. The reaction mixture was then poured into water and extracted with ethyl acetate. The organic extracts were washed with brine and dried with Na2SO4. After removal of the solvent under reduced pressure, the residue was purified by silica gel column chromatography (ethyl acetate/n-hexane = 1/3). The collected material was further purified by recrystallization from CH2Cl2/n-hexane to give 7 as a white solid (53 mg, 75%). Mp 151–154 °C; 1H NMR (300 MHz, acetone-d6): δ = 8.03 (d, J = 8.4 Hz, 4H), 7.81 (d, J = 9.0 Hz, 2H), 7.74 (dd, J = 8.6 Hz, 2.1 Hz, 2H), 7.63 (d, J = 8.4 Hz, 4H), 7.35–7.33 (m, 6H), 7.22 (d, J = 2.7 Hz, 2H), 6.99–6.98 (m, 4H), 3.88 (s, 6H), 1.24 (s, 9H) ppm; 13C NMR (75 MHz, CDCl3): δ = 166.9, 151.5, 145.1, 143.5, 141.8, 139.0, 136.2, 130.3, 130.1, 128.7, 127.9, 127.3, 127.1, 126.9, 125.9, 125.3, 81.8, 58.6, 52.1, 27.9 ppm; IR (KBr): 2950, 1720, 1608, 1478, 1329, 1280 cm−1; LRMS (EI): m/z (%): 524 (100) [M − 177]+, 601 (38) [M − 100]+, 701 (9) [M]+; HRMS (ESI+, quadrupole): m/z calcd for C46H39NO6 [M + Na]: 724.2670, found: 724.2676.2,7-Bis(4-methoxycarbonylphenyl)-9,9-diphenyl-9,10-dihydroacridine (1)
Concentrated hydrochloric acid was added to a solution of 7 (53 mg, 0.08 mmol) in ethyl acetate and the mixture was refluxed for 1 h. The mixture was then cooled to room temperature and neutralized with a K2CO3 solution. The mixture was poured into water and extracted with ethyl acetate. The organic extracts were washed with brine and dried with Na2SO4. After removal of the solvent under reduced pressure, the residue was purified by silica gel column chromatography (ethyl acetate/n-hexane = 1/2). The collected material was further purified by recrystallization from CH2Cl2/n-hexane to give 1 as a pale yellow solid (27 mg, 56%). Mp > 300 °C; 1H NMR (300 MHz, acetone-d6): δ = 8.81 (s, 1H), 7.98 (d, J = 8.4 Hz, 4H), 7.62 (dd, J = 8.3 Hz, 2.0 Hz, 2H), 7.56 (d, J = 8.7 Hz, 4H), 7.33–7.10 (m, 14H), 3.87 (s, 6H) ppm; 13C NMR (75 MHz, CDCl3): δ = 167.0, 145.5, 145.3, 139.5, 132.1, 130.2, 130.1, 129.2, 128.4, 128.0, 127.9, 126.7, 126.3, 126.1, 114.3, 57.0, 52.0 ppm; IR (KBr): 3342, 2947, 1820, 1602, 1481, 1434, 1284, 1186, 1114 cm−1; LRMS (EI): m/z (%): 524 (100) [M − C6H5]+, 601 (28) [M]+; HRMS (EI): m/z calcd for C41H31NO4: 601.2253, found: 601.2268.2,7,9,9-Tetraphenyl-9,10-dihydroacridine-10(9H)-carboxylic acid tert-butyl ester (8)
8 was obtained by a method similar to 7 and was obtained as a white solid (yield: 77%). Mp 242–246 °C; 1H NMR (300 MHz, acetone-d6): δ = 7.75 (d, J = 7.5 Hz, 2H), 7.63 (dd, J = 7.7 Hz, 2.0 Hz, 2H), 7.48 (d, J = 7.8 Hz, 2H), 7.41–7.30 (m, 12H), 7.14 (d, J = 2.7 Hz, 2H), 6.98–6.96 (m, 4H), 1.22 (s, 9H) ppm; 13C NMR (75 MHz, CDCl3): δ = 151.7, 143.8, 141.7, 140.7, 138.3, 137.3, 130.3, 128.7, 127.8, 127.1, 127.0, 127.0, 126.9, 125.6, 125.0, 81.4, 58.6, 27.8 ppm; IR (KBr): 3056, 3029, 2969, 2928, 1712, 1600, 1474 cm−1; LRMS (EI): m/z (%): 408 (100) [M − C11H7O2]+, 484 (36) [M − Boc]+, 585.3 (18) [M]+; HRMS (EI, quadrupole): m/z calcd for C31H21N [M − C11H7O2]: 407.1674, found: 407.1683.2,7,9,9-Tetraphenyl-9,10-dihydroacridine (3)
3 was obtained by a method similar to 1 and was obtained as a white solid (yield: 95%). Mp 120 °C; 1H NMR (300 MHz, acetone-d6): δ = 8.57 (s, 1H), 7.51 (d, J = 2.3 Hz, 2H), 7.42–7.07 (m, 22H) ppm; 13C NMR (75 MHz, acetone-d6): δ = 147.3, 141.9, 140.6, 133.4, 131.1, 129.6, 129.5, 128.6, 128.5, 127.3, 127.2, 126.9, 126.7, 115.3, 57.9 ppm; IR (KBr): 3392, 3056, 1602, 1474, 1303 cm−1; LRMS (MALDI, matrix: SA): 485 [M]+; E.A. (%): found: C 91.22, H 5.46, N 2.62, calcd for C37H27N: C 91.51, H 5.60, N 2.88.2,4-Dibromo-N-(2,4-dibromophenyl)-N-(4-methoxybenzyl) aniline (10)
A solution of 2,2′,4,4′-tetrabromodiphenylamine (969 mg, 2.0 mmol) and NaH (54 mg, 2.2 mmol, 1.1 eq.) in DMF was stirred for 1 h. The mixture was then added to 4-methoxybenzyl chloride (289 μl, 2.1 mmol, 1.06 eq.) and stirred for 14 h at room temperature. Water was added to the mixture and the precipitate was filtered. The crude product was purified by silica gel column chromatography (CH2Cl2/n-hexane = 1/2). The collected material was further purified by recrystallization from CH2Cl2/n-hexane to give 9 as a white solid (1.09 g, 90%). Mp 195–198 °C; 1H NMR (300 MHz, CDCl3): δ = 7.70 (d, J = 2.4 Hz, 2H), 7.36 (d, J = 8.4 Hz, 2H), 7.26 (dd, J = 8.9 Hz, 2.3 Hz, 2H), 6.79 (d, J = 8.4 Hz, 4H), 4.68 (s, 2H), 3.75 (s, 3H) ppm; 13C NMR (75 MHz, CDCl3): δ = 158.7, 145.8, 136.6, 130.9, 128.7, 128.6, 126.4, 121.9, 117.2, 113.8, 56.0, 55.1 ppm; IR (KBr): 2930, 1509, 1464, 1240, 1175 cm−1; LRMS (MALDI, matrix: SA): 600 [M]+; E.A. (%): found: C 40.01, H 2.63, N 2.20, calcd for C20H15Br4NO: C 39.71, H 2.50, N 2.32.2,8-Dibromo-5-(4-methoxybenzyl)-10,10-diphenyl-5,10-dihydrophenazasiline (11)
To a solution of 10 (605 mg, 1.0 mmol) in 19 ml of Et2O, n-BuLi (2.6 M in n-hexane solution, 0.84 ml, 2.1 mmol, 2.1 eq.) was added dropwise at 0 °C and the mixture was stirred for 0.5 h. SiPh2Cl2 (0.25 ml, 1.2 mmol, 1.2 eq.) was next added to the reaction mixture. The mixture was warmed to room temperature and stirred overnight. Water was added, and the resulting mixture was extracted with ethyl acetate. The organic extracts were washed with brine, dried with Na2SO4 and concentrated. The residue was purified by silica gel column chromatography (CH2Cl2/n-hexane = 1/3). The collected material was further purified by recrystallization from CH2Cl2/n-hexane to give 11 as a white solid (483 mg, 77%). Mp 204–208 °C; 1H NMR (300 MHz, CDCl3): δ = 7.53–7.33 (m, 14H), 7.01 (d, J = 8.4 Hz, 2H), 6.88 (d, J = 8.7 Hz, 2H), 6.82 (d, J = 8.4 Hz, 2H), 5.08 (s, 2H), 3.80 (s, 3H) ppm; 13C NMR (75 MHz, CDCl3): δ = 158.7, 148.9, 137.1, 136.1, 133.5, 132.6, 130.2, 128.5, 128.2, 127.4, 121.7, 118.5, 114.3, 114.0, 55.3, 55.3 ppm; IR (KBr): 3066, 2954, 1509, 1449, 1394, 1291, 1220 cm−1; LRMS (MALDI, matrix: SA): 625 [M]+; E.A. (%): found: C 61.24, H 4.08, N 2.08, calcd for C32H25Br2NOSi: C 61.26, H 4.02, N 2.33.2,8-Dibromo-10,10-diphenyl-5,10-dihydrophenazasiline (12)
Pd black (15 mg) and 11 (44 mg, 0.09 mmol) were placed in a one-necked flask and dichloromethane was then added. Nitrogen displacement was next conducted. The mixture was stirred for 15 h after the H2 displacement. Nitrogen was again displaced. The mixture was then filtered through Celite. Dichloromethane was added to carry out a conventional separation treatment. The organic extracts were washed with brine and dried with Na2SO4. After removal of the solvent under reduced pressure, the residue was purified by silica gel column chromatography (CH2Cl2/n-hexane = 1/3). The collected material was further purified by recrystallization from CH2Cl2/n-hexane to give 12 as a white solid (25 mg, 63%). Mp 254–257 °C; 1H NMR (300 MHz, acetone-d6): δ = 9.01 (s, 1H), 7.60–7.57 (m, 6H), 7.51–7.44 (m, 8H), 7.09 (d, J = 9.0 Hz, 2H) ppm; 13C NMR (75 MHz, CDCl3): δ = 144.8, 137.8, 135.8, 133.9, 133.8, 130.1, 128.2, 117.4, 115.9, 112.8 ppm; IR (KBr): 3403, 1595, 1457, 1370, 1327, 1231 cm−1; LRMS (MALDI, matrix: SA): 505 [M]+; E.A. (%): found: C 56.61, H 3.41, N 2.64, calcd for C24H17Br2NSi: C 56.82, H 3.38, N 2.76.2,8-Dibromo-10,10-diphenyl-5,10-dihydrophenazasiline-5(10H)-carboxylic acid tert-butyl ester (13)
13 was obtained by a method similar to 6 and was obtained as a white solid. Yield: 89%; Mp 201–204 °C; 1H NMR (300 MHz, CDCl3): δ = 7.54–7.36 (m, 16H), 1.15 (s, 9H) ppm; 13C NMR (75 MHz, CDCl3): δ = 152.5, 145.8, 136.2, 136.1, 134.3, 132.6, 130.6, 129.4, 128.3, 120.0, 81.6, 27.8 ppm; IR (KBr): 3069, 2874, 2930, 1714, 1450, 1369, 1312, 1248 cm−1; LRMS (MALDI, matrix: SA): 605 [M]+; E.A. (%): found: C 57.12, H 4.20, N 2.13, calcd for C29H25Br2NO2Si: C 57.34, H 4.15, N 2.31.2,8-Bis(4-methoxycarbonylphenyl)-10,10-diphenyl-5,10-dihydrophenazasiline-5(10H)-carboxylic acid tert-butyl ester (14)
14 was prepared by a method similar to 7 and was obtained as a white solid. Yield: 51%; Mp 120–122 °C; 1H NMR (300 MHz, acetone-d6): δ = 8.04 (d, J = 8.7 Hz, 4H), 7.84–7.83 (m, 4H), 7.74–7.71 (m, 6H), 7.66–7.63 (m, 4H), 7.52–7.45 (m, 6H), 3.88 (s, 6H), 1.26 (s, 9H) ppm; 13C NMR (75 MHz, CDCl3): δ = 166.9, 153.3, 148.5, 145.6, 137.8, 137.0, 133.1, 133.1, 131.5, 131.4, 130.8, 129.9, 129.4, 129.3, 129.2, 127.8, 81.6, 52.3, 28.1 ppm; IR (KBr): 2976, 2950, 1719, 1609, 1457, 1432, 1321, 1278, 1161, 1110 cm−1; LRMS (EI): m/z (%): 540 (20) [M − 177]+, 617 (80) [M − 100]+, 662 (100) [M − 55]+, 717 (11) [M]+; HRMS (ESI+, quadrupole): m/z calcd for C45H39NO6Si [M + Na]: 740.2439, found: 740.2446.2,8-Bis(4-methoxycarbonylphenyl)-10,10-diphenyl-5,10-dihydrophenazasiline (2)
H2SO4 was added to a solution of 14 (100 mg, 0.14 mmol) in MeOH and the mixture was stirred at room temperature for 3 h. The mixture was then neutralized with a K2CO3 solution. The mixture was poured into water and extracted with ethyl acetate. The organic extracts were washed with brine and dried with Na2SO4. After removal of the solvent under reduced pressure, the residue was purified by silica gel column chromatography (ethyl acetate/n-hexane = 1/2). The collected material was further purified by recrystallization from CH2Cl2/n-hexane to give 2 as a pale yellow solid (56 mg, 65%); Mp 272–274 °C; 1H NMR (300 MHz, acetone-d6): δ = 9.16 (s, 1H), 8.02 (d, J = 8.7 Hz, 4H), 7.95 (d, J = 2.1 Hz, 2H), 7.81 (dd, J = 8.7 Hz, 2.1 Hz, 2H), 7.74–7.68 (m, 10H), 7.45–7.42 (m, 6H), 7.29 (d, J = 8.1 Hz, 2H), 3.88 (s, 6H) ppm; 13C NMR (75 MHz, CDCl3): δ = 167.0, 146.2, 145.1, 135.9, 134.9, 134.7, 131.7, 130.1, 129.9, 129.8, 128.1, 128.1, 126.2, 116.2, 114.0, 52.1; IR (KBr): 3423, 1720, 1597, 1460, 1434, 1285 cm−1; LRMS (EI): m/z (%): 540 (20) [M − C6H5]+, 617 (80) [M]+; HRMS (ESI−, quadrupole): m/z calcd for C40H31NO4Si [M − H]: 616.1950, found: 616.1962.Acknowledgements
This work was partly supported by a Grant-in-Aid for Scientific Research (No. 25620066) from MEXT, Japan, and the MEXT-Supported Program for the Strategic Research Foundation at Private Universities, 2012–2016.References
- (a) J. R. Lakowicz, in Principles of Fluorescence Spectroscopy, Springer Science + Business, New York, 2010 Search PubMed; (b) B. Valeur, in Molecular Fluorescence, Princeples and Applications, Wiley-VCH, New York, 2002 Search PubMed; (c) A. P. de Silva, H. Q. N. Gunaratne, T. Gunnlaugsson, A. J. M. Huxley, C. P. McCoy, J. T. Rademacher and T. E. Rice, Chem. Rev., 1997, 97, 1515 CrossRef CAS PubMed; (d) K. A. Willets, S. Y. Nishimura, P. J. Schuck, R. J. Twieg and W. E. Moerner, Acc. Chem. Res., 2005, 38, 549 CrossRef CAS PubMed; (e) M. S. T. Gonçalves, Chem. Rev., 2009, 109, 190 CrossRef PubMed; (f) R. W. Sinkeldam, N. J. Greco and Y. Tor, Chem. Rev., 2010, 110, 2579 CrossRef CAS PubMed; (g) Z. Yang, J. Cao, Y. He, J. H. Yang, T. Kim, X. Peng and J. S. Kim, Chem. Soc. Rev., 2014, 43, 4563 RSC.
- G. Saroja, T. Soujanya, B. Ramachandram and A. Samanta, J. Fluoresc., 1998, 8, 405 CrossRef CAS.
- (a) G. Weber and F. J. Farris, Biochemistry, 1979, 18, 3075 CrossRef CAS PubMed; (b) A. Balter, W. Nowak, W. Pawelkiewicz and A. Kowalczyk, Chem. Phys. Lett., 1988, 8, 405 Search PubMed.
- G. Loving and B. Imperiali, J. Am. Chem. Soc., 2008, 130, 13630 CrossRef CAS PubMed.
- (a) M. E. Vazquez, J. B. Blanco and B. Imperiali, J. Am. Chem. Soc., 2005, 127, 1300 CrossRef CAS PubMed; (b) A. R. Katritzky and T. Narindoshvili, Org. Biomol. Chem., 2009, 7, 627 RSC.
- (a) S. Fery-Forgues, J.-P. Fayet and A. Lopez, J. Photochem. Photobiol., A, 1993, 70, 229 CrossRef CAS; (b) B. E. Cohen, A. Pralle, X. Yao, G. Swaminath, C. S. Gandhi, Y. N. Jan, B. K. Kobilka, E. Y. Isacoff and L. Y. Jan, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 965 CrossRef CAS PubMed; (c) P. D. Zoon and A. M. Brouwer, ChemPhysChem, 2005, 6, 1574 CrossRef CAS PubMed; (d) Z. Lu, S. J. Lord, H. Wang, W. E. Moerner and R. J. Twieg, J. Org. Chem., 2006, 71, 9651 CrossRef CAS PubMed; (e) S. J. Load, Z. Lu, H. Wang, K. A. Willets, P. J. Schuck, H.-I. D. Lee, S. Y. Nishimura, R. J. Twieg and W. E. Moerner, J. Phys. Chem. A, 2007, 111, 8934 CrossRef PubMed; (f) K. Baathulaa, Y. Xu and X. Qian, J. Photochem. Photobiol., A, 2010, 216, 24 CrossRef CAS; (g) A. Fakhari M. and S. E. Rokita, Chem. Commun., 2011, 47, 4222 RSC; (h) E. Benedetti, L. S. Kocsis and K. M. Brummond, J. Am. Chem. Soc., 2012, 134, 12418 CrossRef CAS PubMed; (i) A. G. Gilani, M. Moghadam and M. S. Zakerhamidi, Dyes Pigm., 2012, 92, 1052 CrossRef; (j) L. Giordano, V. V. Shvadchak, J. A. Fauerbach, E. A. Jares-Erijman and T. M. Jovin, J. Phys. Chem. Lett., 2012, 3, 1011 CrossRef CAS PubMed; (k) Y. Kubota, Y. Sakuma, K. Funabiki and M. Matsui, J. Phys. Chem. A, 2014, 118, 8717 CrossRef CAS PubMed; (l) A. Tigreros, A. Ortiz and B. Insuasty, Dyes Pigm., 2014, 111, 45 CrossRef CAS; (m) S. Kato, T. Furuya, M. Nitani, N. Hasebe, Y. Le, Y. Aso, T. Yoshihara, S. Tobita and Y. Nakamura, Chem. – Eur. J., 2015, 21, 3115 CrossRef CAS PubMed.
- (a) S. Yamaguchi and K. Tamao, Bull. Chem. Soc. Jpn., 1996, 69, 2327 CrossRef CAS; (b) S. Kyushin, M. Ikarugi, M. Goto, H. Hiratsuka and H. Matsumoto, Organometallics, 1996, 15, 1067 CrossRef CAS; (c) S. Satoh, H. Suzuki, Y. Kimata and A. Kuriyama, Synth. Met., 1996, 79, 97 CrossRef CAS; (d) S. Yamaguchi, S. Akiyama and K. Tamao, Organometallics, 1998, 17, 4347 CrossRef CAS; (e) H. J. Tracy, J. L. Mullin, W. T. Klooster, J. A. Martin, J. Haug, S. Wallace, I. Rudloe and K. Watts, Inorg. Chem., 2005, 44, 2003 CrossRef CAS PubMed; (f) M. Shimizu, H. Tatsumi, K. Mochida, K. Oda and T. Hiyama, Chem. – Asian J., 2008, 3, 1238 CrossRef CAS PubMed; (g) Y. Yabusaki, N. Ohshima, H. Kondo, T. Kusamoto, Y. Yamanoi and H. Nishihara, Chem. – Eur. J., 2010, 16, 5581 CrossRef CAS PubMed; (h) T. Agou, M. D. Hossain and T. Kawashima, Chem. – Eur. J., 2010, 16, 368 CrossRef CAS PubMed; (i) M. Shimizu and T. Hiyama, Synlett, 2012, 973 CrossRef CAS.
- (a) M. Yao, S. Asakura, M. Abe, H. Inoue and N. Yoshioka, Cryst. Growth Des., 2005, 5, 413 CrossRef CAS; (b) M. Yao, H. Inoue and N. Yoshioka, Chem. Phys. Lett., 2005, 402, 11 CrossRef CAS.
- R. Suzuki, R. Tada, Y. Miura and N. Yoshioka, J. Mol. Struct., 2016, 1106, 399 CrossRef CAS.
- T. L. Andrew and T. M. Swager, J. Org. Chem., 2011, 76, 2976 CrossRef CAS PubMed.
- S. Kajigaeshi, T. Kakinami, K. Inoue, M. Kondo, H. Nakamura, M. Fujikawa and T. Okamoto, Bull. Chem. Soc. Jpn., 1988, 61, 597 CrossRef CAS.
- H. Gilman and E. A. Zuech, J. Am. Chem. Soc., 1960, 82, 2522 CrossRef CAS.
- M. E. Lee, H. M. Cho, C. H. Kim and W. Ando, Organometallics, 2001, 20, 1472 CrossRef CAS.
- C. Reichardt, Chem. Rev., 1994, 94, 2319 CrossRef CAS.
- Y. Niko, S. Kawauchi and G. Konishi, Chem. – Eur. J., 2013, 19, 9760 CrossRef CAS PubMed.
- A. S. Klymchenko and A. P. Demchenko, Phys. Chem. Chem. Phys., 2003, 5, 461 RSC.
- O. A. Kucherak, L. Richert, Y. Mély and A. S. Klymchenko, Phys. Chem. Chem. Phys., 2012, 14, 2292 RSC.
- (a) E. L. Lippert, Organic Molecular Photophysics, Wiley, New York, 1975 Search PubMed; (b) N. Mataga and T. Kubota, Molecular Interactions and Electronic Spectra, Dekker, New York, 1970 Search PubMed.
- S. I. van Dijk, P. G. Wiering, C. P. Groen, A. M. Brouwer, J. W. Verhoeven, W. Schuddeboom and J. M. Warman, J. Chem. Soc., Faraday Trans., 1995, 91, 2107 RSC.
- A. K. Dutta, K. Kamada and K. Ohta, J. Photochem. Photobiol., A, 1996, 93, 57 CrossRef CAS.
- S. Kyushin, Y. Ishikita, H. Matsumoto, H. Horiuchi and H. Hiratsuka, Chem. Lett., 2006, 35, 64 CrossRef CAS.
- (a) Z. R. Grabowski, K. Rotkiewicz and W. Rettig, Chem. Rev., 2003, 103, 3899 CrossRef PubMed; (b) H. Kotaka, G. Konishi and K. Mizuno, Tetrahedron Lett., 2010, 51, 181 CrossRef CAS.
- O. A. Kucherak, P. Didier, Y. Mély and A. S. Klymchenko, J. Phys. Chem. Lett., 2010, 1, 616 CrossRef CAS.
- S. Sasaki, Y. Niko, A. S. Klymchenko and G. Konishi, Tetrahedron, 2014, 70, 7551 CrossRef CAS.
- E. Yamaguchi, C. Wang, A. Fukazawa, M. Taki, Y. Sato, T. Sasaki, M. Ueda, N. Sasaki, T. Higashiyama and S. Yamaguchi, Angew. Chem., Int. Ed., 2015, 54, 4539 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Analytical and spectral date, details of computational data and 1H and 13C NMR spectra are available. CCDC 1417594 (1) and 1417595 (2). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5nj02839f |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |