
Tungstosulfonic acid as an efficient solid acid catalyst for acylal synthesis for the protection of the aldehydic carbonyl group†
Reddi Mohan Naidu
Kalla
,
Mi Ri
Kim
,
Yu Na
Kim
and
Il
Kim
*
BK21 PLUS Centre for Advanced Chemical Technology, Department of Polymer Science and Engineering, Pusan National University, Busan 609-735, Republic of Korea. E-mail: ilkim@pusan.ac.kr
First published on 16th November 2015
Abstract
Tungstosulfonic acid (TSA) has been found to be an efficient solid acid catalyst for the protection of aldehydic carbonyl groups by geminal diacetate (acylal) formation following the nucleophilic addition of acetic anhydride under neat conditions as well as in a solvent. The TSA catalyst is fully characterized by infrared spectroscopy, wide-angle X-ray scattering analysis, and scanning electron microscopy with energy dispersive X-ray spectroscopy. The deprotection of acylals to corresponding aldehydes has also been investigated under the similar conditions. The catalyst can be reused seven times without a significant loss of activity. In addition, no chromatographic separations are needed to obtain the desired products. This method is a green approach for the chemoselective protection of aldehydes in the presence of ketones.
1. Introduction
Heterogeneous solid acid (HSA) catalysts have a variety of advantages over liquid acid catalysts and have received extensive attention in synthetic organic chemistry as a result of their financial and environmental benefits, which include simple product separation, possible recycling, and reduced liberation of toxic residues into the atmosphere.1 The ability to deploy HSA catalysts under neat experimental conditions is an important benefit in industrial chemistry. Sulfonic-acid-supported heterogeneous catalysts drive difficult organic transformations faster and employ mild reaction conditions. These catalysts enable feasible and cost-effective synthetic procedures. Because solid catalysts can be separated from reaction mixtures by simple filtration, they can be reused a number of times, which reduces the risk of discharging poisonous reaction residues into the environment compared with conventional homogeneous catalysts. In recent years there has been considerable research activity in the use of sulfonic-acid-supported catalysts in synthetic organic reactions. Sulfonic-acid-supported catalysts have attracted attention because of their many advantages, which include high thermal stability, low toxicity, high efficiency, good selectivity, low expense, reusability, and ease of separation of the catalyst from the reaction mixture.2,3 Tungstosulfonic acid (TSA) is a class of heterogeneous acid catalysts and the chemical structures of tungstic acid (TA) and TSA are shown in Fig. 1.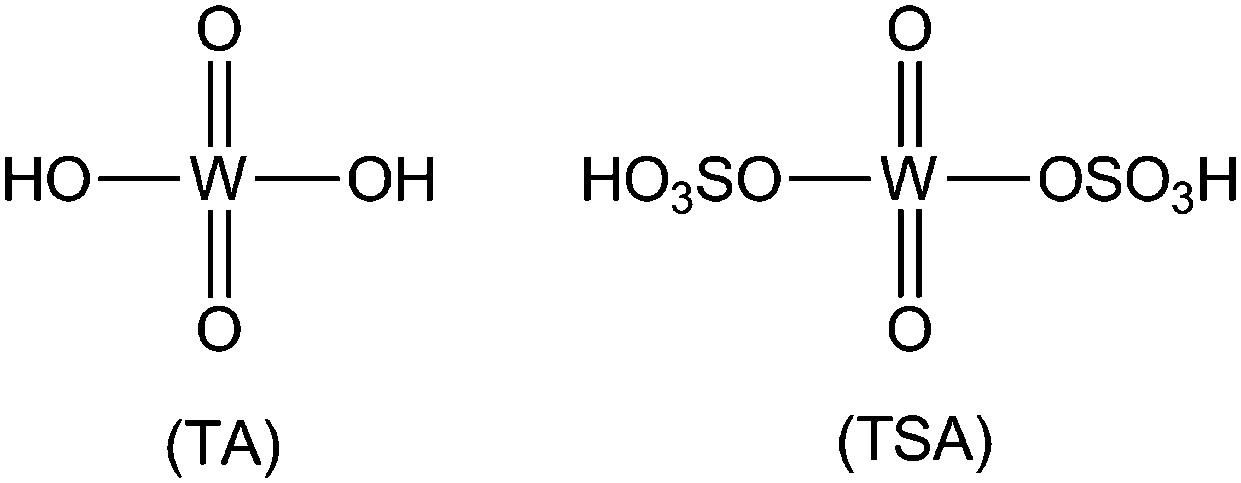 | ||
| Fig. 1 The chemical structures of TA and TSA. | ||
Selective protection of aldehydic carbonyl groups by conversion to their corresponding acylals is an important component of multistep organic syntheses. A crucial property of the acylals formed in this process is their stability in neutral and basic media.4 Reagents available for the protection of aldehydic carbonyl groups include ethanedithiol,5 alcohols,6 2-mercaptoethanol,7 acetic anhydride,8 and trialkyl orthoformate.9 Among all of them acetic anhydride is easy to handle and avoids the offensive odour. Typically, the formation of acylals is promoted by strong protic acids such as NH2SO3H,10 KHSO4,11 and H2SO412,13 and Lewis acids such as ZrCl4,14 AlCl3,15 and Bi(OTf)3.16 From economic and environmental points of view the use of heterogeneous catalysts such as HClO4·SiO2,17 MCM-41-SO3H,18 and zeolite bet supported sulfonic acid (BEA–SO3H)19 has also been reported. However, Lewis acids are susceptible to moisture, and the other catalysts exhibit disadvantages that may include the requirement of a long reaction time, high catalyst loading, high temperature, and microwave or ultrasound stimulation.10 Therefore, the development of an economical protocol employing an easily accessible low toxicity solid acid catalyst and the ability to proceed under neat conditions is greatly preferred for the synthesis of acylals. Synthetic heterocyclic compounds are most important in the fields of organic and medicinal chemistry because of their broad range of pharmacological applications.
In various research laboratories and chemical industries enormous quantities of organic solvents are used and are wasted all over the world. The development of neat (solvent-free) conditions can play an increasingly vital role in synthetic organic chemistry, from not only a practical but also an ecological point of view.20 The most significant goals of “green chemistry” include atom economy, prevention of waste, use of renewable feedstock, use of catalytic reagents, reduction of expenses, easy workup and fast reaction rates.21 Furthermore, the reduction of by-products and the reduction of evaluated toxic gas during the reaction are also an important factor in green chemistry. The most important goals of neat chemistry are to reduce the use of toxic organic solvents, toxic reagents, and laborious work-up procedures associated with the synthesis of various organic compounds.22–29
To the best of our knowledge in the open literature there have been no reports on the tungstosulfonic acid (TSA) catalysed synthesis of acylals. Considering this green chemistry in mind we report herein the use of TSA as a catalyst for the conversion of aldehydes to acylals using acetic anhydride as a protecting agent, which is one of the efficient ways of protecting the aldehyde group. The TSA catalysed reaction was found to be an environmentally benign, solvent-free, inexpensive, non-volatile and non-corrosive process. In addition the heterogeneous TSA catalyst was easy to prepare and recycle.30
2. Results and discussion
In this work 25 acylals were synthesized by using the environmentally friendly TSA catalyst under neat conditions. Among them six compounds, 7, 8, 9, 14, 15 and 21, are new compounds. The method offers many advantages over other literature procedures in terms of yield, ease of preparation, and recyclability of the catalyst. Moreover, we studied the deprotection of acylals under solvent-free conditions. The deprotection of acylals to their corresponding aldehydes proceeds in good yields in the presence of 2.5 mol% of TSA.2.1. Chemistry
In general the protection of aldehydes is very important one during multistep synthesis. Furthermore, the protection of aldehydes by reaction with acetic anhydride is impossible to proceed without the use of a catalyst. The preparation of acylals usually requires high catalyst loadings, long reaction times, and exotic reagents to promote the formation of the desired product. To avoid these problems we performed acylal formation reaction in the presence of TSA that is inexpensive, easy to prepare, and recyclable under mild and neat reaction conditions (Scheme 1).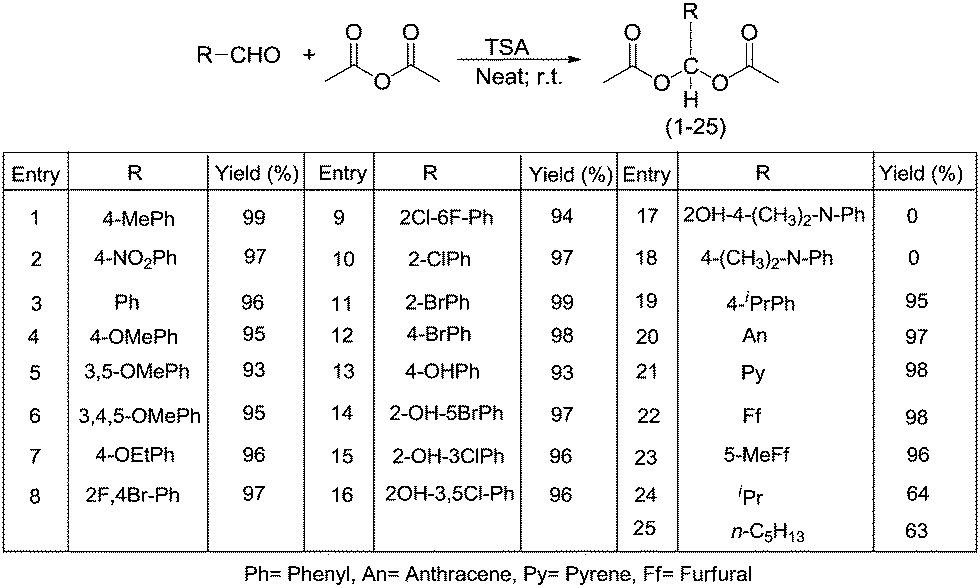 | ||
| Scheme 1 Synthesis of a series of acylals (1–25). | ||
As a first step we investigated acylal synthesis using several inexpensive and easily prepared solid-supported acid catalysts including silicatungstic acid (STA),31 SiO2·B(OH)2,32 KHSO4·SiO2,33 and SiO2·SO3H34 under neat conditions. The results are summarized in Table 1. All catalysts gave acceptable yields except STA. In addition, we attempted the reaction using tungstic acid as a catalyst, but no desired product was formed after 24 h. The reaction was also carried out using ClSO3H as a catalyst; however, the yield was low and the separation of the catalyst from the reaction mixture was difficult. Subsequently we thought that the low yields could be improved by combining TA and ClSO3H to form tungstosulfonic acid, which could be applied to the synthesis of acylals and would be easily separated from the reaction mixture. Pleasantly, the reactions proceeded efficiently in high yields. The synergistic properties of TSA resulted in good yields compared to those of TA or ClSO3H alone.
| Entry | Catalyst | Catalyst amount (mol%) | Time (h) | Yieldb (%) |
|---|---|---|---|---|
| a Reaction conditions: p-methyl benzaldehyde = 1 mmol, acetic anhydride = 1 mmol, and TSA = 2.5 mol%. b Isolated yield measured gravimetrically. c No reaction. | ||||
| 1 | Catalyst free | — | 24 | nrc |
| 2 | STA31 | 7 | 2 | 35 |
| 3 | SiO2·B(OH)232 | 5 | 0.3 | 75 |
| 4 | KHSO4·SiO233 | 5 | 0.5 | 78 |
| 5 | SiO2·SO3H34 | 5 | 0.6 | 88 |
| 6 | TA | 5 | 24 | nrc |
| 7 | ClSO3H | 1 | 0.1 | 75 |
| 8 | TSA | 1 | 0.5 | 84 |
| 9 | TSA | 1.5 | 0.1 | 91 |
| 10 | TSA | 2 | 0.1 | 96 |
| 11 | TSA | 2.5 | 0.03 | 99 |
| 12 | TSA | 3 | 0.03 | 99 |
To compare the effectiveness of neat versus solvent reaction conditions for the synthesis of 1 was performed using TSA as a catalyst in different solvents (Table 2). When the reaction was conducted in protic solvents such as isopropanol (IPA), acetic acid (AcOH), methanol (MeOH), ethanol (EtOH), or water (H2O), the reaction proceeded slowly and provided reduced product yields (Table 2, entries 2–6). However, when aprotic solvents such as toluene, 1,4-dioxane, chloroform (CHCl3), or dichloromethane (CH2Cl2) were used, the reaction proceeded faster and resulted in higher product yields (Table 2, entries 7–10).
| Entry | Solvent | Time (min) | Yield (%) |
|---|---|---|---|
| a Reaction conditions: p-methyl benzaldehyde = 1 mmol, acetic anhydride = 1 mmol, and TSA = 2.5 mol% at room temperature. | |||
| 1 | Solvent-free | 2 | 99 |
| 2 | IPA | 40 | 62 |
| 3 | AcOH | 45 | 64 |
| 4 | MeOH | 56 | 50 |
| 5 | EtOH | 60 | 65 |
| 6 | H2O | 75 | 60 |
| 7 | Toluene | 35 | 86 |
| 8 | 1,4-Dioxane | 25 | 90 |
| 9 | CHCl3 | 23 | 89 |
| 10 | CH2Cl2 | 18 | 91 |
To establish the optimum catalyst level of TSA the model reaction was carried out using 1, 1.5, 2, 2.5, and 3 mol% of neat TSA at room-temperature (r.t). The product yields were 86%, 91%, 96%, 99%, and 99%, respectively (Table 1, entries 8–12). Increasing the amount of catalyst beyond 2.5 mol% offered no further enhancement of the reaction.
We next explored the general applicability of the reaction conditions employing a catalytic amount of TSA in the synthesis of acylals from various aromatic, heteroaromatic, and aliphatic aldehydes (Table 3). Results were obtained within 2–9 min. We found the reaction to be well-suited to compounds containing halide, nitro, cyano, methoxy, and ethoxy functionalities. In the case of hydroxy benzaldehyde and salicylaldehydes the corresponding triacetates were formed (13–16). It is possible to monitor the protection reaction of aldehydes using acetic anhydride visually. In case of aromatic aldehydes a clear solution is observed upon mixing of the aldehyde and anhydride; addition of the catalyst to the reaction mixture results in the formation of a solid (after stirring for a few minutes) indicating the completion of the reaction. Exceptions to this rule are the 4-ethoxy-and 4-isopropyl-benzaldehyde derivatives (7 and 19), in which case liquids are obtained as indicated by thin layer chromatography (TLC). Aliphatic aldehydes (24 and 25) also formed liquids and were identified by TLC. In the case of p-dimethylamino salicylaldehyde and p-(N,N-dimethylamino)benzaldehyde TSA was notable to catalyse the conversion to the corresponding acylals (17 and 18).
| Entry | R in R-CHO | Yield (%) | Time (min) | m.p. (°C) |
|---|---|---|---|---|
| a Reaction conditions: p-methyl benzaldehyde = 1 mmol and acetic anhydride = 1 mmol. | ||||
| 1 | 4-Me-C6H4 | 99 | 2 | 63–65 |
| 2 | 4-NO2-C6H4 | 97 | 5 | 124–125 |
| 3 | C6H5 | 96 | 4 | 43–44 |
| 4 | 4-OMe-C6H4 | 95 | 6 | 80–82 |
| 5 | 3,5-OMe-C6H3 | 93 | 8 | 93–95 |
| 6 | 3,4,5-OMe-C6H2 | 95 | 7 | 111–113 |
| 7 | 4-OEt-C6H4 | 96 | 5 | Oil |
| 8 | 2-F-4-Br-C6H3 | 97 | 5 | 50–52 |
| 9 | 2-Cl-6-F-C6H3 | 94 | 4 | 92–94 |
| 10 | 2-Cl-C6H4 | 97 | 3 | 56–57 |
| 11 | 2-Br-C6H4 | 99 | 2 | 78–79 |
| 12 | 4-Br-C6H4 | 98 | 3 | 81–82 |
| 13 | 4-OH-C6H4 | 93 | 8 | 93–94 |
| 14 | 2-OH-5-Br-C6H3 | 97 | 5 | 92–94 |
| 15 | 2-OH-3-Cl-C6H3 | 96 | 4 | 65–66 |
| 16 | 2-OH-3,5-Cl-C6H2 | 96 | 5 | 72–74 |
| 17 | 2-OH-4-(CH3)2-N-C6H3 | 0 | 10 h | nrc |
| 18 | 4-(CH3)2-N-C6H4 | 0 | 10 h | nrc |
| 19 | 4-iPr-C6H4 | 95 | 4 | Oil |
| 20 | C14H9 (Anthrecene) | 97 | 5 | 200–202 |
| 21 | C14H9 (Pyrene) | 98 | 3 | 66–68 |
| 22 | 2-Furfural(C4H3O) | 98 | 6 | 51–52 |
| 23 | 5-Me-2-Furfural(C4H2O) | 96 | 8 | 50–52 |
| 24 | CH(CH3)2 | 64 | 8 | Oil |
| 25 | C5H13 | 63 | 7 | Oil |
No reaction was observed for acetophenone at room temperature after 24 h as well as at 60 °C after 12 h. No new spot was observed by TLC, and no corresponding protons were observed in the NMR spectrum. Encouraged by this result, we conducted a competitive reaction involving the acylation of p-methyl benzaldehyde in the presence of acetophenone under our standard reaction conditions. We found that p-methyl benzaldehyde was converted to the corresponding acylal, while acetophenone remained unreacted (Scheme 2). This examination indicates that aldehydes are more reactive than ketones most probably due to higher electrophilicity and less steric hindrance of aldehydes than ketones.
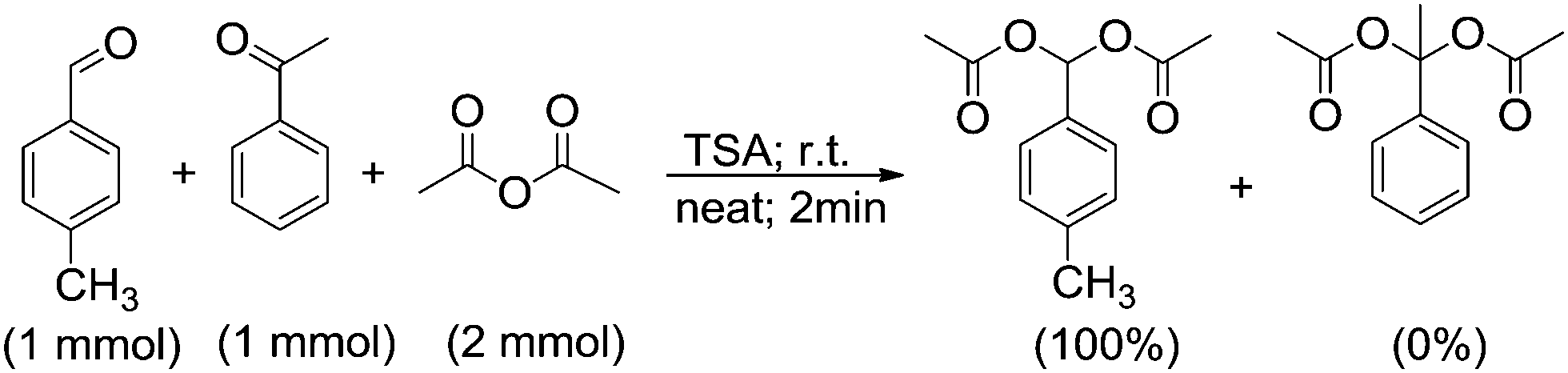 | ||
| Scheme 2 Competitive acylal formation from p-methyl benzaldehyde in the presence of acetophenone. | ||
In addition to the synthetic process we investigated the possible deprotection of product acylals within the same catalytic system by adding water. For this purpose the reaction system was monitored for 2 h after forming acylal 1 from p-methyl benzaldehyde without changing conditions. Conversion of the reaction system from solid to oily liquid in water suggested that the product acylal is deportected (Scheme 3). The resulting mixture was examined by NMR spectroscopy without purification. A 1H NMR spectrum of the resulting mixture before purification shows a singlet at δ 9.91 ppm which is corresponding to an aldehydic proton. In addition there were no peaks between δ 80 and 95 ppm in the 13C NMR spectrum, indicating the absence of Ar–CH carbon. The new peak at around δ 178 ppm clearly indicates the carbonyl carbon. All of these results show that aceticanhydride is converted to acetic acid during the deprotection reaction (Fig. 2).
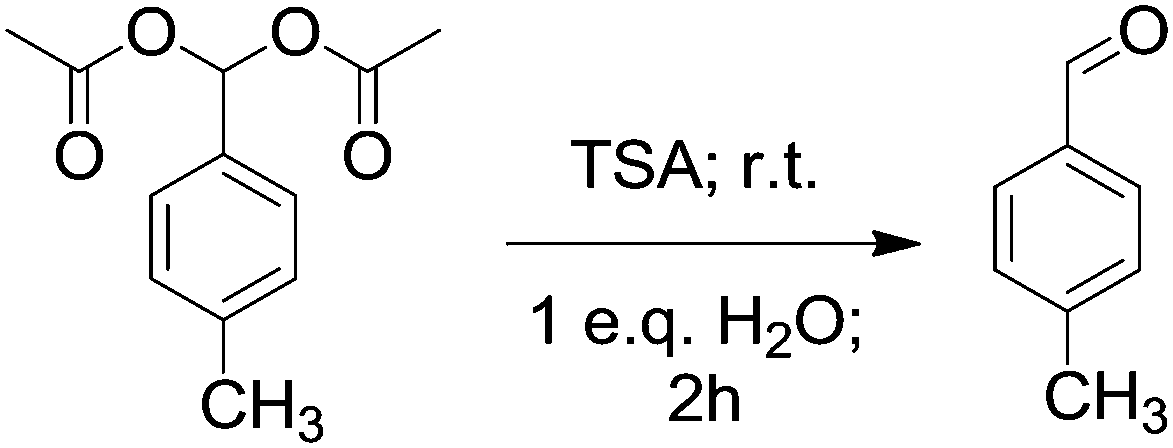 | ||
| Scheme 3 TSA-catalyzed cleavage of acylal 1 to p-methyl benzaldehyde in the presence of water. | ||
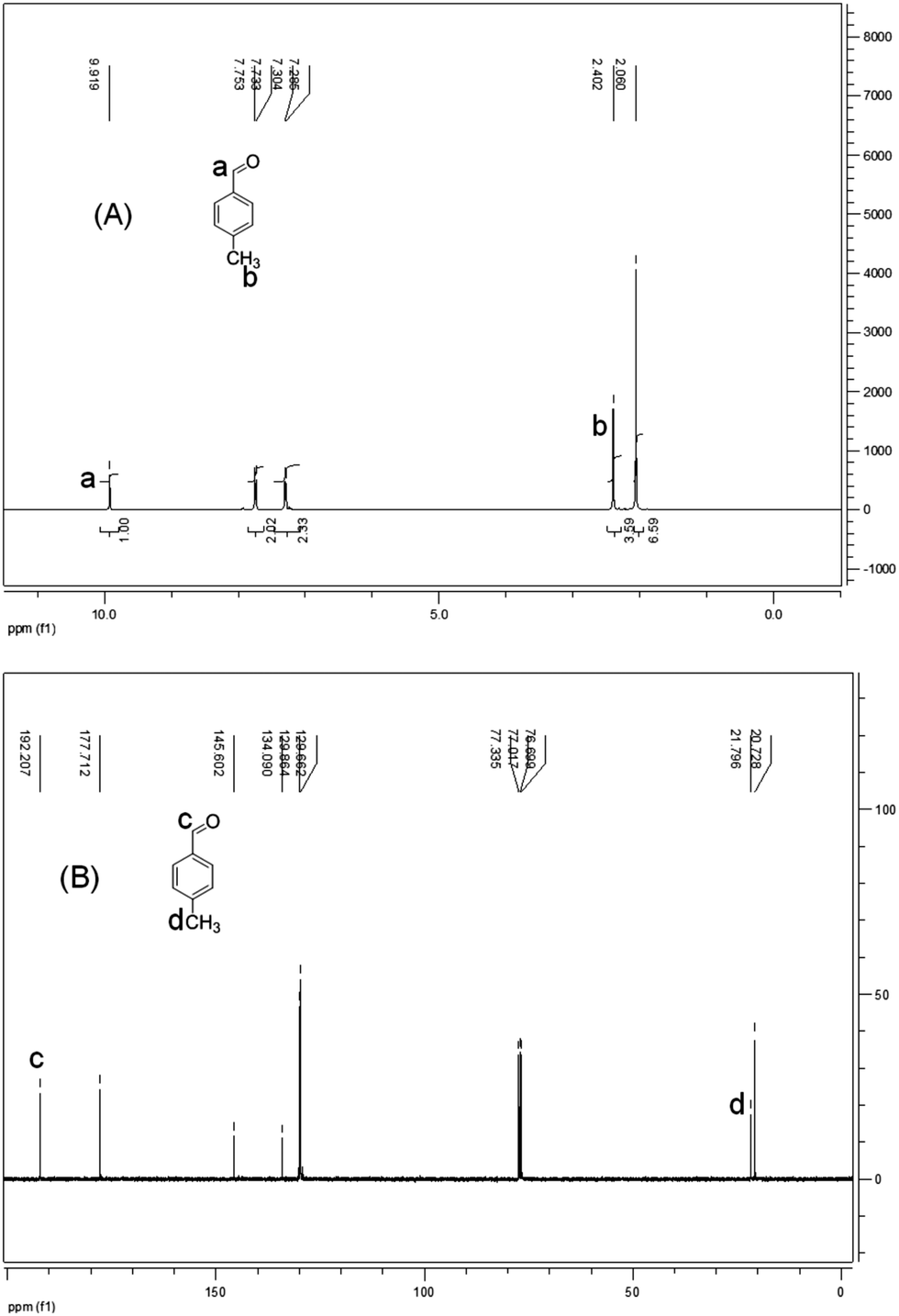 | ||
| Fig. 2 (A) 1H NMR and (B) 13C NMR spectra of a solution mixture after 2 h of reaction. | ||
The performance of TSA compared to that of other recently reported heterogeneous catalysts for the acylation of aldehydes is summarized in Table 4. In 2013 Zareyee35 reported the formation of acylals using a nanosilica material (SBA-15-Ph-PrSO3H); the reaction rate was high, and acetic anhydride was used in excess. Tourani et al.,18 reported the synthesis of acylals using MCM-41-SO3H, wherein the heterogeneous catalyst loading was high; the product yield was low compared to that of TSA. Kannasani et al.36 used NaHSO4–SiO2 as a supported catalyst at high loading with a fourfold excess of acetic anhydride; column chromatography was used for the separation of the acylals. In 2007 Hajipour et al.,37 employed P2O5/Al2O3 as a catalyst for the protection of aldehydes with acetic anhydride; this procedure required a long time. Compared to all the heterogeneous acid catalysts mentioned above, TSA as described herein is superior for the high yield synthesis of acylals, because it requires neither harsh reaction conditions, additional energy input (i.e., microwave or ultra-sonication), nor laborious work-up procedures.
| Entry | Catalyst (mg) | Solvent | Time (min) | Yield (%) | Ref. |
|---|---|---|---|---|---|
| a Reaction conditions: p-methyl benzaldehyde = 1 mmol and acetic anhydride = 1 mmol. | |||||
| 1 | SBA-15Ph-pr-SO3H (1) | Neat | 5 | 100 | 31 |
| 2 | MCM-41-SO3H (10) | Neat | 3 | 95 | 18 |
| 3 | NaHSO4–SiO2 (25%/wt) | Neat | 15 | 94 | 32 |
| 4 | P2O5/Al2O3 (15 mol%) | Neat | 45 | 86 | 33 |
| 5 | TSA (2.5 mol%) | Neat | 2 | 99 | This work |
Reusability of a catalyst is crucial for large-scale operations and from an industrial point of view. Therefore, the reusability of TSA was examined in the synthesis of 1 formed by the reaction of acetic anhydride with p-methyl benzaldehyde. The catalyst was effortlessly recovered by adding chloroform to the reaction mixture. Insoluble TSA was separated by simple filtration, washed twice with chloroform (20 mL), and dried under vacuum at 100 °C. The catalyst displayed good reusability after 7 runs (Fig. 3).
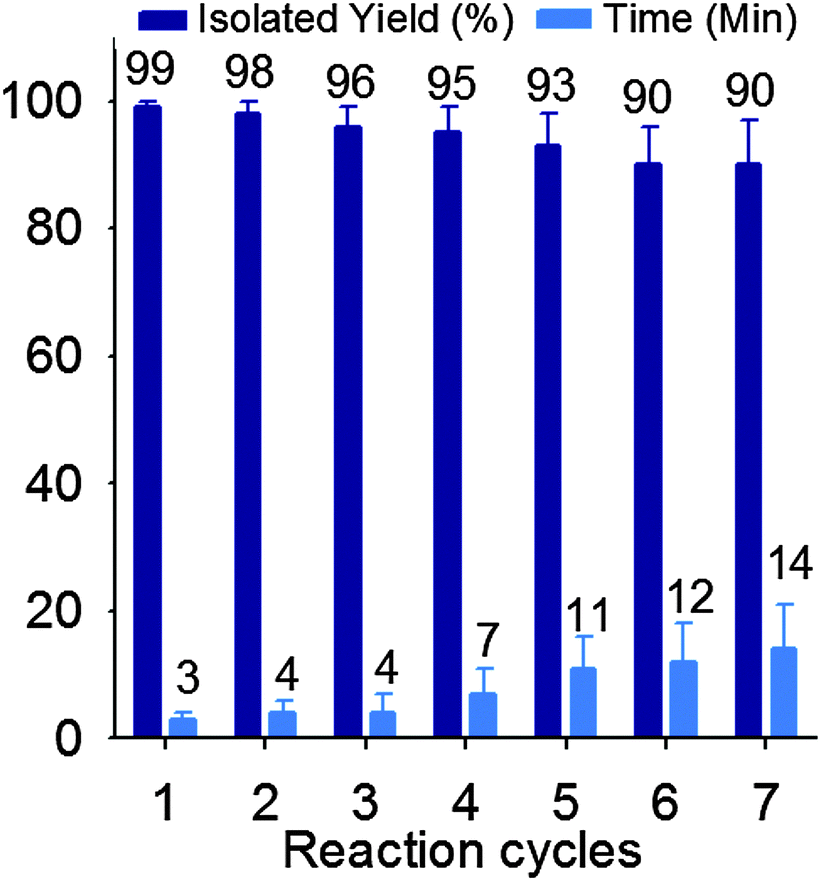 | ||
| Fig. 3 Effect of recycling of TSA on the yield of acylal 1. | ||
Based on the obtained results, the reaction mechanism might be proposed for the protection of aldehydes with acetic anhydride in the presence of TSA (Scheme 4). At first p-methyl benzaldehyde reacts with acidic hydrogen from TSA to form an intermediate benzylideneoxonium ion which reacts with acetic anhydride to form an oxonium ion. This oxonium ion undergoes an intramolecular rearrangement and the proton is rearranged to TSA to form targeted acylals.
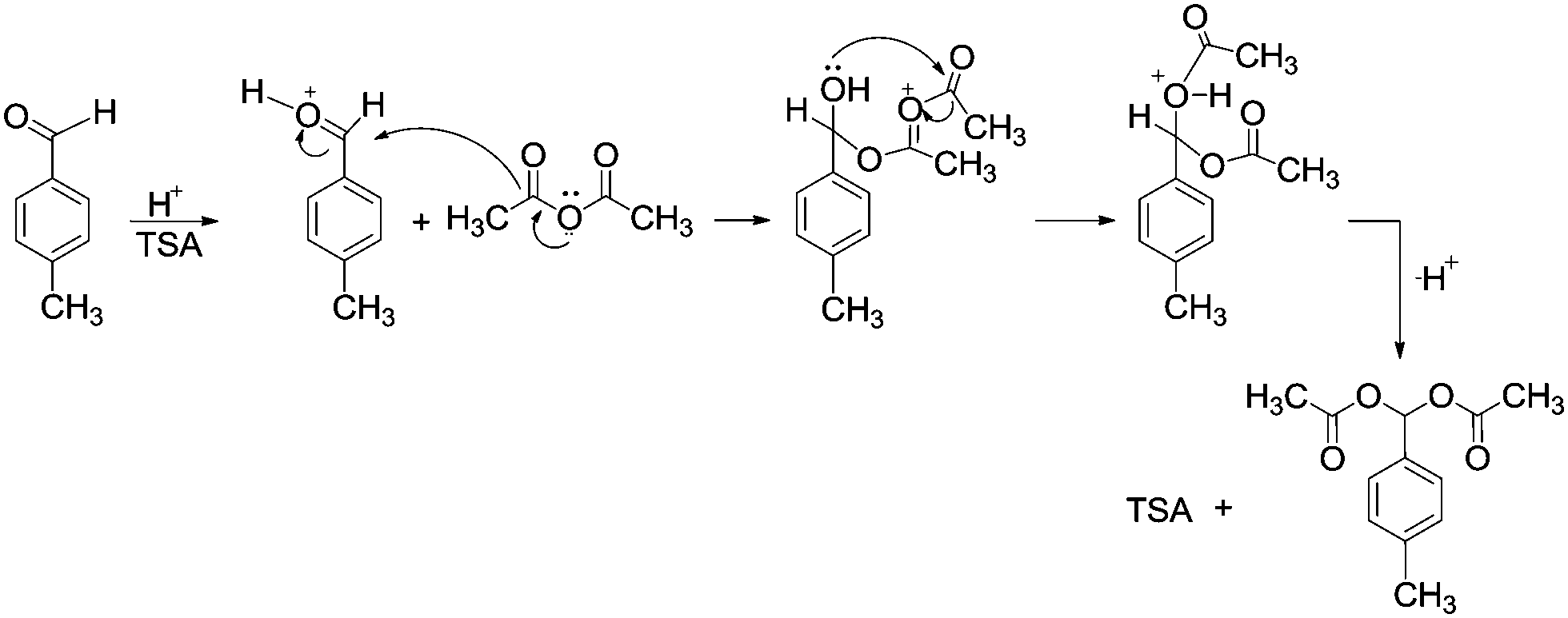 | ||
| Scheme 4 The plausible reaction mechanism for the formation of acylals in presence of TSA. | ||
The structures of all the compounds were confirmed by IR, 1H and 13C NMR spectroscopies. The IR spectra of compounds 1–25 showed the expected regions. In the 1H NMR spectrum, the Ar-CH proton signal appeared as a singlet in the region of δ 9.22–7.56 ppm. The remaining proton signals were observed in the expected regions. In the 13C NMR spectra, the Ar-CH carbon signal appeared at δ 89.5–83.6 ppm confirms the formation of acylals and all known products were identical with those reported in the literature.31 The detailed descriptions of the spectral data for all compounds (1–25) are given in the experimental section.
3. Conclusions
A straightforward, effective and green procedure for the synthesis of acylals catalysed by heterogeneous tungstosulfonic solid acid under neat conditions has been described. This low-cost method has a number of advantages including use of readily available chemicals, high yields, neat reaction conditions (solvent-free), and easy work-up procedures. The procedure provides chemists with a fast and effective method for preparing diversely substituted acylals. The TSA catalyst has been fully characterized by FTIR, XRD, and SEM-EDX to confirm the sulfonation of tungstic acid.4. Experimental
4.1. General
The various substituted aldehydes were purchased from Sigma Aldrich Co. (St. Louis, MO, USA), tungstic acid (TA) and chloro sulfonic acid (Yakuri Pure Chemical Co., Japan). All experiments were carried out under solvent-free conditions. Analytical thin layer chromatography (TLC) was carried out in an ethyl acetate/n-hexane (8/2) mixture on pre-coated silica gel plates (Merck Chem., Germany) and developed by use of iodine. Fourier transform infrared (FT-IR) spectra were recorded on a Shimadzu IR Prestige 21 spectrometer at room temperature. The samples were analyzed as KBr discs in the range of 3500–500 cm−1. The 1H NMR (400 MHz) and 13C NMR (100 MHz) spectra were recorded on a Varian INOVA 400 NMR spectrometer at room temperature. Chemical shift values are reported relative to tetramethylsilane (TMS, Me4Si). The data are presented as follows: chemical shift (ppm), multiplicity (s = singlet, d = doublet, m = multiplet), and coupling constant, J (Hz). The XRD analysis was performed by use of an automatic Philips powder diffractometer with nickel-filtered Cu Kα radiation. The diffraction pattern was collected in the 2θ range of 0–80° in steps of 0.02° with counting times of 2 s step−1. The microstructures of the samples and the energy dispersive X-ray spectra (EDX) were investigated by use of an S-3000 scanning electron microscope (SEM; Hitachi, Japan).4.2. General procedure for the synthesis of acylals 1–25
In a 50 mL round bottom flask TSA (2.5 mol%) was added to a mixture of p-methyl benzaldehyde (1 mmol) and acetic anhydride (1 mmol). The reaction mixture was stirred at room temperature, becoming solid within 2 min. Chloroform was added, and the insoluble catalyst was separated by simple filtration, washed twice with chloroform, and dried at 100 °C for 2 h for reuse. The chloroform filtrate was evaporated under reduced pressure and the solid obtained was washed using petroleum ether (20 mL) three times. Recrystallization from dichloromethane afforded the resulting acylal, 1. All known products afforded spectral and physical data consistent with those reported in the literature. New products were characterized by their IR, 1H NMR, and 13C NMR spectra. Detailed descriptions for all these compounds are given below.4.3. Spectral characterization
Acknowledgements
This work was supported by the Fusion Research Program for Green Technologies through the National Research Foundation of Korea funded by the Ministry of Science, ICT and Future Planning (2012M3C1A1054502) and the Basic Science Research Program through the National Research Foundation of Korea (2015R1D1A1A09057372). The authors also thank the BK21 PLUS Program for partial financial support.Notes and references
- M. Parveen, F. Ahmad, A. M. Malla and S. Azaz, New J. Chem., 2015, 39, 2028–2041 RSC.
- R. M. N. Kalla, H. Park, T. T. K. Hoang and I. Kim, Tetrahedron Lett., 2014, 55, 5373–5376 CrossRef CAS.
- A. R. Kiasat, A. Mouradzadegun and S. J. Saghanezhad, J. Serb. Chem. Soc., 2013, 78, 469–476 CrossRef CAS.
- T. W. Greene and P. G. M. Wuts, Protective Groups in Organic Synthesis, John Wiley and Sons, New York, 3rd edn, 1999 Search PubMed.
- G. Perin, L. G. Mello, C. S. Radatz, L. Savegnago, D. Alves, R. G. Jacob and E. J. Lenardao, Tetrahedron Lett., 2010, 51, 4354–4356 CrossRef CAS.
- S. Madabhushi, K. K. R. Mallu, N. Chinthala, C. R. Beeram and V. S. Vangipuram, Tetrahedron Lett., 2012, 53, 697–701 CrossRef CAS.
- F. Shirini, P. Sadeghzadeh and M. Abedini, Chin. Chem. Lett., 2009, 20, 1457–1460 CrossRef CAS.
- N. G. Khaligh and F. Shirini, J. Mol. Catal. A: Chem., 2011, 348, 20–29 CrossRef CAS.
- A. Roy, M. Rahman, S. Das, D. Kundu, S. K. Kundu, A. Majee and A. Hajra, Synth. Commun., 2009, 39, 590–595 CrossRef CAS.
- T. S. Jin, G. Sun, Y. W. Li and T. S. Li, Green Chem., 2002, 4, 255–256 RSC.
- M. M. Heravi, K. Bakhtiari, S. Taheri and H. A. Oskooie, Green Chem., 2005, 7, 867–869 RSC.
- M. J. Gregory, J. Chem. Soc. B, 1970, 1201–1207 RSC.
- M. Tomita, T. Kikuchi, K. Bessho, T. Hori and Y. Inubushi, Chem. Pharm. Bull., 1963, 11, 1484–1489 CrossRef CAS PubMed.
- G. Smitha and C. S. Reddy, Tetrahedron, 2003, 59, 9571–9576 CrossRef CAS.
- S. Gowravaram, S. Abraham, T. Ramalingam and J. S. Yadav, J. Chem. Res., 2002, 3, 144–147 Search PubMed.
- M. D. Carrigan, K. J. Eash, M. C. Oswald and R. S. Mohan, Tetrahedron Lett., 2001, 42, 8133–8135 CrossRef CAS.
- V. T. Kamble, V. S. Jamode, N. S. Joshi, A. V. Biradara and R. Y. Deshmukha, Tetrahedron Lett., 2006, 47, 5573–5576 CrossRef CAS.
- H. Tourani, M. R. N. Jamal, M. G. Dekamin and M. Amirnejad, C. R. Chim., 2012, 15, 1072–1076 CrossRef CAS.
- R. J. Kalbasi, A. R. Massah and A. Shafiei, J. Mol. Catal. A: Chem., 2011, 335, 51–59 CrossRef CAS.
- D. J. C. Constable, P. J. Dunn, J. D. Hayler, G. R. Humphrey, J. L. LeazerJr, R. J. Linderman, K. Lorenz, J. Manley, B. A. Pearlman and A. Wells, Green Chem., 2007, 9, 411–420 RSC.
- K. Tanaka and F. Toda, Chem. Rev., 2000, 100, 1025–1074 CrossRef CAS PubMed.
- R. M. N. Kalla, J. V. John, H. Park and I. Kim, Catal. Commun., 2014, 57, 55–59 CrossRef CAS.
- R. M. N. Kalla, M. R. Kim and I. Kim, Tetrahedron Lett., 2015, 56, 717–720 CrossRef CAS.
- R. M. N. Kalla, H. R. Lee, J. Cao, J. W. Yoo and I. Kim, New J. Chem., 2015, 39, 3916–3922 RSC.
- R. M. N. Kalla, S. J. Byeon, M. S. Heo and I. Kim, Tetrahedron, 2013, 69, 10544–10551 CrossRef CAS.
- R. M. N. Kalla, J. S. Choi, J. W. Yoo b, S. J. Byeon, M. S. Heo and I. Kim, Eur. J. Med. Chem., 2014, 76, 61–66 CrossRef CAS PubMed.
- K. R. M. Naidu, B. Satheesh Krishna, M. A. Kumar, P. Arulselvan, S. K. Ibrahim and O. Lasekan, Molecules, 2012, 17, 7543–7555 CrossRef CAS PubMed.
- K. R. M. Naidu, S. I. Khalivulla, S. Rasheed, S. Fakurazi, P. Arulselvan, O. Lasekan and F. Abas, Int. J. Mol. Sci., 2013, 14, 1843–1853 CrossRef CAS PubMed.
- K. R. M. Naidu, S. I. Khalivulla, P. C. R. Kumar and O. Lasekan, Org. Commun., 2012, 5, 150–159 CAS.
- S. Khodabakhshi and B. Karami, Tetrahedron Lett., 2014, 55, 7136–7139 CrossRef CAS.
- B. Karami, V. Ghashghaee and S. Khodabakhshi, Catal. Commun., 2012, 20, 71–75 CrossRef CAS.
- M. Parveen, F. Ahmad, A. M. Malla and S. Azaz, New J. Chem., 2015, 39, 2028–2041 RSC.
- A. Goswami, R. N. Das and N. Borthakur, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 2007, 46, 1893–1895 Search PubMed.
- M. A. Zolfigol, Tetrahedron, 2001, 57, 9509–9511 CrossRef CAS.
- D. Zareyeea, S. M. Moosavi and A. Alaminezhad, J. Mol. Catal. A: Chem., 2013, 378, 227–231 CrossRef.
- R. K. Kannasani, V. V. S. Peruri and S. R. Battula, Chem. Cent. J., 2012, 6, 136–139 CrossRef CAS PubMed.
- A. R. Hajipour, A. Zareib and A. E. Ruohoa, Tetrahedron Lett., 2007, 48, 2881–2884 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR Spectras for the new compounds. See DOI: 10.1039/c5nj02697k |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |