
Nonanebis(peroxoic acid) mediated efficient and selective oxidation of sulfide†
Eknath M.
Gayakwad
,
Vilas V.
Patil
and
Ganapati S.
Shankarling
*
Institute of Chemical Technology, Department of Dyestuff Technology, Nathalal Parikh Road, Matunga, Mumbai, Maharashtra, India. E-mail: gsshankarling@gmail.com; gs.shankarling@gmail.com
First published on 21st October 2015
Abstract
Two efficient and rapid protocols for the selective oxidation of sulfide in the presence of various oxidizable functionalities such as –CHO, –CH2–OH, alkene and tert amine were investigated, using nonanebis(peroxoic acid) as an oxidant. The acetonitrile based system offers selective oxidation to sulfoxide and sulfone, while the water based system gives selective sulfone formation with a high conversion and 90–100% selectivity. Recovery and recycling of the parent acid of up to 75–80% was achieved in the water based protocol.
Introduction
The growing interest in and applications of sulfoxide and sulfone have encouraged investigation into new methodologies of their synthesis. Sulfoxide and sulfone are two important sulphur compounds which are widely used in the synthesis of fine chemicals, ligands1 in asymmetric catalysis, and oxo transfer reagents.2 The recent applications of π-conjugated heterocyclic compounds containing vinyl sulfone and sulfoxide3 have exhibited a wide variety of optical, electrical and optoelectric properties4 and have drawn much attention due to their potential applications in organic optoelectronics.5Ample oxidising systems have been reported for this oxidation purpose. Various transition metals catalysts such as Ti,6 Mo,7 Fe,8 V,9 W,10 Rh,11 Ru,12 Sc,13 Zr14 and Mn,15 Ce16 under homogeneous and heterogeneous conditions were used with hydrogen peroxide as an oxidant to carry out this transformation. Hydrogen peroxide shows a slow reactivity toward the oxidation of sulfide,17 for which reason it is either used in excess or in combination with a metal catalyst. Dioxirane is an efficient and mild oxygen transfer agent which works under both neutral and non nucleophilic conditions.18–22 Dieva23et al. have reported the use of chiral dioxirane for the synthesis of chiral sulfoxides. The oxidation of sulfide to sulfoxide and sulfone has been broadly studied using azohydroperoxides.24–27 These peroxides are highly reactive and exhibit a better oxygen transfer ability. Their reactivity is on a par with that of peracids. Though dioxirane and azohydroperoxides are effective in oxidizing various sulfides to the corresponding sulfoxides and sulfones, they suffer from major drawbacks such as instability and a shock sensitive and explosive nature. Azohydroperoxides in particular are highly explosive under dry conditions, and therefore it is essential to store them in the dark under slightly moist conditions in benzene at −60 °C temperature. Though the above systems offer recycling of the catalyst and low waste generation, in most cases the reactions need to perform at a high temperature for a longer time,28–31 and require halogenated solvents, promoters13,32 or co-catalysts. In addition to this, over-oxidation occurs and in some cases metal residue remains in the product. Enzymes,33 supported reagents,34 cyanuril chloride,35 silver nitrite with TBHP36 and phosphorous oxychloride37 are the other oxidizing agents used to carry out this transformation.
As a continuation of our research to explore the applications of aliphatic diperoxy acids in organic synthesis,38–40 here we report two efficient protocols for the selective oxidation of sulfide in acetonitrile and water as solvents using nonanebis(peroxoic acid). The diperoxy acid used here is stable at room temperature, easy to handle and non-shock sensitive in nature which was conformed by DSC analysis. It is noticeable that we have recovered 75–80% of the parent acid and recycled it for up to three cycles.
Results and discussion
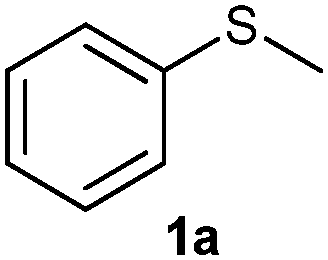
The reaction conditions were optimised using methyl phenyl sulfide 1a (MPS) (0.8 mmol, 1 equiv.) as a substrate and acetonitrile as a solvent at room temperature (Table 1). The effect of the oxidant on MPS was studied by varying its molar ratio from 0.5 to 1.6 equiv. (Table 1, entries 1–9). It was observed that 0.7 equiv. of the oxidant gives 94% of 2a in 7 min (Table 1, entry 3) while 1.5 equiv. of the oxidant gives 100% of 3a in 15 min (Table 1, entry 8). Along with acetonitrile, other solvents such as methanol, ethanol and ethyl acetate also show a higher conversion, but give a mixture of 2a and 3a (Table 1, entries 17–19). On the other hand, halogenated solvents such as dichloromethane, ethylene dichloride, chloroform (Table 1, entries 20–22) and acetic acid (Table 1, entry 23) demonstrate a good conversion as well as a good selectivity. Considering the hazards of using halogenated solvents and acetic acid we decided on acetonitrile as the best solvent for this reaction.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Solvent | Oxidant equiv. | Time (min) | Conv.b (%) | Selectivityb (%) | |
| 2a | 3a | |||||
| a Reaction conditions: 1a (0.8 mmol, 1 equiv.), solvent (3 mL), oxidants: nonanebis(peroxoic acid); temperature: 30–32 °C (room temperature). b Conversion and selectivity determined by GC with area normalization; DCM: dichloromethane; DCE: dichloroethane. c Reaction temperature: 50–55 °C. | ||||||
| 1 | Acetonitrile | 0.5 | 360 | 55 | 55 | — |
| 2 | 0.65 | 60 | 87 | 87 | — | |
| 3 | 0.7 | 7 | 100 | 94 | 6 | |
| 4 | 0.75 | 30 | 100 | 83 | 17 | |
| 5 | 1 | 120 | 100 | 59 | 41 | |
| 6 | 1.25 | 90 | 100 | 27 | 73 | |
| 7 | 1.4 | 90 | 100 | 12 | 88 | |
| 8 | 1.5 | 15 | 100 | — | 100 | |
| 9 | 1.6 | 15 | 100 | — | 100 | |
| 10 | Water | 0.7 | 12 h | 80 | 24 | 56 |
| 11c | Water | 0.7 | 25 | 100 | 76 | 24 |
| 12c | Water | 1.5 | 25 | 100 | — | 100 |
| 13 | Toluene | 0.7 | 120 | 82 | 52 | 30 |
| 14 | Hexane | 0.7 | 120 | 15 | 2 | 13 |
| 15 | Acetone | 0.7 | 20 | 71 | 69 | 2 |
| 16 | DMF | 0.7 | 20 | 59 | 41 | 8 |
| 17 | Methanol | 0.7 | 20 | 100 | 77 | 23 |
| 18 | Ethanol | 0.7 | 20 | 100 | 81 | 19 |
| 19 | Ethyl acetate | 0.7 | 20 | 97 | 85 | 12 |
| 20 | DCM | 0.7 | 20 | 100 | 98 | 2 |
| 21 | DCE | 0.7 | 20 | 100 | 94 | 6 |
| 22 | Chloroform | 0.7 | 20 | 100 | 97 | 3 |
| 23 | Acetic acid | 0.7 | 20 | 100 | 98 | 2 |
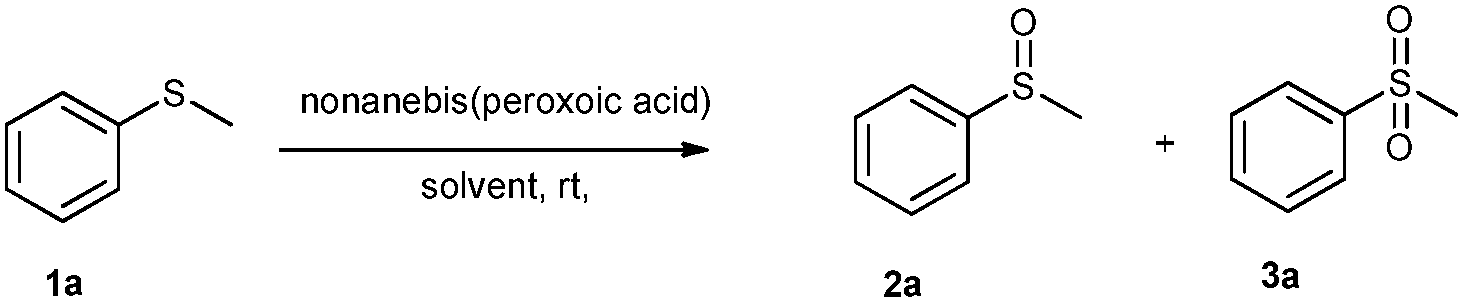
Acetonitrile plays a vital role in this reaction. The reaction proceeds in the homogeneous phase which allows the proper mixing of the substrate with peracid. We also believe that the nitrogen atom of the –CN stabilizes the partial positive charge on the sulfur atom in the transition state (as shown in the mechanism).41–44
The rate of the addition of peracid plays a vital role in higher selectivity for sulfoxide. When the oxidation of 1a to 2a was carried out by adding nonanebis(peroxoic acid) (0.7 equivalents) in one portion, exotherm was observed which further oxidizes the 2a formed in the reaction to 3a. The reaction resulted in the formation of 85% of 2a and 15% of 3a, whereas a slow addition of peracid over 2 to 3 min gives 94% of 2a and only 6% of 3a. Thus from these results, it was clear that the slow addition of peracid favours a higher selectivity for 2a.
During solvent free reaction conditions the strong exotherm observed was due to the vigorous reaction between the peracid and sulfide (which results in the rapid decomposition of the peracid). Also the improper mixing of the reaction mass leads to a mixture of products. The oxidation of 1a to 2a under solvent free conditions gives a mixture of 2a (29%) and 3a (32%), along with unreacted 1a (32%). Similarly, the oxidation of 1a to 3a under similar conditions results in a mixture of 2a (19%) and 3a (81%).
During the solvent study, we observed that when water was used as a solvent, 56% of 3a was obtained which was much higher than in all other oxidants under the sulfoxide formation conditions (Table 1, entry 10). Out of curiosity, we performed the same reaction at 50–55 °C and varied the oxidant equiv. During this, it was found that at 0.7 equiv. of oxidant, 76% of 2a was obtained in 25 min while at 1.5 equiv. of oxidant, 100% of 3a was obtained in 25 min (Table 1, entries 11 & 12). This indicates that at a higher temperature, selective sulfone formation occurs in water. With these studies in hand, we optimized two protocols; one with acetonitrile for 2a and 3a using 0.7 and 1.5 equiv. of oxidant at room temperature, and the second with water for 3a using 1.5 equiv. of oxidant at 50–55 °C.
It is well reported that sulfoxide is less nucleophilic in nature, due to which the oxidation of sulfoxide to sulfone require a longer time.42,45 Also, the rate of oxidation of sulfoxide to sulfone was lower when peracids were used as an oxidant. The reaction rate was enhanced either by adding an excess of oxidant or incorporating a metal catalyst along with an oxidant as an activator for the oxidant.45
The reaction conditions optimized using acetonitrile as a solvent were further used to investigate the effect of other oxidizing agents on the oxidation of 1a in to 2a (Table 2, entries 1–11). Amongst these oxidants, a higher conversion was obtained for 50% H2O2 and m-CPBA, but both failed to give a high selectivity for 2a (Table 2, entries 1 and 3). Other oxidants were found to give a poor conversion even after keeping the reaction at room temperature for 2 h (Table 2, entries 2 and 4–9). The study with the other diperoxy acids under the present conditions failed to give a high conversion, but offered a high selectivity for 2a in 2 h (Table 2, entries 10 and 11). On the other hand, when we performed the oxidation of 1a in to 3a in water using these diperoxy acids, it was observed that hexanebis(peroxoic acid) gives 100% conversion while dodecanebis(peroxoic acid) gives only 20% conversion to 3a (Table 2, entries 12 and 13).
| Entry | Oxidant | Conv.b (%) | Selectivityb (%) | |
|---|---|---|---|---|
| 2a | 3a | |||
a Reaction conditions: 1a (0.8 mmol, 1 equiv.), other oxidants (1.4 equiv.), bisperoxy acids (0.7 equiv.), temperature: 30–32 °C (room temperature), acetonitrile was used as solvent for entries 1–11 (3 mL), time: 20 min.
b Conversion and yield determined by GC with the area normalization method.
c Time: 2 h.
d Solvent used: acetonitrile![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :methanol (3:2).
e Water (3 mL) was used as solvent, temperature 50–55 °C. :methanol (3:2).
e Water (3 mL) was used as solvent, temperature 50–55 °C.
|
||||
| Method-A | ||||
| 1c | 50% H2O2 | 85 | 66 | 19 |
| 2c | Oxone | 40 | 37 | 3 |
| 3c | m-CPBA | 99 | 79 | 20 |
| 4c,d | Sodium perborate | 2 | 2 | — |
| 5c | tert Butyl hydrogen peroxide | 33 | 33 | — |
| 6c | Urea hydrogen peroxide | Traces | — | — |
| 7c | Ammonium persulphate | Traces | — | — |
| 8c | Performic acid | 08 | 08 | — |
| 9c | Peracetic acid | 19 | 19 | — |
| 10c | Hexanebis(peroxoic acid) | 32 | 32 | — |
| 11c | Dodecanebis(peroxoic acid) | 56 | 56 | — |
| Method-B | ||||
| 12e | Hexanebis(peroxoic acid) | 100 | — | 100 |
| 13e | Dodecanebis(peroxoic acid) | 20 | — | 20 |
It was observed that the higher conversion and selectivity for the oxidation of sulfide to sulfoxide and sulfone is solubility dependent. A higher conversion and selectivity is observed when the reaction proceeds under homogeneous conditions. Hexanebis(peroxoic acid) is completely insoluble while dodecane(bisperoxoic acid) is partially soluble in acetonitrile (Method-A). Due to this, both peracids show a lower conversion to 2a even after keeping the reaction for 2 h, but the more nucleophilic nature of sulfide over sulfoxide favours a high selectivity for sulfoxide (Table 2, entries 10 and 11).
In order to investigate other peracids for the oxidation of sulfide to sulfone in water at a temperature of 50–55 °C (Method-B), we carried out reactions using hexanebis(peroxoic acid) and dodecanebis(peroxoic acid) under similar conditions. It was observed that the water soluble nature of hexanebis(peroxoic acid) favours 100% conversion to sulfone, whereas a lower conversion was observed for dodecanebis(peroxoic acid) which is insoluble in water (Table 2, entries 12 and 13).
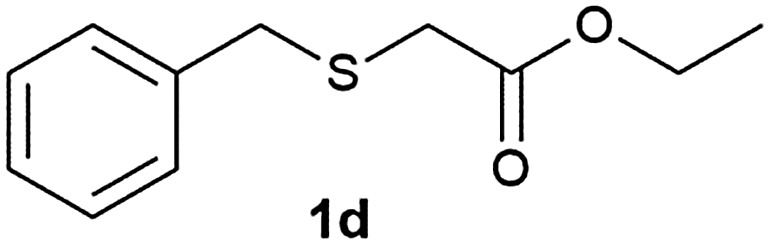
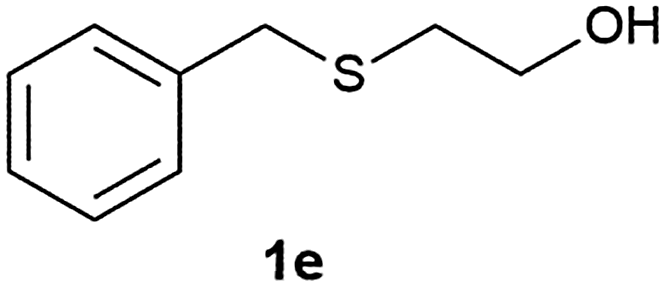
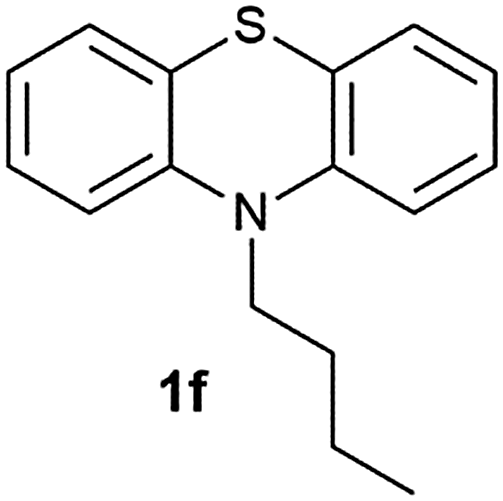
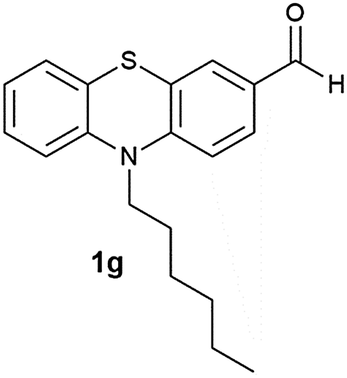
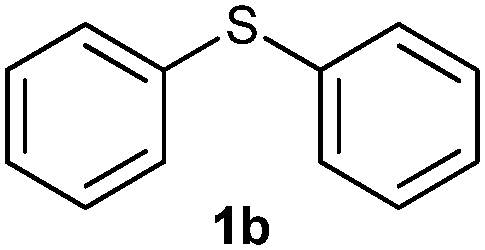
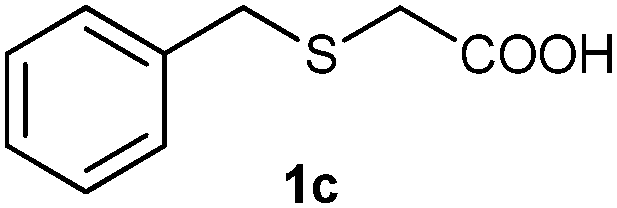
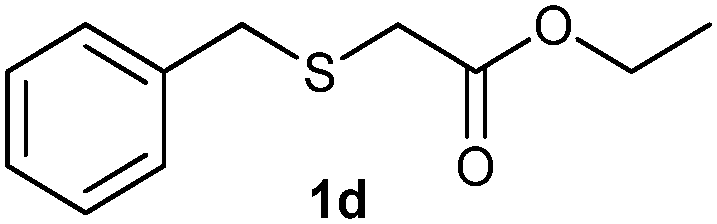
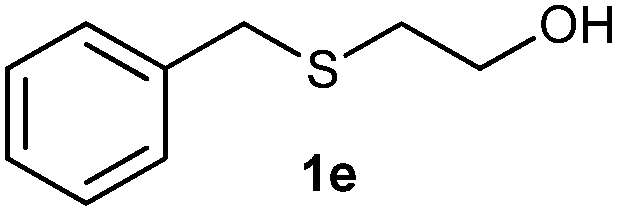
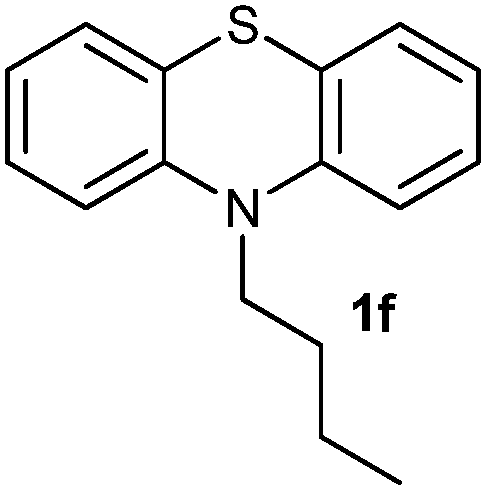
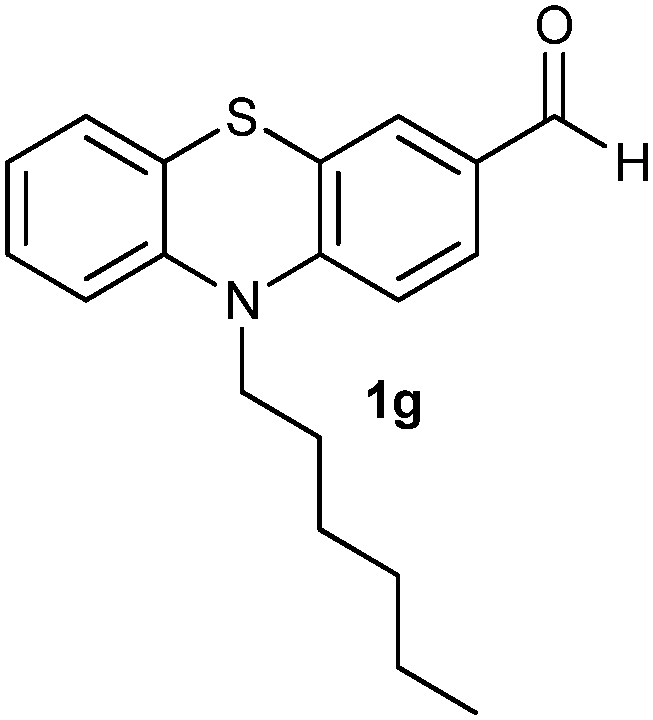
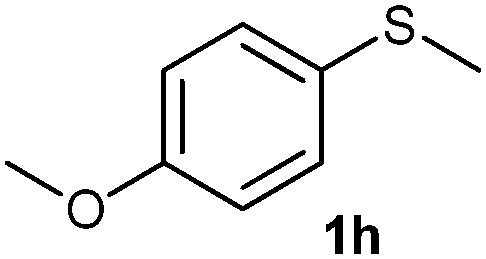
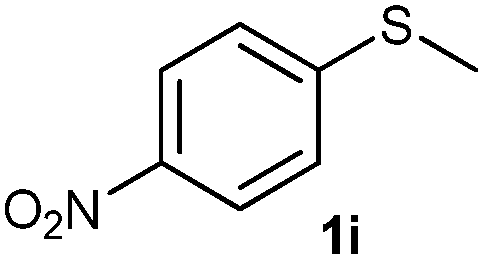
After optimizing the reaction conditions for both methods, we investigated the substrate scope using various aryl–alkyl and diaryl sulfides bearing oxidizable functionalities (Table 3, entries 1–9). The sulfoxide formation occurred in 5 to 15 min and sulfone formation occurred in 15 to 60 min using Method-A. When using Method-B, sulfone formation occurred in 25 min. Bigi et al. have reported that the oxidation of 1b to 2b and 3b occurred in 24 h.46 In our case, when using Method-A, the formation of 2b occurred in 12 min and 3b in 15 min, whereas by using Method-B, the formation of 3b was observed in 25 min (Table 3, entry 2). For the substrate bearing acid group 1c, no oxidation product of the acid was observed in either method (Table 3, entry 3). Similar results were obtained for the substrate bearing alcohol, tert amine (Table 3, entries 5 and 6). It was noticeable that for the oxidation of substrate 1g to 3g by using Method-A 60 min was required, whereas the same transformation occurred in 25 min by using Method-B (Table 3, entry 7). Also in both methods, traces of acid formation was observed. The influence of electron donating and withdrawing substrates on the rate of reaction was not observed for either protocol. The substrate bearing electron donating as well as the withdrawing groups were smoothly oxidized to the corresponding sulfoxide and sulfone in 15 and 25 min respectively by Method-A. On the other hand it took 25 min to produce the corresponding sulfone using Method-B (Table 3, entries 8 and 9).
| Method-A | |||||
|---|---|---|---|---|---|
| Entry | Substrate | Time (min) | Yieldb (%) | ||
| Sulfoxide | Sulfone | Sulfoxide | Sulfone | ||
| a Reaction conditions: Method-A sulfide (1 equiv.), acetonitrile (3 mL), nonanebis(peroxoic acid): 0.7 equiv. for sulfoxide, 1.5 equiv. for sulfone, temperature: 30–32 °C (room temperature). b Yield – isolated yield, Method-B sulfide (1 equiv.), water (3 mL), nonanebis(peroxoic acid): 1.5 equiv., temperature 50–55 °C. | |||||
| 1 |
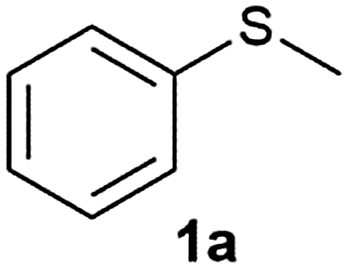
|
7 | 15 | 93 | 87 |
| 2 |
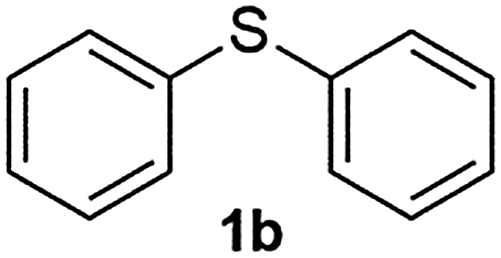
|
12 | 15 | 87 | 83 |
| 3 |
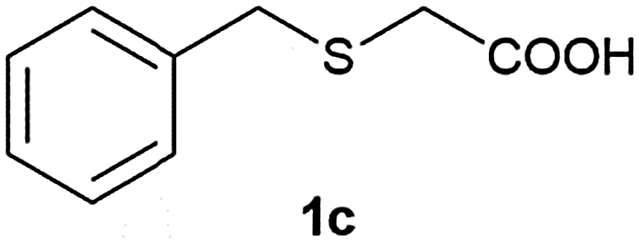
|
12 | 15 | 89 | 85 |
| 4 |
|
7 | 20 | 88 | 98 |
| 5 |
|
13 | 20 | 89 | 86 |
| 6 |
|
8 | 15 | 85 | 75 |
| 7 |
|
5 | 60 | 88 | 80 |
| 8 |
|
15 | 25 | 92 | 94 |
| 9 |
|
15 | 25 | 90 | 95 |
| Method-B | |||
|---|---|---|---|
| Entry | Substrate | Time (min) | Yieldb (%) |
| Sulfone | Sulfone | ||
| 1 |
|
25 | 95 |
| 2 |
|
25 | 90 |
| 3 |
|
25 | 88 |
| 4 |
|
25 | 95 |
| 5 |
|
25 | 87 |
| 6 |
|
25 | 80 |
| 7 |
|
25 | 80 |
| 8 |
|
25 | 95 |
| 9 |
|
25 | 96 |
Various oxidizing systems were reported for the chemoselective oxidation of sulfide, such as urea hydrogen peroxide with a Mn-complex,47 sodium perborate and sodium percarbonate with an Amberlyst support under solvent free conditions,48 ammonium nitrate and citric acid with MBr,49 IBX with TEAB as a catalyst,50and oxone under solvent free conditions.51 It was observed that in almost all cases (except oxone) a catalyst was used for the chemoselective oxidation of sulfide. To investigate the chemoselectivity, we have also carried out some competitive reactions (Scheme 1) for the oxidation of 1a to 2a in the presence of the substrate bearing oxidizable groups such as benzyl alcohol, styrene and aliphatic and aromatic tert amine using Method-A. When the GC analysis was performed for these reactions, selective 2a formation was observed with a 91 to 98% selectivity. No oxidation product of alcohol, double bond or tert amine was observed. Thus the present approach offers the chemoselective oxidation of sulfide under catalyst free conditions.
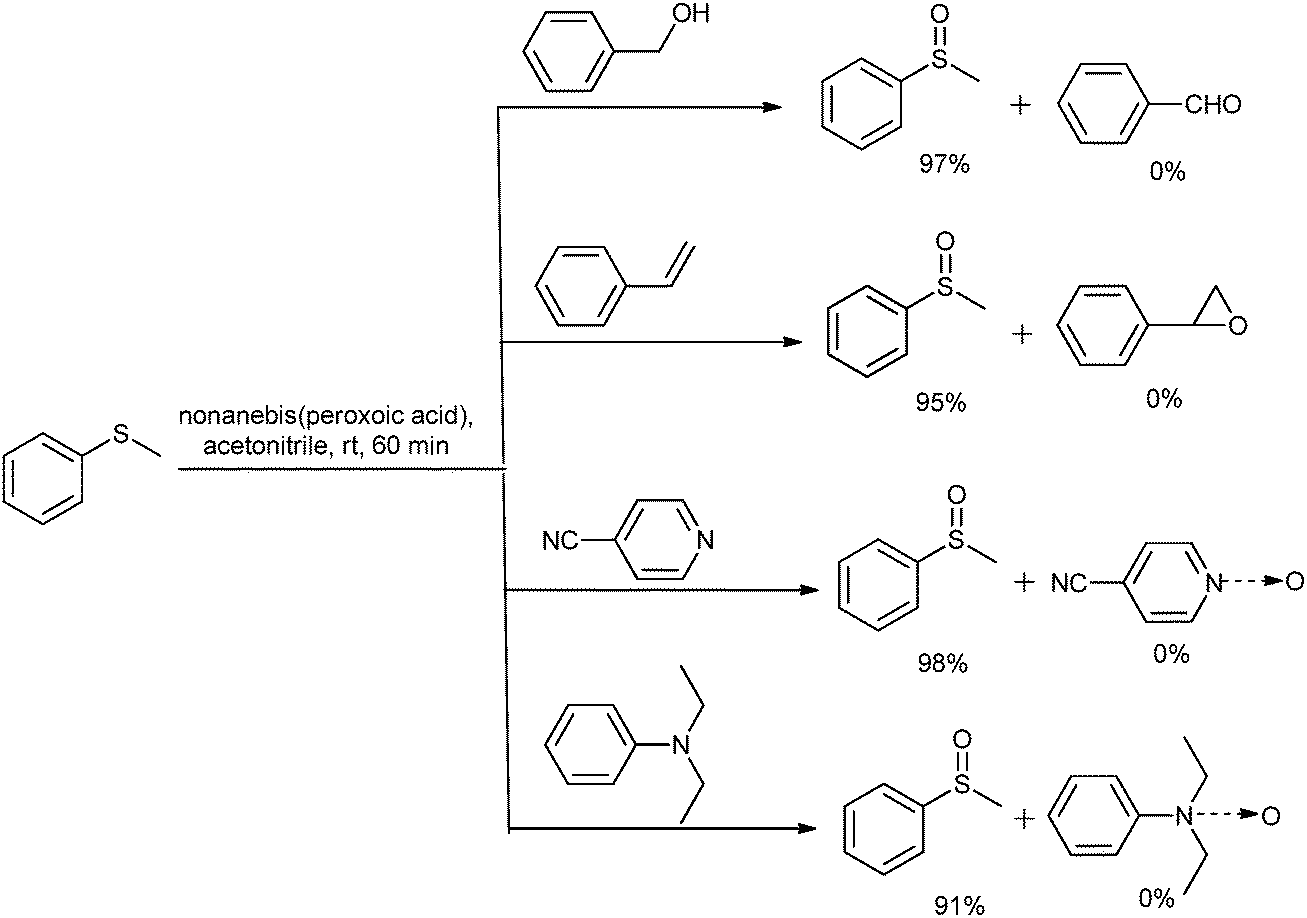 | ||
| Scheme 1 Chemoselective reaction. | ||
The recyclability study was carried out on 5 g of 1a (Table 4). The main problem we were facing while using these diperoxy acid was the recovery and recycling of the parent acid. Here we succeeded in recovering 75 to 80% of the parent acid which was further used up to a third cycle using Method-B. The detailed results are depicted in Table 4. The diperoxy acid prepared using the recovered parent acid was analysed using iodometric titration for % active oxygen content. The results obtained were consistent and in good agreement with the reported active oxygen content value. In all cycles, 100% selectivity for 3a was observed with a 92 to 95% isolated yield. Similarly, we performed the same study using Method-A, where only 66% recovery of the parent acid was obtained (Table 4, entry 1).
| Entry | No. of cycles | % AOC of oxidantd | Isolated yield | Isolated yield | Selectivitye (%) |
|---|---|---|---|---|---|
| Azealic acid | Sulfone | Sulfone | |||
| a Reaction conditions: 1a (1 equiv.), nonane(bis peroxoic)acid (1.5 equiv.), temperature: 50–55 °C, solvent: water. b Peracid used was synthesised from recovered azealic acid. c Solvent: acetonitrile. d AOC – active oxygen content determined using iodometric titration. e Selectivity determined by GC. | |||||
| 1c | Fresh | 14.3 | 66 | 89 | 100 |
| 2 | Fresh | 14.3 | 75 | 95 | 100 |
| 3b | First | 14.1 | 77 | 94 | 100 |
| 4b | Second | 14.2 | 80 | 92 | 100 |
| 5b | Third | 14.1 | 79 | 93 | 100 |
Plausible mechanism
A plausible mechanism41–43,52,53 for the oxidation of sulfide to sulfoxide and sulfone is shown in Fig. 1. We believe that the presence of intra-molecular hydrogen bonding between the carbonyl oxygen and hydrogen increases the electrophilic nature of the peroxy oxygen which is then attacked by the nucleophilic sulfide atom. This leads to the formation of positive charge on the sulfur atom from sulfide.52 This partial positive charge on sulfur may be stabilized by the electron donating nature of the nitrogen from acetonitrile. The parent acid, nonanedioic acid, formed during the reaction was recovered and recycled further by converting it to nonanebis(peroxoic acid).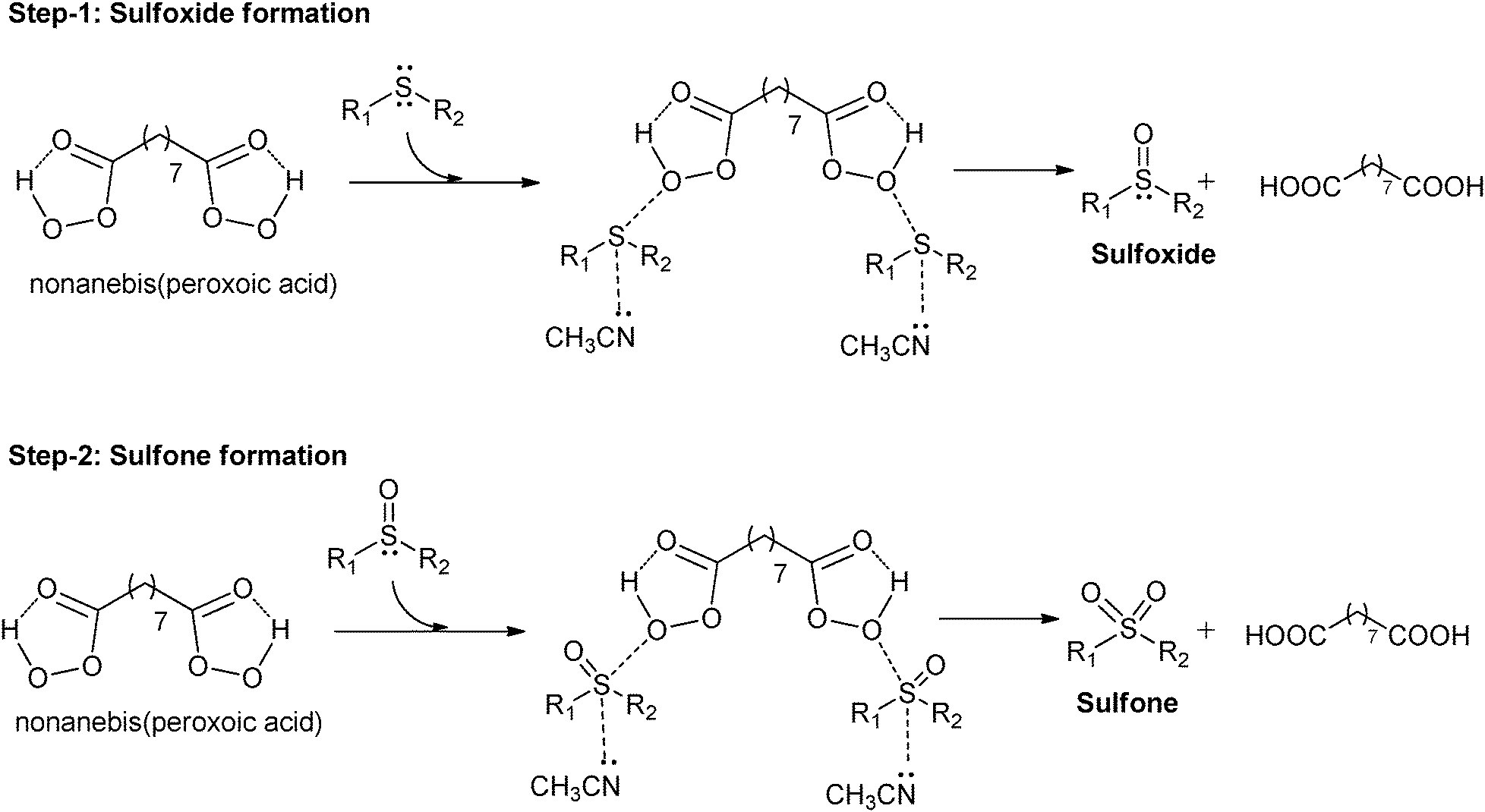 | ||
| Fig. 1 Plausible mechanism for sulfoxide and sulfone formation. | ||
Conclusion
In conclusion, nonanebis(peroxoic acid) was found to be effective for oxidizing various sulfides to the corresponding sulfoxides and sulfones. The developed protocol was found to be compatible with various oxidizable groups. In acetonitrile, the reaction smoothly proceeded at room temperature in 5–25 min, whereas in water the reaction needed to be carried out at 50–55 °C to give full selectivity for sulfone in 25 min. Nonanedioic acid generated at the end of the reaction in Method-B was effectively recovered and reused for up to three cycles by converting it to nonanebis(peroxoic acid).Experimental
General
Common reagent grade chemicals were purchased from Alfa Aesar, Spectrochem, Sigma Aldrich and Sd fine chemicals. FT-IR spectra were recorded on a Bomen Hartmann and Braun MB-Series FT-IR spectrometer. GC-MS was carried out with a GC-MS-QP 2010 instrument. The 1H NMR spectroscopic data were recorded with an Agilent 500 MHz spectrometer with CDCl3 and DMSO as the solvent. GC analysis was carried out with a Thermo scientific, column-TR-1, 30 m × 0.25 mm, ID × 0.25 μm film, FID detector and with a sample size of 0.11 μL.Procedure for the preparation of nonanebis(peroxoic acid)
Nonanebis(peroxoic acid) was synthesized and its purity was determined from its active oxygen content using an iodometric titration method.38,54General procedure for oxidation of sulfide to sulfoxide and sulfone
:hexane as an eluting system.
:2). 2 mL of 50% potassium iodide solution was added, and it was immediately stoppered and placed in an ice bath for 5 min. 50 mL of distilled water was added, and it was titrated against 0.1 M sodium thiosulfate solution. Starch indicator was added towards the endpoint. The endpoint is dark brown to colourless.38
Spectroscopic data of compounds
(1) (Methylsulfinyl)benzene (2a):55 pale yellow oil; yield: 93%; 1H NMR (500 MHz, CDCl3): δ = 7.56–7.53 (m, 2H), 7.44–7.37 (m, 3H), 2.62 (s, 3H); IR: ν/cm−1 = 1080; GC-MS (EI, 70 eV): m/z, [M]+ = 140.(2) (Methylsulfonyl)benzene (3a):56 white solid; yield: Method-A: 87%, Method-B: 95%; mp 88–90 °C; 1H NMR (500 MHz, CDCl3): δ = 7.95–7.93 (m, 2H), 7.67–7.63 (m, 1H), 7.58–7.55 (m, 2H), 3.04 (s, 3H); IR: ν/cm−1 = 1142, 1282; GC-MS (EI, 70 eV): m/z, [M]+ = 156.
(3) Sulfinyldibenzene (2b):57 white solid; yield: 87%; mp 68 °C; 1H NMR (500 MHz, CDCl3) δ 7.65 (m, 4H), 7.50–7.41 (m, 6H); IR: ν/cm−1 = 1035, 1085; GC-MS (EI, 70 eV): m/z, [M]+ = 202.
(4) 2-(Benzylsulfinyl)acetic acid (2c):58 white solid; 89% yield; mp 124–126 °C; 1H NMR (500 MHz, DMSO): δ 13.12 (s, 1H, –COOH), 7.30–7.39 (m, 5H), 4.24–4.21 (d, 2H), 4.23–4.12 (d, J = 10 Hz, 1H, SOCH2COOH), 4.03–4.05 (d, J = 15 Hz, 1H, ArCH2SO), 3.84–3.81 (d, J = 15 Hz, 1H, ArCH2SO), 3.54–3.52 (d, J = 10 Hz, 1H, SOCH2COOH); IR: ν/cm−1 = 1069, 1698; MS calculated m/z = 198.2, observed m/z = 196.85 [M − 1].
(5) 2-(Benzylsulfonyl)acetic acid (3c):58 white solid; yield: Method-A: 85%, Method-B: 88%; mp 138 °C; 1H NMR (500 MHz, DMSO) δ 7.40 (m, 5H), 4.62 (s, 2H), 4.15 (s, 2H); IR: ν/cm−1 = 1137, 1299, 1697; GC-MS (EI, 70 eV): m/z, [M]+ = 214.
(6) Ethyl-2-(benzylsulfonyl)acetate (3d):59 white solid; yield: Method-A: 98%, Method-B: 95%; mp 42 °C; 1H NMR (500 MHz, CDCl3): δ 7.39–7.41 (m, 3H), 7.48–7.50 (m, 2H), 4.51 (s, 2H,); 4.28–4.32 (q, 2H), 3.77 (s, 2H), 1.33–1.36 (t, 3H); IR: ν/cm−1 = 1138, 1290, 1716; GC-MS (EI, 70 eV): m/z, [M]+ = 242.
(7) 2-(Benzylsulfinyl) ethanol (2e):60 yellowish liquid; yield: 89%; 1H NMR (500 MHz, CDCl3) δ 7.31 (m, 5H), 4.07 (s, 2H), 4.05–3.98 (m, 2H), 3.28 (s, 1H), 2.87–2.70 (m, 2H); IR: ν/cm−1 = 1088, 3448; GC-MS (EI, 70 eV): m/z, [M]+ = 184.
(8) 2-(Benzylsulfonyl) ethanol (3e):61 white solid; yield: Method-A: 86%, Method-B: 87%; mp 66 °C; 1H NMR (500 MHz, CDCl3) δ 7.48–7.37 (m, 5H), 4.35 (s, 1H), 4.09 (dd, 2H), 3.11–3.07 (t, 2H), 2.51 (s, br, 1H); IR: ν/cm−1 = 1280, 1249, 3425; GC-MS (EI, 70 eV): m/z, [M]+ = 200.
(9) 10-Butyl-10H-phenothiazine 5-oxide (2f):62 white solid; yield: 85%, mp 130 °C; 1H NMR (500 MHz, CDCl3) δ 7.94 (dd, 2H), 7.65–7.59 (m, 2H), 7.42 (d, 2H), 7.25 (dd, 2H), 4.26–4.19 (t, 2H), 1.95 (m, 2H), 1.62–1.52 (m, 2H), 1.07 (t, 3H); IR: ν/cm−1 = 1023; GC-MS (EI, 70 eV): m/z, [M]+ = 271.
(10) 10-Butyl-10H-phenothiazine 5,5-dioxide (3f):63 white solid; yield: Method-A: 75%, Method-B: 80%; mp 148 °C; 1H NMR (500 MHz, CDCl3) δ 8.13 (dd, 2H), 7.65–7.59 (m, 2H), 7.35 (d, 2H), 7.27 (dd,2H), 4.20–4.13 (t, 2H), 1.92 (m, 2H), 1.56–1.46 (m, 2H), 1.03 (t, 3H); IR: ν/cm−1 = 1376, 1279; GC-MS (EI, 70 eV): m/z, [M]+ = 287.
(11) 10-Hexyl-10H-phenothiazine-3-carbaldehyde-5-oxide (2g): white solid; yield: 88%; mp 138 °C; 1H NMR (500 MHz, CDCl3) δ 10.01 (s, 1H), 8.44 (d, J = 2.0 Hz, 1H) 8.14 (dd, J = 8.9, 2.0 Hz, 1H), 7.99 (dd, J = 7.7, 1.6 Hz, 1H), 7.69 (ddd, J = 8.8, 7.3, 1.7 Hz, 1H), 7.50 (t, J = 8.8 Hz, 2H), 7.38–7.34 (m, 1H), 4.30–4.25 (m, 2H), 2.04–1.95 (m, 2H), 1.60–1.52 (m, 2H), 1.47–1.35 (m, 4H), 0.94 (t, J = 7.1 Hz, 3H); IR: ν/cm−1 = 1026, 1679; MS calculated m/z = 327.4, observed m/z = 328.02 (M + 1).
(12) 10-Hexyl-10H-phenothiazine-3-carbaldehyde-5,5-dioxide (3g): white solid; yield: Method-A: 80%, Method-B: 80%; mp 178 °C; 1H NMR (500 MHz, CDCl3) δ 10.01 (s, 1H), 8.602–8.598 (d, 1H, J = 2), 8.16–8.13 (m, 1H, J = 8, 1.5 Hz), 7.702–7.667 (m, 1H, J = 9, 2 Hz), 7.453–7.359 (m, 3H), 4.233–4.201 (t, 2H), 1.977–1.914 (m, 2H), 1.530–1.470 (m, 2H), 1.400–1.339 (m, 4H), 0.930–0.901 (t, 3H); IR: ν/cm−1 = 1135, 1284; MS calculated m/z = 343.4, observed m/z = 344.13 (M + 1).
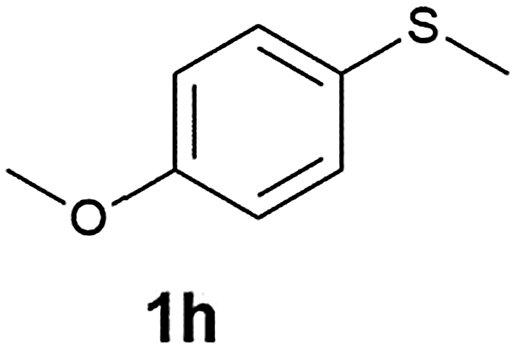
(13) 1-Methoxy-4-(methylsulfinyl)benzene (2h):34 colourless oil; yield: 92%, 1H NMR (500 MHz, CDCl3) δ 7.58–7.55 (m, 2H), 7.02–6.98 (m, 2H), 3.82 (s, 3H), 2.68 (s, 3H); IR: ν/cm−1 = 1088, GC-MS (EI, 70 eV): m/z, [M]+ = 170.1.
(14) Methoxy-4-(methylsulfonyl)benzene (3h):34 white crystals; yield: Method-A: 94%, Method-B: 95%; mp 120 °C; 1H NMR (500 MHz, CDCl3) δ 7.90–7.84 (d, 2H), 7.05–7.00 (d, 2H), 3.89 (s, 3H), 3.03 (s, 3H). IR: ν/cm−1 = 1139, 1291; GC-MS (EI, 70 eV): m/z, [M]+ = 186.
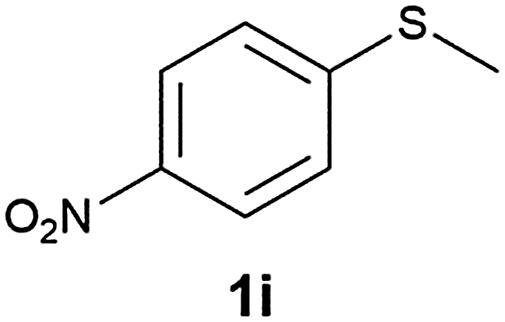
(15) 1-(Methylsulfinyl)-4-nitrobenzene (2i):34 pale yellow solid; yield: 90%; mp 150 °C; 1H NMR (500 MHz, CDCl3) δ 8.40 (d, J = 8.5 Hz, 2H), 7.84 (d, J = 8.5 Hz, 2H), 2.80 (s, 3H); IR: ν/cm−1 = 1085; GC-MS (EI, 70 eV): m/z, [M]+ = 185.
(16) 1-(Methylsulfonyl)-4-nitrobenzene (3i):34 pale yellow solid; yield: Method-A: 95%, Method-B: 96%; mp 140 °C; 1H NMR (500 MHz, CDCl3) δ 8.45–8.41 (d, 2H), 8.19–8.14 (d, 2H), 3.13 (s, 3H); IR: ν/cm−1 = 1149, 1287; GC-MS (EI, 70 eV): m/z, [M]+ = 201.
Acknowledgements
The authors are thankful to the UGC-GREEN TECH and UGC-CAS for providing financial assistance, and MS Indoco Remedies Ltd, Rabale, Navi Mumbai for recording the EI-MS spectra.References
- C.-Y. Lai, W.-L. Mak, E. Y. Y. Chan, Y.-K. Sau, Q.-F. Zhang, S. M. F. Lo, I. D. Williams and W.-H. Leung, Inorg. Chem., 2003, 42, 5863–5870 CrossRef CAS.
- V. Y. Kukushkin, Coord. Chem. Rev., 1995, 139, 375–407 CrossRef CAS.
- M. Monçalves, D. da S. Rampon, P. H. Schneider, F. S. Rodembusch and C. da C. Silveira, Dyes Pigm., 2014, 102, 71–78 CrossRef.
- C.-J. Zheng, J. Wang, J. Ye, M.-F. Lo, X.-K. Liu, M.-K. Fung, X.-H. Zhang and C.-S. Lee, Adv. Mater., 2013, 25, 2205–2211 CrossRef CAS.
- J. Ye, Z. Chen, M.-K. Fung, C. Zheng, X. Ou, X. Zhang, Y. Yuan and C.-S. Lee, Chem. Mater., 2013, 25, 2630–2637 CrossRef CAS.
- J. Gao, H. Guo, S. Liu and M. Wang, Tetrahedron Lett., 2007, 48, 8453–8455 CrossRef CAS.
- F. R. Sensato, R. Custodio, E. Longo, V. S. Safont and J. Andres, J. Org. Chem., 2003, 68, 5870–5874 CrossRef CAS.
- A. M. I. Jayaseeli and S. Rajagopal, J. Mol. Catal. A: Chem., 2009, 309, 103–110 CrossRef CAS.
- J. Orive, E. S. Larrea, R. Fernández de Luis, M. Iglesias, J. L. Mesa, T. Rojo and M. I. Arriortua, Dalton Trans., 2013, 42, 4500–4512 RSC.
- K. Kamata, K. Yonehara, Y. Sumida, K. Hirata, S. Nojima and N. Mizuno, Angew. Chem., Int. Ed., 2011, 50, 12062–12066 CrossRef CAS.
- H. Q. N. Gunaratne, M. A. McKervey, S. Feutren, J. Finlay and J. Boyd, Tetrahedron Lett., 1998, 39, 5655–5658 CrossRef CAS.
- T.-H. Chen, Z. Yuan, A. Carver and R. Zhang, Appl. Catal., A, 2014, 478, 275–282 CrossRef CAS.
- M. Matteucci, G. Bhalay and M. Bradley, Org. Lett., 2003, 5, 235–237 CrossRef CAS.
- M. Bonchio, G. Licini, S. Mantovani, G. Modena and W. A. Nugent, J. Org. Chem., 1999, 64, 1326–1330 CrossRef CAS.
- M. Bagherzadeh, R. Latifi, L. Tahsini and M. Amini, Catal. Commun., 2008, 10, 196–200 CrossRef CAS.
- B. Rama Raju, S. Sarkar, U. Chandramoulali Reddy and A. K. Saikia, J. Mol. Catal. A: Chem., 2009, 308, 169–173 CrossRef CAS.
- S. Kumar, S. Verma, S. L. Jain and B. Sain, Tetrahedron Lett., 2011, 52, 3393–3396 CrossRef CAS.
- W. Adam and L. Hadjiarapoglou, Tetrahedron Lett., 1992, 33, 469–470 CrossRef CAS.
- T. R. Boehlow, P. Christopher Buxton, E. L. Grocock, B. A. Marples and V. L. Waddington, Tetrahedron Lett., 1998, 39, 1839–1842 CrossRef CAS.
- M. E. González-Núñez, R. Mello, J. Royo, J. V Ríos and G. Asensio, J. Am. Chem. Soc., 2002, 124, 9154–9163 CrossRef.
- S. Colonna and N. Gaggero, Tetrahedron Lett., 1989, 30, 6233–6236 CrossRef CAS.
- A. Levai, ARKIVOC, 2003, 2003, 14 Search PubMed.
- S. A. Dieva, R. M. Eliseenkova, Y. Y. Efremov, D. R. Sharafutdinova and A. A. Bredikhin, Russ. J. Org. Chem., 2006, 42, 12–16 CrossRef CAS.
- A. L. Baumstark and P. C. Vasquez, J. Phys. Org. Chem., 1988, 1, 259–265 CrossRef CAS.
- A. L. Baumstark, Bioorg. Chem., 1986, 14, 326–343 CrossRef CAS.
- T. Tezuka, M. Iwaki and Y. Haga, J. Chem. Soc., Chem. Commun., 1984, 325–326 RSC.
- A. L. Baumstark and P. C. Vasquez, J. Org. Chem., 1983, 48, 65–69 CrossRef CAS.
- S. Velusamy, A. V Kumar, R. Saini and T. Punniyamurthy, Tetrahedron Lett., 2005, 46, 3819–3822 CrossRef CAS.
- M. Hirano, J. Tomaru and T. Morimoto, Bull. Chem. Soc. Jpn., 1991, 64, 3752–3754 CrossRef CAS.
- Y.-J. Chen and Y.-P. Huang, Tetrahedron Lett., 2000, 41, 5233–5236 CrossRef CAS.
- M. Rahimizadeh, G. Rajabzadeh, S.-M. Khatami, H. Eshghi and A. Shiri, J. Mol. Catal. A: Chem., 2010, 323, 59–64 CrossRef CAS.
- K. Bahrami, M. M. Khodaei and M. Sheikh Arabi, J. Org. Chem., 2010, 75, 6208–6213 CrossRef CAS.
- R. Pievo, M. Gullotti, E. Monzani and L. Casella, Biochemistry, 2008, 47, 3493–3498 CrossRef CAS.
- C. Yang, Q. Jin, H. Zhang, J. Liao, J. Zhu, B. Yu and J. Deng, Green Chem., 2009, 11, 1401–1405 RSC.
- K. Bahrami, M. M. Khodaei and S. Sohrabnezhad, Tetrahedron Lett., 2011, 52, 6420–6423 CrossRef CAS.
- R. Das and D. Chakraborty, Synthesis, 2011, 277–280 CAS.
- M. M. Khodaei, K. Bahrami and Y. Tirandaz, J. Sulfur Chem., 2009, 30, 581–584 CrossRef CAS.
- V. V. Patil and G. S. Shankarling, Beilstein J. Org. Chem., 2014, 10, 921–928 CrossRef.
- V. V Patil, E. M. Gayakwad and G. S. Shankarling, New J. Chem., 2015, 39, 6677–6682 RSC.
- V. V. Patil and G. S. Shankarling, J. Org. Chem., 2015, 80, 7876–7883 CrossRef CAS.
- R. Curci, R. DiPrete, J. O. Edwards and G. Modena, J. Org. Chem., 1970, 35, 740–745 CrossRef CAS.
- P. Hanson, R. A. A. J. Hendrickx and J. R. Lindsay Smith, Org. Biomol. Chem., 2008, 6, 762–771 CAS.
- B. Yu, A.-H. Liu, L.-N. He, B. Li, Z.-F. Diao and Y.-N. Li, Green Chem., 2012, 14, 957–962 RSC.
- A. V Malkov, N. Derrien, M. Barłóg and P. Kočovský, Chem. – Eur. J., 2014, 20, 4542–4547 CrossRef.
- J. H. Ramsden, R. S. Drago and R. Riley, J. Am. Chem. Soc., 1989, 111, 3958–3961 CrossRef CAS.
- F. Bigi, A. Corradini, C. Quarantelli and G. Sartori, J. Catal., 2007, 250, 222–230 CrossRef CAS.
- S. Rayati, F. Nejabat and S. Zakavi, Inorg. Chem. Commun., 2014, 40, 82–86 CrossRef CAS.
- M. V. Gomez, R. Caballero, E. Vazquez, A. Moreno, A. de la Hoz and A. Diaz-Ortiz, Green Chem., 2007, 9, 331–336 RSC.
- A. Ghorbani-Choghamarani and S. Rezaei, J. Chin. Chem. Soc., 2009, 56, 251–254 CrossRef CAS.
- V. G. Shukla, P. D. Salgaonkar and K. G. Akamanchi, J. Org. Chem., 2003, 68, 5422–5425 CrossRef CAS.
- G. Cravotto, D. Garella, D. Carnaroglio, E. C. Gaudino and O. Rosati, Chem. Commun., 2012, 48, 11632–11634 RSC.
- K. R. Pellarin, M. S. McCready and R. J. Puddephatt, Dalton Trans., 2013, 42, 10444–10453 RSC.
- H. Veisi, F. H. Eshbala, S. Hemmati and M. Baghayeri, RSC Adv., 2015, 5, 10152–10158 RSC.
- W. E. Parker, L. P. Witnauer and D. Swern, J. Am. Chem. Soc., 1957, 79, 1929–1931 CrossRef CAS.
- A. R. Hajipour and I. Mohammadpoor-baltork, Phosphorus, Sulfur Silicon Relat. Elem., 2000, 164, 145–151 CrossRef CAS.
- C. Shen, J. Xu, W. Yu and P. Zhang, Green Chem., 2014, 16, 3007–3012 RSC.
- J. N. Moorthy, K. Senapati and K. N. Parida, J. Org. Chem., 2010, 75, 8416–8421 CrossRef CAS.
- X. Ning, Y. Guo, X. Wang, X. Ma, C. Tian, X. Shi, R. Zhu, C. Cheng, Y. Du, Z. Ma, Z. Zhang and J. Liu, J. Med. Chem., 2014, 57, 4302–4312 CrossRef CAS.
- I. Shahak and E. D. Bergmann, Isr. J. Chem., 1970, 8, 589–593 CrossRef CAS.
- S. Hussain, D. Talukdar, S. K. Bharadwaj and M. K. Chaudhuri, Tetrahedron Lett., 2012, 53, 6512–6515 CrossRef CAS.
- K. E. Bashford, A. L. Cooper, P. D. Kane, C. J. Moody, S. Muthusamy and E. Swann, J. Chem. Soc., Perkin Trans. 1, 2002, 1672–1687 RSC.
- F.-D. I. Monica Toşa, C. Paiz, C. Majdik, L. Poppe, P. Kolonits, I. A. Silberg and L. Novák, Heterocycl. Commun., 2001, 7, 277 Search PubMed.
- H. Wunderlich, W. Lugenheim, A. Stark and G. Detrekoi, Pharmazie, 1966, 21, 57–58 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5nj02616d |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |