
Tungsten oxide by non-hydrolytic sol–gel: effect of molecular precursor on morphology, phase and photocatalytic performance†
Emma
Oakton
,
Georges
Siddiqi
,
Alexey
Fedorov
and
Christophe
Copéret
*
ETH Zürich, Department of Chemistry and Applied Biosciences, Vladimir Prelog Weg 2, CH-8093 Zürich, Switzerland. E-mail: ccoperet@inorg.chem.ethz.ch; Fax: +41 44 633 1325; Tel: +41 44 633 9394
First published on 30th October 2015
Abstract
A practical gram-scale synthesis of high surface area blue tungsten oxide via a non-hydrolytic sol–gel method is reported. The use of diisopropyl ether in 1,2-dichloroethane at 180 °C transforms W(VI) and W(IV) chloride precursors into WOx materials of cube or rod-like morphology respectively. These oxides are characterised by a range of methods including TEM, N2 adsorption–desorption, XRD, XAS, EPR, UV-Vis DRS, elemental analysis and conductivity measurements. Treatment of WOx under N2 causes a decrease in surface area and increase in electrical conductivity. Prepared materials are investigated as catalysts for oxygen evolution by photocatalytic water splitting.
Introduction
High surface area tungsten oxide has potential applications in fabrication of electrochromic devices,1,2 gas sensors,3–5 photocatalysts,6,7 and low temperature proton exchange fuel cells (PEFCs).8,9 In particular, tungsten oxide is a widely studied oxidation catalyst for the degradation of organic compounds and photocatalytic oxygen evolution6,10–12 with potential for energy generation, waste remediation, and other applications.13,14Due to this wide variety of potential applications of tungsten oxide materials, investigation of a range of synthetic routes is vital.15 Previously mesoporous tungsten oxide was prepared by a sol–gel route, reacting a variety of alcohols with WCl6 in the presence of steam.16 Non-hydrolytic sol–gel (NHSG) is an alternative route to metal oxides that uses anhydrous organic alcohols or aprotic oxygen donors.17 This approach offers many advantages, including the ability to control oxide particle size and shape by the nature of oxygen donor. In addition, slower reaction rates for NHSG often lead to crystalline materials.18 A variety of publications have discussed how the alteration of NHSG reaction conditions can affect the resulting oxide morphology.19,20 The texture of metal oxides, including crystallinity, surface area and particle shape, is an important parameter influencing, for example, photocatalytic performance.11,21,22 Mesoporous blue tungsten oxide was prepared by reaction of WCl6 in a 1-butanol/tert-butanol mixture, where the alcohol ratio influences porosity.19 The investigation of precursor influences on morphology has also shown some interesting results, where the use of W(OiPr)65 or WCl620 in combination with benzyl alcohol provided nanowires or nanoplatelets, respectively. In the latter reported case, the addition of 4-tert-butylcatechol triggered the formation of rod-like structures.20 In place of alcohols, diisopropyl ether has been found to be a very efficient aprotic oxygen donor, which reacts with metal chlorides to yield a broad range of metal oxides,23–28 including homogeneous mixed oxides.29–31 The preparation of pure tungsten oxide from WCl6 and diisopropyl ether has been reported, although this route involves highly toxic CS2 solvent.32
Here we utilise NHSG chemistry with diisopropyl ether taken in excess to investigate the effect of tungsten chloride precursor oxidation state (4+ vs. 6+) on the morphology and phase of blue tungsten oxide. TEM imaging shows a distinct change in particle shape and X-ray powder diffraction indicates materials prepared from a W(IV) precursor form significantly oxygen deficient phase, supported by W L3-edge XANES. This information is used to rationalise the differences in photocatalytic oxygen evolution activity of these materials before and after post-synthetic treatment.
Results and discussion
Tungsten oxide materials were prepared by the reaction of WCl6 or WCl4(DME) (DME = 1,2-dimethoxyethane) with diisopropyl ether in 1,2-dichloroethane at 180 °C to give WOx(VI) or WOx(IV), respectively. The TEM images of these two WOx materials show a notable difference in morphology, with WOx(VI) forming cubes around 20–50 nm in size and WOx(IV) forming rods approximately 20–100 nm in width as well as some amorphous-like material (Fig. 1). The N2 adsorption–desorption data of both samples are similar, showing a type II isotherm, indicating minimal microporosity, some mesoporosity and mainly macroporosity. The BET surface areas of both materials are comparable with values of 85 and 90 m2 g−1 for WOx(VI) and WOx(IV) respectively (Fig. 1). Both WOx(VI) and WOx(IV) are blue in colour, which is consistent with the presence of oxygen defects and thus electron rich tungsten centres.33,34 This is supported by the increased electrical conductivity of WOx(VI) and WOx(IV) (1.5 × 10−4 and 1.0 × 10−4 S cm−1, respectively) in comparison to commercial WO3, (4.5 × 10−7 S cm−1). XRD shows a significant difference between these WOx materials, with the XRD powder diffraction pattern of WOx(VI) matching that of tetragonal WO3, whilst WOx(IV) is consistent with an oxygen deficient phase distinguishable from WO3, such as WO2.65, W18O49 or WO2.90 (Fig. 1). These substoichiometric phases have highly similar powder patterns, making a more accurate assignment difficult. WO3 is composed of WO6 octahedral units connected by corner atoms. Removal of oxygen from this structure results in vacancy sites, the presence of which can trigger the formation of edge sharing octahedra.35 WO3 can be substoichiometric until WO2.98 and therefore the defects in WOx(VI) are likely low in concentration and randomly distributed.36 In contrast, distinct oxygen deficient phases have highly ordered arrays of edge sharing octahedra.37 This characterisation shows that a different phase of tungsten oxide, with a significantly lower oxygen stoichiometry, is formed from the W(IV) precursor. The presence of paramagnetic species consistent with the formation of oxygen defects was confirmed by EPR spectroscopy with a signal at g = 1.8 for both samples.38 Another signal at g = 2.0 present in both materials likely arise from remaining organic functionalities on the oxide surface after the NHSG synthesis.39 Given that these WOx materials were prepared from precursors with different oxidation states, the bulk oxidation state of W in the as-prepared oxides was determined by X-ray Absorption Near Edge Spectroscopy and compared to commercial WO3 and WO2 (Fig. 2 and Table S3, ESI†). XANES characterisation of WOx(VI) shows that its white line is shifted by −0.35 eV compared to WO3, while WOx(IV) is shifted further by −0.75 eV. Though small, this shift to lower energies indicates that the W species present in these as-synthesised WOx samples are more reduced than bulk WO3, and that WOx(IV) is more reduced than WOx(VI).40 These XANES results are in agreement with the aforementioned XRD and EPR results.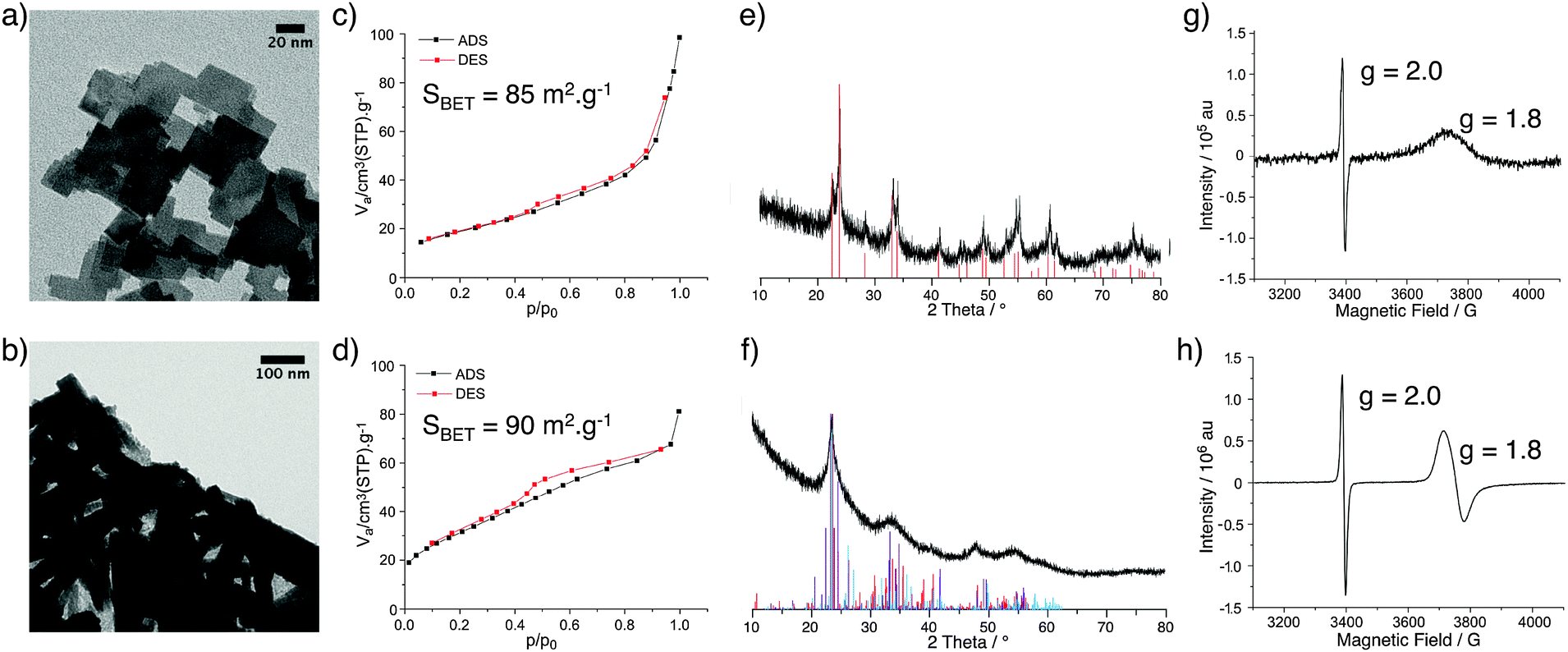 | ||
| Fig. 1 TEM micrographs of WOx(VI) (a) and WOx(IV) (b); N2 adsorption–desorption isotherms of WOx(VI) (c) and WOx(IV) (d); XRD diffraction pattern of WOx(VI) with reference pattern of tetragonal WO3 (red lines) (e) and XRD diffraction pattern of WOx(IV) (f) with reference patterns of WO2.65 (purple), W18O49 (red) and WO2.90 (blue) (f); EPR spectra of WOx(VI) (g) and WOx(IV) (h). | ||
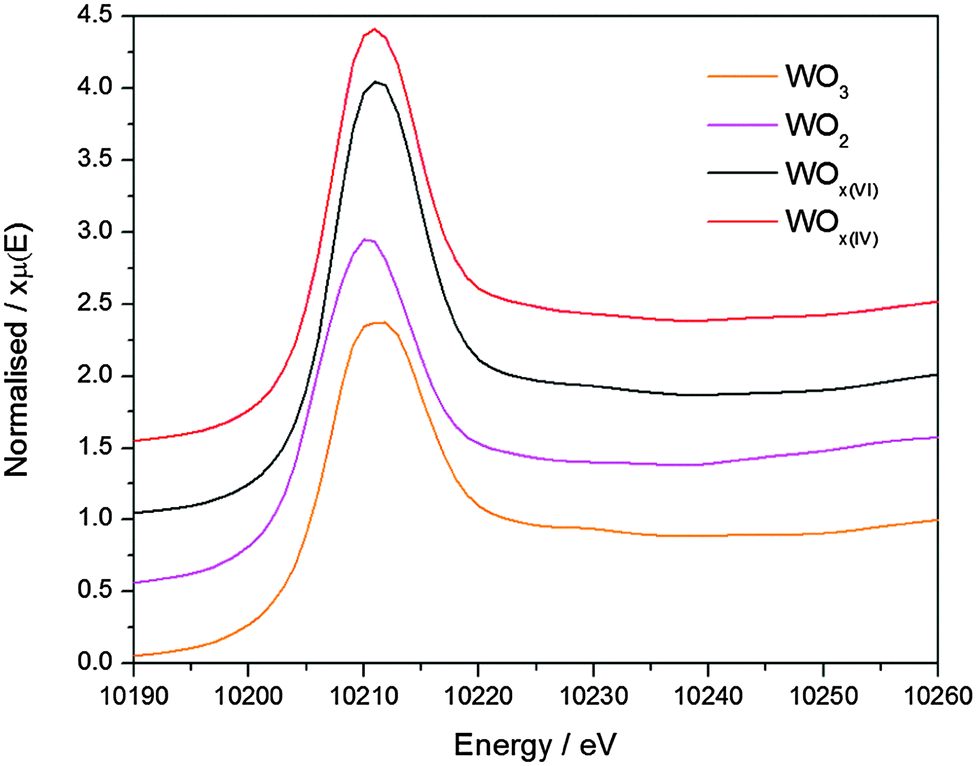 | ||
| Fig. 2 XANES characterisation of WO3 (orange), WOx(VI) (black), WOx(IV) (red) and WO2 (pink). | ||
The treatment of both WOx(VI) and WOx(IV) under N2 flow at 500 °C for 3 h produced dark blue and dark blue/black materials respectively and the characterisation of these samples is summarised in Fig. 3. TEM images of WOx(VI)–N2 show that the previously cubic particles have lost edge sharpness and sintered. The WOx(IV)–N2 particles retain the rod-like shape of WOx(IV) and appear to have decreased in width. N2 adsorption–desorption measurements indicate particle sintering leading to a decrease in surface area (90 vs. 46 m2 g−1). An analogous decrease in surface area is observed for WOx(VI)–N2 (85 vs. 42 m2 g−1). In the case of WOx(VI)–N2, there is a shift in the XRD powder diffraction pattern, matching that of monoclinic WO3 (Fig. 3). Interestingly the EPR spectra show an absence of any W(V) signal around g = 1.8, although the g = 2.0 signal remains. The lack of the EPR W(V) signal is likely due to the increased spin concentration leading to spin pairing and/or faster spin relaxation. This increased presence of oxygen defects is supported by electronic conductivity measurements, which show a higher electronic conductivity for both samples after N2 treatment (4.1 × 10−3 and 4.0 × 10−2 S cm−1 for WOx(VI)–N2 and WOx(IV)–N2 respectively), in accord with the literature.19 The EPR signal at g = 2.0 ascribed to organic radicals is in accord with minor amounts of carbon by elemental analysis of nitrogen-treated samples (see ESI† for details).
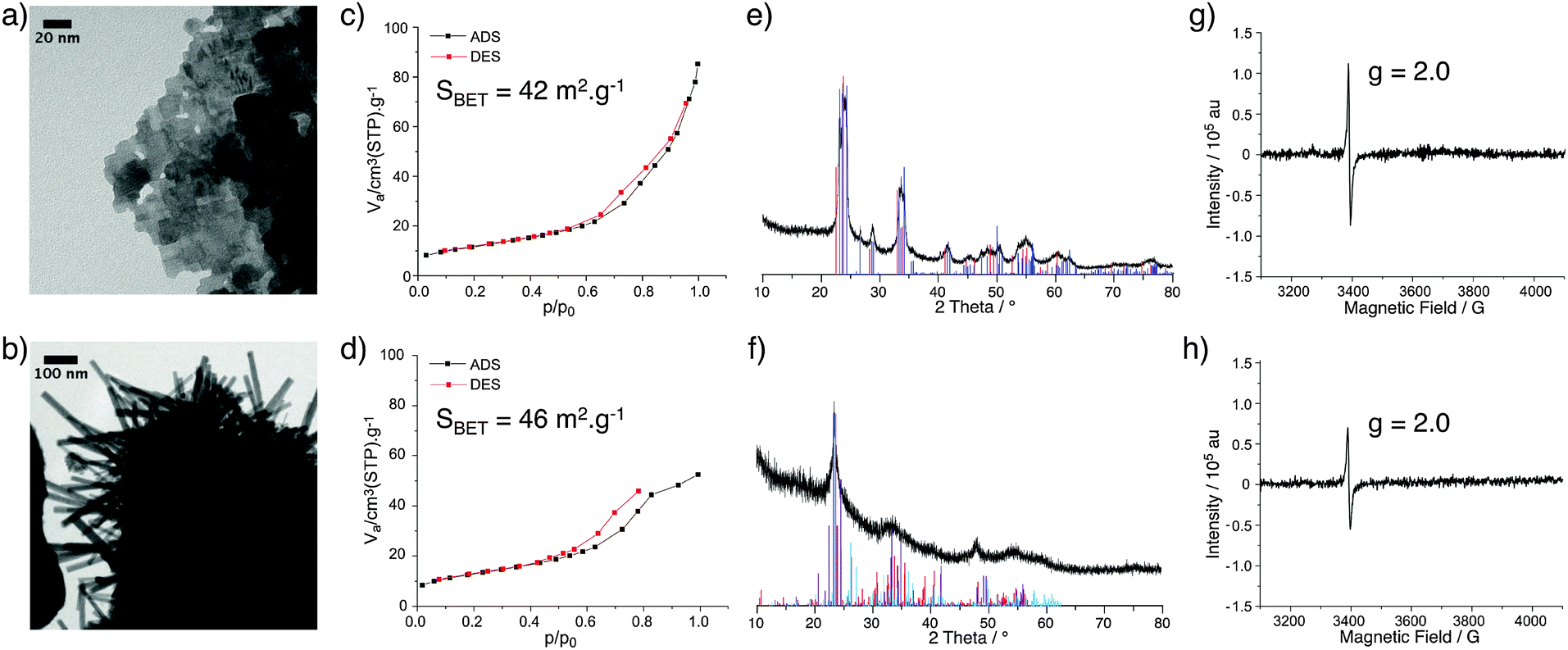 | ||
| Fig. 3 TEM micrographs of WOx(VI)–N2 (a) and WOx(IV)–N2 (b); N2 adsorption–desorption isotherms of WOx(VI)–N2 (c) and WOx(IV)–N2 (d); XRD diffraction pattern of WOx(VI)–N2 with reference pattern of tetragonal (red) and monoclinic (blue) WO3 (e) and XRD diffraction pattern of WOx(IV)–N2 with reference patterns of WO2.65 (purple), W18O49 (red) and WO2.90 (blue) (f); EPR spectra of WOx(VI)–N2 (g) and WOx(IV)–N2 (h). | ||
Characterisation of the synthesised materials before and after N2 treatment by UV-vis DRS spectroscopy is shown in Fig. 4. Variations in phase and morphology between the WOx samples hamper direct comparisons between their UV-vis spectra.41 However, adsorptions between 400 and 800 nm support the presence tungsten centres with different oxidation states formed due to oxygen defects.38 The prepared WOx materials absorb less in the UV region as commercial WO3. Comparing the adsorption edges indicates an increase in the optical band gap energy when moving from WO3 to the NHSG synthesised oxides.
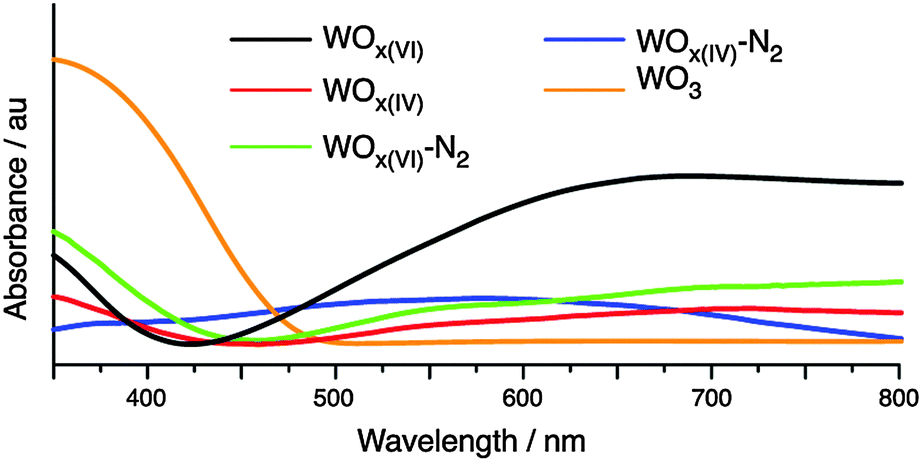 | ||
| Fig. 4 UV-vis DRS spectra of WOx materials. | ||
The performance of these materials as catalysts in photocatalytic oxygen evolution was then studied and compared to commercial WO3 (Fig. 5). The characterisation of this reference oxide can be found in the supporting information. The results show that the commercial WO3 sample has the highest activity, reaching 5.3 × 10−3 mol min−1 g−1 after 1.2 h. Interestingly, despite their relatively low surface area, both N2 treated samples reach higher activities than their as-prepared counterparts, with WOx(VI)–N2 achieving a maximum rate of 3.3 × 10−3 mol min−1 g−1 after 0.5 h. It is important to note that whilst all NHSG prepared materials exhibited lower activities than WO3 during the first 2.5 h, they show less deactivation with both nitrogen treated samples outperforming WO3 between 2.5 and 4.5 h. WOx(VI) and WOx(IV) show minimal activity.
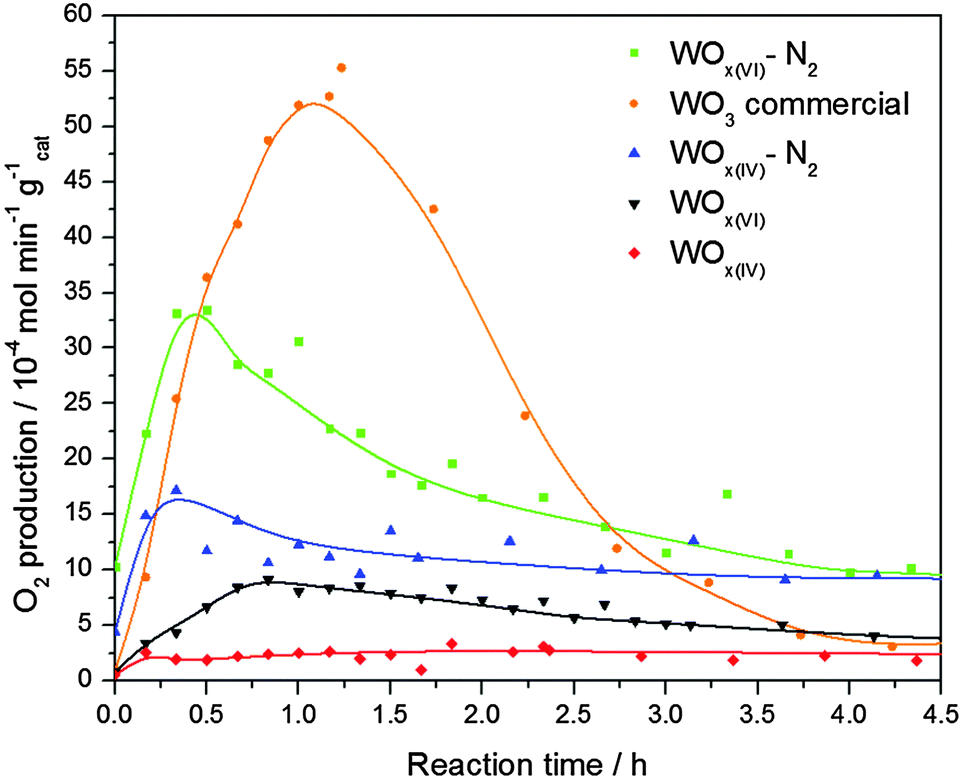 | ||
| Fig. 5 Photocatalytic oxygen evolution of selected catalysts normalised to mass of catalyst. | ||
The previous analysis has shown that the NHSG prepared materials contain oxygen defects. The reaction rates of these defected catalysts are lower than that of WO3, probably because the defects act as electron–hole pair recombination sites and hence quench photocatalytic activity.42 Indeed the activity of the W18O49 phase is expected to be lower than WO3 due to increased amount of reduced W(IV) and W(V) centres.43 Additionally, UV-vis DRS measurements suggest that WO3 has the smallest optical band gap, which is advantageous in photocatalysis, hence its increased activity. Comparing the NHSG prepared materials, the enhanced activity of the N2 treated samples can be explained by an increase in particle size, as electron–hole pair recombination is also known to occur at faster rates on smaller tungsten oxide particles.44 The large increase in activity for WOx(VI)–N2 is attributed to the tetragonal-monoclinic transformation, as monoclinic WO3 is known to have a higher activity towards oxygen evolution than other WO3 phases.45
Conclusions
In conclusion we report a simple method for the preparation of high surface area blue tungsten oxide by the reaction of WCl6 and WCl4(DME) with diisopropyl ether in 1,2-dichloroethane. Substoichiometric tungsten oxides are obtained, with different morphologies depending on the starting precursor. EPR spectroscopy, UV-vis DRS spectroscopy and electrical conductivity measurements suggest the presence of oxygen defects in both materials. Treatment under N2 increases electronic conductivity, indicating an increase in defects. Reduction of the surface area by the N2 treatment improved activity for the photocatalytic evolution of oxygen from water, despite an increased amount of defects. The lower rate of deactivation for the WOx(VI)–N2 and WOx(IV)–N2 catalysts provided activities higher than the reference WO3 for an extended catalytic test. Future work will involve the preparation of doped WOx materials by NHSG methods and their application in photocatalysis.Experimental
General
All experiments were conducted under argon atmosphere using standard Schlenk and glove box techniques unless otherwise stated. Solvents were dried over alumina column (MB SPS-800, MBraun), stored over 4 Å molecular sieves and degassed before use, with the exception of 1,2-dichloromethane and diisopropyl ether, which were dried and distilled over CaH2 and Na respectively. WCl6 (99.99%) was purchased from Strem Chemicals. WCl4(DME) was prepared according to literature.46 Digestion vessels from the Parr Instrument Company were used for NHSG synthesis. Commercial WO3 and WO2 were purchased from Sigma Aldrich.XRD diffractograms were recorded using a Stoe STADI P diffractometer (Cu Kα radiation, λ = 1.54051 Å). TEM images were collected with a Philips CM12 transmission electron microscope. N2 adsorption–desorption analysis was conducted using a BEL-Mini device supplied by BEL Japan Inc with ca. 500 mg of each sample. Results were fitted using BEL-Master programme and BET theory. EPR samples were prepared in an Ar filled glove-box by suspending ca. 10 mg of solid in 0.5 mL toluene in a quartz Young EPR tube and freezing in liquid N2. Continuous Wave (CW) EPR spectra were recorded on a Bruker EMX X-Band spectrometer (9.5 GHz microwave frequency) at 110 K. Electrical conductivity values were determined from powder disks under 0.625 MPa pressure with thicknesses in the range 100–1000 μm. For commercial WO3 conductivity values were determined using Electrical Impedance Spectroscopy. In the case of all other samples, DC measurements were used; where the current was recorded for a range of voltages (0.001–0.1 V) and conductivity subsequently calculated using Ohm's law. Electrical conductivity was determined for three pellet thicknesses and averaged to give the values reported above. UV-Visible DRS Spectroscopy was conducted under ambient conditions using a Cary UV-vis-NIR spectrophotometer, for clarity the spectra were smoothed using the Smooth function of OriginPro programm from OriginLab. For characterisations conducted under ambient conditions, samples were used immediately after removal from the glove box. No colour change was noted for any sample during analysis. XANES measurements at W LIII-edge were performed at the SuperXAS beamline at the Swiss Light Source (Paul Scherrer Institute, Villigen, Switzerland). The SLS is a third generation synchrotron, which operates under top up mode, 2.4 GeV electron energy, and a current of 400 mA. The SuperXAS beamline is positioned on one of three super-bent ports. The incident beam was collimated by Si-coated mirror at 2.8 mrad, monochromatized using a double crystal Si(111) monochromator, and focused with Rh coated toroidal mirror (at 2.8 mrad) down to 100 × 100 μm. The beam intensity was of 4–5 × 1011 ph s−1. We collected XANES data in transmission mode at 298 K under Ar atmosphere.
Representative procedure for the preparation of WOx(VI)
In an Ar filled glove-box, WCl6 (3.96 g, 0.01 mol) was loaded into a digestion vessel with a capacity of 125 mL. 1,2-Dichloroethane (31 mL) was added, followed by excess diisopropyl ether (14 mL, 0.1 mol). After closing, the vessel was removed from the glove box and heated to 180 °C for 3 days using a conventional oven, then allowed to cool overnight. The blue solid obtained was filtered on a glass frit in air, washed with EtOH (3 × 20 mL) and dried overnight under high vacuum (10−5 mbar) at 140 °C. The solid was then stored in an Ar filled glove-box (2.0 g, ca. 85% yield).Preparation of WOx(IV)
WCl4(DME) (4.05 g, 0.01 mol) was reacted with excess diisopropyl ether (14 mL, 0.1 mol) in 1,2-dichloroethane (31 mL) as described above to afford 2.0 g of dark blue material, with a yield of ca. 85%.N2 treatment of WOx
WOx (ca. 2 g) was loaded into a gas flow reactor and heated to 500 °C (2 °C min−1) under N2 flow (120 mL min−1) and kept at this temperature for 3 h. The solid was then evacuated using high vacuum (10−5 mbar) and stored in an Ar filled glove box.Photocatalysis
The photoreactor consisting of a 500 mL jacketed reactor with a 220 mm jacketed quartz immersion well was purchased from Ace Glass, Inc. A quartz mercury low pressure UV arc lamp (450 W, Ace Glass, Inc.) is used to illuminate the photocatalysts. All joints of the reactor are lubricated with Teflon grease (Krytox®), as other greases are observed to decompose and release volatiles when under UV illumination. O2 production is monitored using a micro gas-chromatograph (490 Micro GC, Agilent Technologies, AG) with a 1 m CP-COX column used to separate O2. An oil-filled bubbler is used to prevent backflow of ambient air into either the micro GC or the reactor. Photo-catalytic tests are performed under a flow (26 mL min−1) of N2, with a T-junction going to the microGC allowing on-line sampling of the gases evolved. Photocatalysis is performed within a sealed (non-reflective) light-tight enclosure, and the reactor and UV lamp are cooled with 20 °C cooling water. Agitation is provided via a Teflon stir bar spinning at 435 rpm.Prior to starting photocatalysis, the reactor is filled with a 0.1 M AgNO3 solution, and 100–600 mg of catalyst is loaded. The suspension is then degassed under dynamic vacuum (ca. 20 mbar) for 30 min, followed by de-aeration with bubbling N2 (26 mL min−1) for 1 h. GC analysis of the gas stream is free of oxygen prior to starting each photocatalytic test.
During photocatalysis, the micro GC is programmed to sample the outlet gas stream on regular intervals, and the UV light is turned on at the same moment of the first GC sample. Once photo-catalysis begins the reactor enclosure is not opened and gas production is regularly monitored.
Quantification of the O2 production is done via TCD, which is calibrated prior to use. O2 production was calculated by the following route:
v = volumetric flowrate (mL min−1)
c = concentration (vol%)
n = molar flowrate (mol min−1)
Known quantities: vN2 (N2 flowrate, 26 mL min−1) and cO2 (concentration of O2 measured by TCD, vol%), P = atmospheric pressure 1.01 bar, T = 300 K.
Calculating volumetric O2 flow:
| vO2(mL) = vN2·cO2 |
Converting volumetric O2 flow to molar O2 flow using the ideal gas law:
| nO2 (mol min−1) = PvO2/RT |
This number is then divided the mass of catalyst used.
Acknowledgements
The authors would like to thank CCEM and Umicore AG for funding this project. The group of T. J. Schmidt (PSI Villigen, Switzerland) is acknowledged for insightful discussions and access to electrical conductivity set up. In addition we would like to thank ScopeM for the use of their electron microscopy facilities.Notes and references
- W. Cheng, E. Baudrin, B. Dunn and J. I. Zink, J. Mater. Chem., 2001, 11, 92–97 RSC.
- L. Liang, J. Zhang, Y. Zhou, J. Xie, X. Zhang, M. Guan, B. Pan and Y. Xie, Sci. Rep., 2013, 3, 1936 Search PubMed.
- G. Eranna, B. C. Joshi, D. P. Runthala and R. P. Gupta, Crit. Rev. Solid State Mater. Sci., 2004, 29, 111–118 CrossRef CAS.
- O. Berger, T. Hoffmann and W.-J. Fischer, J. Mater. Sci.: Mater. Electron., 2004, 15, 483–493 CrossRef CAS.
- J. Polleux, A. Gurlo, N. Barsan, U. Wiemar, M. Antonietti and M. Niederberger, Angew. Chem., Int. Ed., 2006, 45, 261–265 CrossRef CAS PubMed.
- R. Abe, H. Takami, N. Murakami and B. Ohtani, J. Am. Chem. Soc., 2008, 130, 7780–7781 CrossRef CAS PubMed.
- S. S. K. Ma, K. Maeda, R. Abe and K. Domen, Energy Environ. Sci., 2012, 5, 8390–8397 CAS.
- H. Chhina, S. Campbell and O. Kesler, J. Electrochem. Soc., 2007, 154, B533–B539 CrossRef CAS.
- D. L. Wang, C. V. Subban, H. S. Wang, E. Rus, F. J. DiSalvo and H. D. Abruna, J. Am. Chem. Soc., 2010, 132, 10218–10220 CrossRef CAS PubMed.
- G. R. Bamwenda and H. Arakawa, Appl. Catal., A, 2001, 210, 181–191 CrossRef CAS.
- Z.-G. Zhao and M. Miyauchi, Angew. Chem., Int. Ed., 2008, 47, 7051–7055 CrossRef CAS PubMed.
- G. Liu, J. Han, X. Zhou, L. Huang, F. Zhang, X. Wang, C. Ding, X. Zheng, H. Han and C. Li, J. Catal., 2013, 307, 148–152 CrossRef CAS.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- S. Jeon and K. Yong, J. Mater. Chem., 2010, 20, 10146–10151 RSC.
- H. Zeng, J. Z. Ou, M. S. Strano, R. B. Kaner, A. Mitchell and K. Kalantar-Zadeh, Adv. Funct. Mater., 2011, 21, 2175–2196 CrossRef.
- G. Orsini and V. Tricoli, J. Mater. Chem., 2010, 20, 6299–6308 RSC.
- A. Vioux, Chem. Mater., 1997, 9, 2292–2299 CrossRef.
- M. Niederberger, Acc. Chem. Res., 2007, 40, 793–800 CrossRef CAS PubMed.
- G. Orsini and V. Tricoli, J. Mater. Chem., 2011, 21, 14530–14542 RSC.
- J. Polleux, M. Antonietti and M. Niederberger, J. Mater. Chem., 2006, 3969–3975 RSC.
- H. Kato, K. Asakura and A. Kudo, J. Am. Chem. Soc., 2003, 125, 3082–3089 CrossRef CAS PubMed.
- N. Balazs, K. Mogyorosi, D. F. Sranko, A. Pallagi, T. Alapi, A. Oszko, A. Dombi and P. Sipos, Appl. Catal., B, 2008, 84, 356–362 CrossRef CAS.
- P. H. Mutin and A. Vioux, Chem. Mater., 2009, 21, 582–596 CrossRef CAS.
- D. P. Debecker and P. H. Mutin, Chem. Soc. Rev., 2012, 41, 3624–3650 RSC.
- R. J. P. Corriu, D. Leclercq, P. Lefevre, P. H. Mutin and A. Vioux, J. Mater. Chem., 1992, 2, 673–674 RSC.
- S. Acosta, R. J. P. Corriu, D. Leclercq, P. Lefevre, P. H. Mutin and A. Vioux, J. Non-Cryst. Solids, 1994, 170, 234–242 CrossRef CAS.
- P. Arnal, R. J. P. Corriu, D. Leclercq, P. H. Mutin and A. Vioux, Chem. Mater., 1997, 9, 694–698 CrossRef CAS.
- L. Bourget, R. J. P. Corriu, D. Leclercq, P. H. Mutin and A. Vioux, J. Non-Cryst. Solids, 1998, 242, 81–91 CrossRef CAS.
- D. P. Debecker, V. Hulea and P. H. Mutin, Appl. Catal., A, 2013, 451, 192–206 CrossRef CAS.
- R. J. P. Corriu, D. Leclercq, P. Lefevre, P. H. Mutin and A. Vioux, Chem. Mater., 1992, 4, 961–963 CrossRef CAS.
- D. P. Debecker, R. Delaigle, K. Bouchmella, P. Eloy, E. M. Gaigneaux and P. H. Mutin, Catal. Today, 2010, 157, 125–130 CrossRef CAS.
- P. Arnal, R. J. P. Corriu, D. Leclercq, P. H. Mutin and A. Vioux, Mater. Res. Soc. Symp. Proc., 1994, 346, 339–344 CrossRef CAS.
- M. Gillet, C. Lemire, E. Gillet and K. Aguir, Surf. Sci., 2003, 532, 519–525 CrossRef.
- C. G. Granqvist, Appl. Phys. A: Solids Surf., 1993, 57, 3–12 CrossRef.
- J. G. Allpress, R. J. D. Tilley and M. J. Sienko, J. Solid State Chem., 1971, 3, 440–451 CrossRef CAS.
- E. Gerbert and R. J. Ackermann, Inorg. Chem., 1966, 5, 136–142 CrossRef.
- K. Viswanathan, K. Brandt and E. Salje, J. Solid State Chem., 1981, 36, 45–51 CrossRef CAS.
- A. Chemseddine, R. Morineau and J. Livage, Solid State Ionics, 1983, 9–10, 357–361 CrossRef CAS.
- A. Aboulaich, B. Boury and P. H. Mutin, Eur. J. Inorg. Chem., 2011, 3644–3649 CrossRef CAS.
- O. Y. Khyzhun, J. Alloys Compd., 2000, 305, 1–6 CrossRef CAS.
- J. Liu, O. Margeat, W. Dachraoui, X. Liu, M. Fahlman and J. Ackermann, Adv. Funct. Mater., 2014, 24, 6029–6037 CrossRef CAS.
- S. B. Rawal, S. Bera and W. I. Lee, Catal. Lett., 2012, 142, 1482–1488 CrossRef CAS.
- C. Guo, S. Yin, M. Yan, M. Kobayashi, M. Kakihana and T. Sato, Inorg. Chem., 2012, 51, 4763–4771 CrossRef CAS PubMed.
- F. Amano, E. Ishinaga and A. Yamakata, J. Phys. Chem. C, 2013, 117, 22584–22590 CAS.
- G. Xin, W. Guo and T. Ma, Appl. Surf. Sci., 2009, 256, 165–169 CrossRef CAS.
- C. Persson and C. Andersson, Inorg. Chim. Acta, 1993, 203, 235–238 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Representative bright field TEM image of commercial WO3, XRD powder diffraction pattern of commercial WO3, N2 adsorption–desorption isotherm of commercial WO3, surface area and conductivity data and elemental analysis data for the NHSG materials before and after N2 treatment. See DOI: 10.1039/c5nj01973g |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |