
Design of dual stimuli responsive polymer modified magnetic nanoparticles for targeted anti-cancer drug delivery and enhanced MR imaging†
Dipsikha
Bhattacharya
*ab,
Birendra
Behera
c,
Sumanta Kumar
Sahu
d,
Rajakumar
Ananthakrishnan
a,
Tapas Kumar
Maiti
c and
Panchanan
Pramanik
*ef
aDepartment of Chemistry, Indian Institute of Technology Kharagpur, West Bengal, India. E-mail: dipsikha.chem@gmail.com; pramanik1946@gmail.com; Fax: +91-522-2628227; Tel: +91-522-2620107
bNanotherapeutics Laboratory, CSIR-Indian Institute of Toxicology Research, Lucknow, India
cDepartment of Biotechnology, Indian Institute of Technology Kharagpur, West Bengal, India
dDepartment of Applied Chemistry, Indian School of Mines, Dhanbad, India
eDepartment of Nano-science and Nanotechnology, GLA University, Mathura, UP 281406, India
fMCKV Institute of Engineering, Howrah, 711204, India
First published on 19th October 2015
Abstract
Herein, we report a strategic design of dual temperature and pH responsive polymer integrated magnetic nanohybrids comprising of smart block copolymers and mixed ferrite nanoparticles (MFNPs) for efficient anti-cancer drug delivery and magnetic resonance imaging (MRI). Citrate stabilized mixed ferrite nanoparticles (CA-MFNPs) were intelligently modified with the dual responsive polyethyleneimine (PEI) cross-linked Pluronic F127 copolymer via the EDC/NHS method. In order to accomplish cancer targeting and imaging capability, both the folic acid (FA) and rhodamine isothiocyanate (RITC) were tethered to the nanoparticles via intricate chemical approaches. These FA targeted nanohybrids were further entrapped with doxorubicin (DOX) and their release pattern was investigated. These DOX loaded FA targeted nanoparticles (DOX–FA–Poly-MFNPs) demonstrated high drug payload and encapsulation efficiency i.e. 4.6% and 92.0%, respectively. It is shown that at the lower pH/higher temperature i.e. acidic pH (5.0) and at body temperature (37 °C), the DOX–FA–Poly-MFNPs exhibited enhanced release of DOX (drug release value ∼53%) while retaining their stealthy structure under physiological conditions (drug release value ∼12%), exhibiting an apparent thermo/pH controlled drug release pattern. The folic acid receptor (FAR) specific endocytosis to cancer cells (human cervix adenocarcinoma i.e. HeLa) in comparison to normal immortalized keratinocytes (HaCaT) cells were demonstrated via fluorescence microscopy and magnetic resonance imaging (MRI). Furthermore, these DOX–FA–Poly-MFNPs displayed effective therapeutic activity evaluated by a cytotoxicity assay and cell cycle analysis in HeLa cells. Therefore, these dual responsive mixed ferrite nanoparticles may serve as promising theranostic agents for in vivo cancer therapy.
1. Introduction
Since the last decade, cancer research has envisioned that malignant cells exhibit an enhanced temperature and acidic microenvironment due to their rapid growth, providing a handle for pathological cell treatment with controlled drug release properties. This concept has stimulated researchers towards the development of thermo, pH and dual stimuli-responsive polymeric nanomaterials (micelles, vesicles, and gels) for cancer treatment.1,2 Among them, dual pH and thermo-responsive nanocarriers have been striving forward as next generation therapeutic nanomedicines because they can intelligently trigger their release in a stimuli responsive manner with minimal toxicity, improved therapeutic efficacy and reduced dosage to healthy cells.3–6 Inorganic nanoparticles such as silica7,8 and magnetic nanoparticles (MNPs) in forms of iron oxide as well as metal doped mixed ferrite nanoparticles integrated with dual responsive polymeric functionalities have gained emerging attention for a myriad of biomedical applications in simultaneous diagnosis and therapy.9–15 Of late, there is a stride of developing intelligent theranostic systems using stimuli responsive polymers as delivery carriers and MNPs as MR imaging agents which can offer an improved target-guided delivery of therapeutics with a synergistic package of specific targeting,15 MR based diagnostic imaging16 and stimuli triggered delivery efficacy.17,18In the past decade, pH-responsive polymer embedded MNP nanocarriers have attracted profound attention as therapeutic carriers since there is a significant discrepancy in pH values in normal tissues (7.4)19 and the acidic environment of both the extracellular (pH ∼ 6.8) and endosomal environments (∼5.0–5.5) of tumor cells.20,21 Additionally, temperature-responsive MNP nanocarriers21 (exhibiting lower critical solution temperature – LCST) have also been widely studied because of their inherent capability to generate heat in the presence of an applied magnetic field (AMF)22 which can trigger the release of drugs from the polymeric carrier in tumor cells. To date, very limited reports are available on dual-responsive polymer modified MNPs harnessing both the pH23,24 and thermo responsive25–31 eminences of the synthesized polymer to MNPs.29–32 In addition, it is also noteworthy that in most of the cases, poly(N-isopropylacrylamide) (PNIPAAm) or poly(N-vinylcaprolactam) (PVCL) has been used as thermo sensitive polymers.29–32 Recently, our lab has also reported dual responsive MNPs as drug delivery carriers utilizing PNIPAAm as the surface activating polymer.31 Of late, Patra et al. also reported a dual responsive polymer (poly N-isopropylacrylamide-co-poly glutamic acid) coated mixed ferrite nanosystems for drug delivery applications.33
Among the block copolymers, triblock copolymer pluronic, consisting of both hydrophobic poly(propylene oxide) (PPO) and hydrophilic poly(ethylene oxide) (PEO) blocks, has been widely acclaimed as thermo responsive delivery nanocarriers for both in vivo and in vitro drug delivery applications.32,34–38 Similarly, polyethylene imine (PEI), a highly acclaimed pH responsive polymer,39 has already been used in various modifications for drug/gene delivery applications.38–40 Recently, Liu et al. has reported Pluronic crosslinked PEI (PF127-PEI) block copolymer stabilized magnetic nanosystems as thermoresponsive drug delivery carriers,27 however, as par our knowledge is concerned, no one has embarked on the development of PF127-PEI tethered MNPs as dual delivery systems.
In addition, along with the concern of delivery of therapeutics, the development of enhanced MR based diagnostic imaging for cancer monitoring is also of prime concern to visualize, tract and monitor the disease at its earliest stages. MNPs, especially Mn doped mixed ferrite nanoparticles (MFNPs), have drawn attention as ultrasensitive T2 contrast agents to enhance the sensitivity of non-invasive MRI imaging.41 In light of this advantage, we envisioned that these PEI-PF127 polymer tethered MFNPs as theranostic nanocarriers would be of great interest, due to their potential advantage in delivering therapeutics in a intelligent manner distinguishing normal and pathological cells, and triggered release of cargos in a stimuli responsive manner along with enhanced monitoring efficacy.
To achieve the target, a strategic design for the fabrication of dual responsive mixed ferrite nanohybrids is proposed and followed by further integration/encapsulation with a targeting agent (FA), fluorescence imaging dye i.e. rhodamine B isothiocyante (RITC) and doxorubicin (DOX) as the anticancer drug. In particular, dual responsive polymer i.e. PEI-PF127 was synthesized using a simple conjugation approach and latter tethered to citrate stabilized mixed ferrite MFNPs (CA-MFNPs). Thereafter, FA and RITC were conjugated to aminated PEI-PF127-MFNPs (Poly-MFNPs) to endow the carrier with cancer targeting and optical imaging ability. Finally, a therapeutic agent, DOX molecules, has been encapsulated inside the hydrophobic cores of the nanocarriers to construct the targeted nanomedicine. Since most tumor sites usually exhibit a more acidic environment or a higher temperature, the dual pH and thermo-responsive releasing efficacy of the DOX entrapped nanoparticles can trigger enhanced drug release at a tumor region than a normal one. The resulting nanocomposites have several advantages such as high drug entrapment efficiency, enhanced FAR targeting and stimuli triggered cargo release ability to acidic cancer cells along with the MR assisted intracellular tracking ability. As far our knowledge is concerned, this is the first attempt of a dual responsive polymer modified targeted nanomedicine (DOX–FA–Poly-MFNPs) with cancer targeting, stimuli responsive drug delivery and enhanced MR imaging ability.
2. Results and discussion
2.1. Synthesis and characterization of dual responsive theranostic mixed ferrite nanoparticles
The goal of this work was to design and develop a dual pH and temperature responsive polymer modified targeted nanomedicine (DOX–FA–Poly-MFNPs) for the targeted delivery of the anticancer drug doxorubicin (DOX) in a stimuli triggered fashion and to achieve enhanced MR imaging. The step wise illustration involved in the fabrication of this magnetic nanomedicine has been shown in Scheme 1.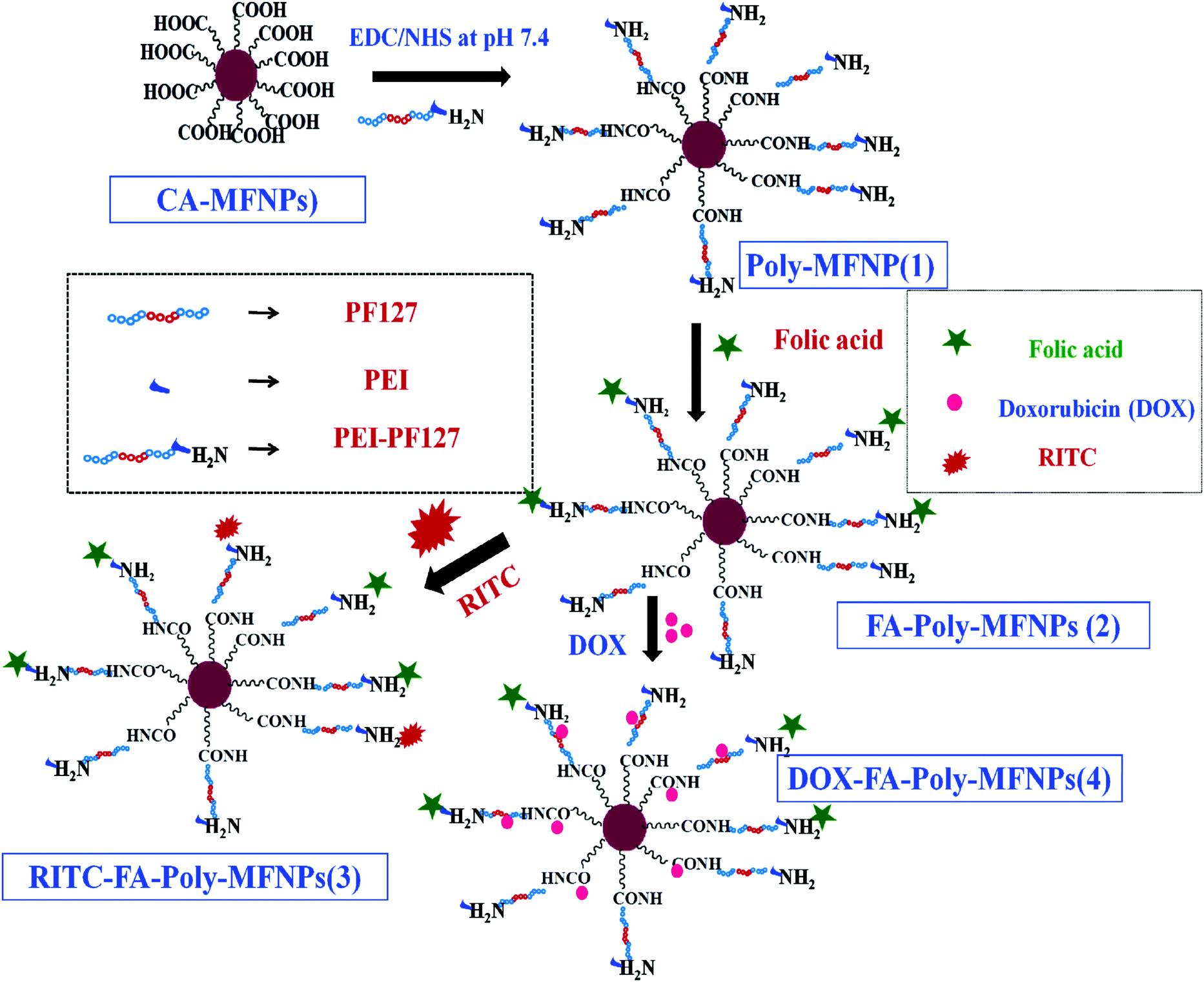 | ||
| Scheme 1 Schematic illustration of the fabrication of dual pH and temperature responsive Poly-MFNPs and their subsequent conjugation with folic acid, RITC and DOX to fabricate a multifunctional nanomedicine. | ||
To fabricate dual responsive mixed ferrite particles described as PEI-PF127-MFNPs or Poly-MFNPs (1), monodispersed mixed ferrite nanoparticles (MFNPs) were synthesized by an aqueous co-precipitation approach in the presence of trisodium citrate. For the synthesis of dual responsive copolymers, Pluronic F127 (PF127) has been chosen as the thermo responsive polymer because of its lower LCST facilitating the self assembly formation around body temperature due to the temperature responsive hydration/dehydration phenomena.27 Polyethyleneimine (PEI) was chosen because of its manifold role of pH responsive nature due to its well known proton sponge effect and also its functional amine donating efficacy.42 Amine functionalities of the dual responsive PEI-PF127 copolymer were covalently tethered to the citrate stabilized mixed ferrite MFNPs (CA-MFNPs) using EDC/NHS chemistry.43 Furthermore, the cancer targeting property was introduced into these Poly-MFNPs through folic acid (FA) tethering via the EDC/NHS approach.44 Both of the FA targeted (FA–Poly-MFNPs) and non targeted (Poly-MFNPs) were also labelled with RITC by covalent linking45 utilizing some of the residual surface amine groups imparting RITC–Poly-MFNPs and RITC–FA–Poly-MFNPs respectively.
The high resolution X-ray diffraction pattern of CA-MFNPs (Fig. S1 in the ESI†) depicted the broadening of peaks with small crystalline sizes of the nanoparticles (an average core size of about 4–6 nm) using the Debye–Scherrer formula for spherical particles. The size distribution and morphology of these Poly-MFNPs (1) were examined by transmission electron microscopy (TEM) analysis. Fig. 1(A and B) displays the TEM images of CA-MFNPs and Poly-MFNPs (1). Fig. 1(A) demonstrated that before polymeric modification, the size of the CA-MFNPs were in between 8 ± 2 nm whereas after modification, these Poly-MFNPs showed a size enhancement as well as the presence of a thin polymer layer depicting their successful modification (∼12 nm) [Fig. 1(B)].
 | ||
| Fig. 1 (A) and (B) TEM images of CA-MFNPs and Poly-MFNPs. | ||
However, some agglomeration appeared in the case of Poly-MFNPs due to their high magnetization and surface energy, although it was comparatively lower with respect to CA-MFNPs. Uniform hydrodynamic sizes (HDs) and surface charges of these Poly-MFNPs (1) at physiological pH were observed at around 80 ± 10 nm (PDI ∼ 0.213) and 14.5 mV reflecting good stabilization of these Poly-MFNPs. After FA modification, the moderate increase in HD of FA–Poly-MFNPs (2) (∼91 nm) and decrease in positive charge (−12.5 mV at pH 7.4) demonstrated that these FA–Poly-MFNPs were functionalized with carboxylate groups that endowed enhanced hydrogen bonding efficacy compared to 1 nanoparticles. [A detailed description of DLS and zeta potential of these nanoconjugates is included in the ESI† in Table S1 and Fig. S2(A).]
The pH responsive nature of our nanoconjugates (Poly-MFNPs and FA–Poly-MFNPs) was validated by measuring their effective hydrodynamic diameters (HDs) and surface charge analysis under various pHs at room temperature (RT). (See Fig. S2(A) i.e. zeta potential vs. pH and (B) i.e. HD change vs. pH in the ESI.†) It was observed that the surface charges and HDs of Poly-MFNPs showed an enhancement from 14.5 mV to 35.5 mV and 81 nm to 113 nm when pH was decreased from 7.4 to 5. These phenomena might be due to the extensive protonation of the PEI residues of Poly-MFNPs due to its proton sponge effect, which resulted in an electrostatic repulsion between the immobilized –NH2 groups of the polymer. Thus, these Poly-MFNPs inevitably underwent swelling under acidic pH ∼ 5, which can eventually accelerate the disruption of cargos entrapped in the nanohybrids.38 Therefore, the results showed that these Poly-MFNPs could endow pH responsive properties. In order to check whether the pH responsive nature was retained or not, these FA–Poly-MFNPs (2) were also thoroughly checked with changes in HD vs. pHs and also zeta potential vs. pHs. It is shown in [Fig. S2(A) and (B), ESI†] that at pH ∼ 5, 2 exhibit reasonable HD changes (105 nm) and high positive charge (+22.5 mV) as compared to their value from physiological pH (91.5 nm and −12.5 mV). It was assumed that at lower pH, the presence of both functional –NH3+ and –CO2H groups on FA–Poly-MFNPs counterbalanced the drastic HD change as compared to Poly-MFNPs (113 nm at pH 5), although displayed the same pH responsive nature. In contrast, with increasing pH, most of their carboxylate groups of 2 exhibit repulsion between the functional –CO2H groups, reflected by their enhanced change in HD and surface charge (103 nm and −19.6 mV at pH 9). This HD change at higher pH was reasonably larger compared to Poly-MFNPs (91 nm). Therefore, it was clear that both of these nanoparticles showed almost similar response at lower pH values while at higher pH, the FA–Poly-MFNPs exhibited considerable discrepancy indicating the presence of –CO2H groups.
Likewise, the thermo responsive nature of our nanohybrids (both Poly-MFNPs and FA–Poly-MFNPs) was illustrated with the variation of HDs under different temperatures at physiological pHs (see Fig. S2(C) in the ESI†). The volume average HDs of Poly-MFNPs underwent a sharp decrease from 85 to 31 nm with the increasing temperature from 2 °C to 45 °C. This temperature responsive phenomenon might be attributed due to the thermally induced self assembly of the PF127 (PEO–PPO–PEO) segment of the copolymer and was harmonized well with the previously reported stimuli responsive Pluronic block copolymers.27,38 At relatively low temperatures, the PF127 chains of Poly-MFNPs were fully extended via interaction with water molecules, facilitating the entrapment of drug molecules into the vector. While with increasing temperature above critical micelle concentration (CMC), a dehydration process of polymer blocks was induced resulting in the effective clustering of PPO blocks.27,46,47 Thus, at the temperature close to the body temperature (37 °C), the polymer shell of Poly-MFNPs got contracted, forming a compact nanostructure, which would reflect in the decrease of the HDs with increasing temperature. As the thermoresponsive nature depends on PF127, it was assumed that after FA modification, as the polymer PF127 segment remains intact, therefore the FA–Poly-MFNPs must exhibit the similar pattern of changes with respect to HD vs. temperature.
The successful consecutive modifications of MFNPs using citric acid (CA-MFNPs), PEI-PF127 as well as FA, were evaluated by FTIR analysis. The FTIR spectra of CA-MFNP, PEI-PF127-MFNPs i.e. Poly-MFNPs (1), bare FA, and FA–PEI-PF127-MFNPs i.e. FA–Poly-MFNPs (2) are shown in Fig. 2. (The FT-IR spectra of activated PF127 and PEI-PF127 are given in Fig. S3, ESI.†) For CA-MFNPs, the broad band, at around 1630 cm−1 and a small peak at around 1715 cm−1 confirmed the successful modification of the mixed ferrite surface with carboxylate groups.48
 | ||
| Fig. 2 FT-IR spectra of CA-MFNPs, PEI-PF127-MFNPs (Poly-MFNPs), FA–PEI-PF127-MFNPs (FA–Poly-MFNPs) and free folic acid (FA). | ||
A strong absorption band of M–O bonds was also observed at 590 cm−1 in this spectrum, indicating the successful formation of MFNPs. The spectrum of Poly-MFNPs (1) showed not only the main characteristic band of M–O but also the characteristic peaks of polymer at 1657 and 1563 cm−1 indicating the formation of the amide band. The bands at 1626 cm−1 and a broad band at 3400 cm−1 validated the presence of primary amine groups on 1.45 Hence, these results portrayed that the PEI-PF127 polymer was successfully immobilized onto the surface of CA-MFNPs. The FTIR spectrum of pure FA was characterized by a number of characteristic bands according to previously reported literature.49 In the FTIR spectrum of FA–Poly-MFNPs (2), the peaks at 1654 cm−1 (bonded C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of –CONH amide band II) and 1564 cm−1 (–NH amine band II) appeared with increased absorbance, generated from the amide bands within the FA structure as well as the amide bonding between FA and amine-functionalized nanoparticles. Both the absence of a band at 1629 cm−1 and the presence of sharp bands at 1687, 1605 and 1401 cm−1 hereby confirmed the successful conjugation of FA to these Poly-MFNPs. Further, to demonstrate the modification of FA on Poly-MFNPs, the folate content on 1 and 2 were conveniently compared with a standard FA absorbance, attributing to folate only (as shown in Fig. S4, ESI†). It is already known that Pure FA showed two peaks at 280 and 360 nm due to n–π* and π–π*transitions, respectively. [Given as Fig. S4 (inset figure) in the ESI.†] After modification with FA, these two peaks were observed with a little shift, signifying the successful modification with folic acid.
O of –CONH amide band II) and 1564 cm−1 (–NH amine band II) appeared with increased absorbance, generated from the amide bands within the FA structure as well as the amide bonding between FA and amine-functionalized nanoparticles. Both the absence of a band at 1629 cm−1 and the presence of sharp bands at 1687, 1605 and 1401 cm−1 hereby confirmed the successful conjugation of FA to these Poly-MFNPs. Further, to demonstrate the modification of FA on Poly-MFNPs, the folate content on 1 and 2 were conveniently compared with a standard FA absorbance, attributing to folate only (as shown in Fig. S4, ESI†). It is already known that Pure FA showed two peaks at 280 and 360 nm due to n–π* and π–π*transitions, respectively. [Given as Fig. S4 (inset figure) in the ESI.†] After modification with FA, these two peaks were observed with a little shift, signifying the successful modification with folic acid.
The formation of the as mentioned surface-modified nanoparticles was further corroborated by Thermal Gravimetric analysis (TGA) measurements. Step-wise weight loss from the nanoparticles with increasing temperature (ESI,† Fig. S5) could be explained depending on the decomposition of the surface functionalities introduced at various stages. To authenticate the feasibility and sensitivity of Poly-MFNPs (1) as enhanced MR imaging nanoprobes, it is crucial that MFNPs should retain their favourable magnetic properties after surface modification. The magnetic properties of CA-MFNPs, Poly-MFNPs and FA–Poly-MFNPs have been evaluated using the vibration sample magnetometry (VSM) analysis (as depicted in Fig. S6, ESI†). The results corroborated that after modification with stimuli responsive polymers, Poly-MFNPs exhibited superparamagnetic nature with reduced magnetization compared to CA-MFNPs signifying their successful modification. Moreover, surface functionalization was performed further and confirmed by XPS studies. A detail explanation of XPS spectra and analysis is given in the ESI† (Fig. S7).
2.2. Fabrication of DOX loaded targeted nanomedicine (DOX–FA–Poly-MFNPs) and their dual responsive drug release phenomena
Doxorubicin (DOX) was employed as a model drug to investigate the loading and release profile of the dual responsive Poly-MFNPs. The DOX loading was thoroughly standardized by varying DOX concentrations (0.2 mg ml−1 to 1 mg ml−1) (data not shown) with 10 mg FA–Poly-MFNPs and the best ratio of DOX![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :FA–Poly-MFNPs obtained was 20:1. The loading content and entrapment efficiency of DOX in FA–Poly-MFNPs were found to be 4.6% and 92.0%, respectively. It was previously reported that DOX can be loaded inside nanocarriers via both hydrophobic/ionic interactions46 as well as covalent pH sensitive bonding.50,51 In our case, entrapment of DOX in FA–Poly-MFNPs (2) was due to the combined hydrophobic and hydrogen bonding interactions between DOX molecules and PEI-PF127. In order to validate the dual responsive property of Poly-MFNPs, in vitro DOX release from DOX–FA–Poly-MFNPs (4) was investigated at varied pHs i.e. physiological and lysosomal pH (pH 7.4 and 5.5) and varied temperatures (25 and 37 °C) [Fig. 3(A)]. The stimuli triggered release mechanism of DOX from DOX–FA–Poly-MFNPs is well represented in Scheme 2.
:FA–Poly-MFNPs obtained was 20:1. The loading content and entrapment efficiency of DOX in FA–Poly-MFNPs were found to be 4.6% and 92.0%, respectively. It was previously reported that DOX can be loaded inside nanocarriers via both hydrophobic/ionic interactions46 as well as covalent pH sensitive bonding.50,51 In our case, entrapment of DOX in FA–Poly-MFNPs (2) was due to the combined hydrophobic and hydrogen bonding interactions between DOX molecules and PEI-PF127. In order to validate the dual responsive property of Poly-MFNPs, in vitro DOX release from DOX–FA–Poly-MFNPs (4) was investigated at varied pHs i.e. physiological and lysosomal pH (pH 7.4 and 5.5) and varied temperatures (25 and 37 °C) [Fig. 3(A)]. The stimuli triggered release mechanism of DOX from DOX–FA–Poly-MFNPs is well represented in Scheme 2.
 | ||
| Fig. 3 (A) Stimuli triggered release pattern of DOX from DOX–FA–Poly-MFNPs under different pH and temperature conditions. (B) Cell cytotoxicity assay of HeLa and HaCaT cells treated with FA–Poly-MFNPs, DOX–Poly-MFNPs and DOX–FA–Poly-MFNPs at different concentrations. Cell toxicity is measured by an MTT assay at 570 nm. | ||
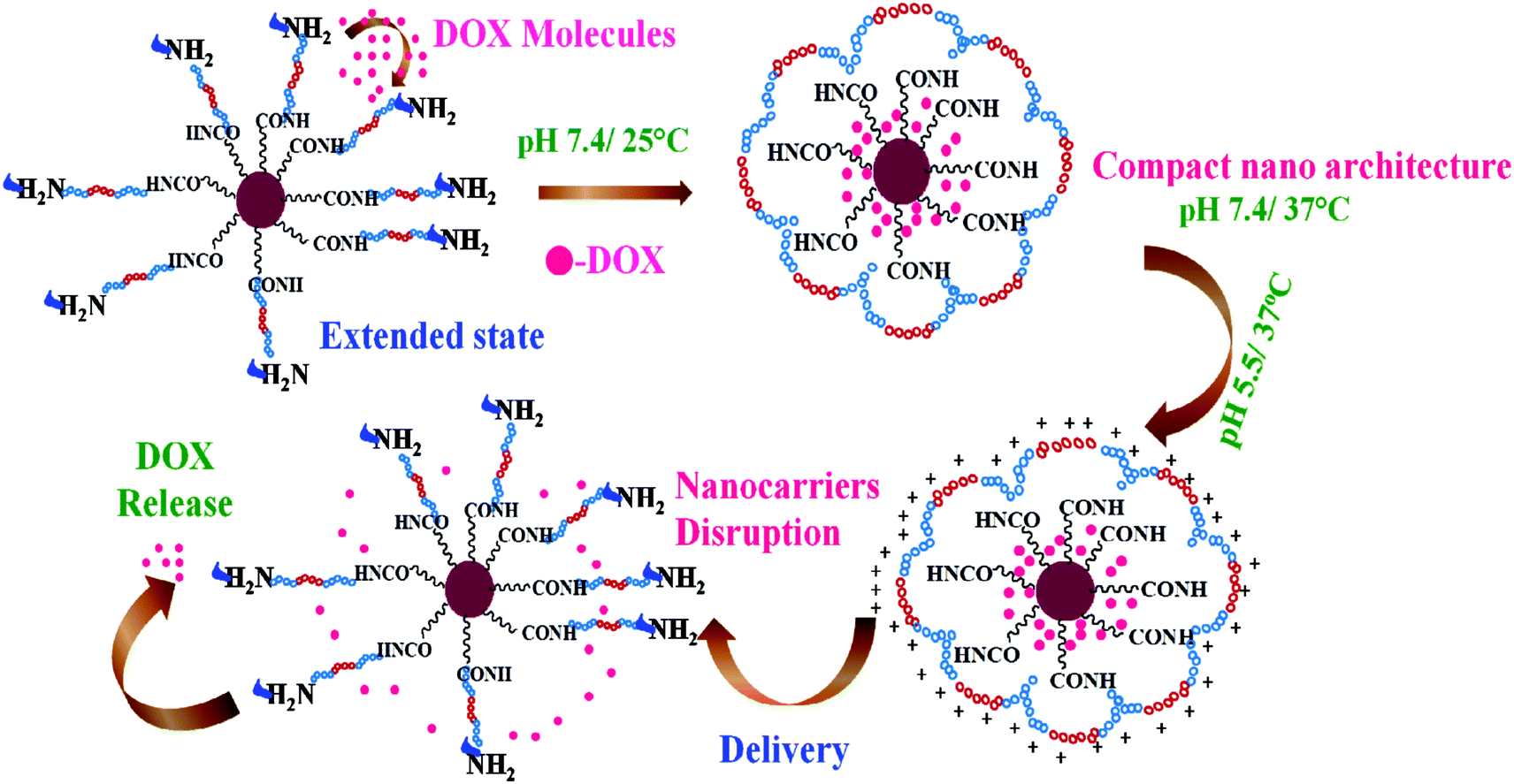 | ||
| Scheme 2 Proposed presentation of dual temperature and pH responsive formation of self assembled Poly-MFNP nanocarriers and the stimuli triggered release of doxorubicin through disruption of Poly-MFNPs at acidic pH and physiological temperature. | ||
For temperature controlled release, at pH 7.4, nearly, 11–12% DOX was released at 37 °C after 24 h, whilst 23% drug was released at 25 °C from DOX–FA–Poly-MFNPs (4). In contrast, at pH 5.5, about 31% of the loaded DOX was released after 24 h at 25 °C, whereas 53% was released at the same time interval at 37 °C. From this, it was speculated that the cumulative drug release was enhanced at lysosomal i.e. acidic pH compared to physiological pH at body temperature. The block copolymer exhibited the combined and complementary role of a stimuli responsive drug delivery carrier where the PF127 segment was merely responsible for the fabrication of thermoresponsive compact Poly-MFNPs at body temperature while PEI plays the pH responsive role. The slower DOX release at the physiological temperature (37 °C) and pH 7.4 authenticated the supreme stability of DOX–FA–Poly-MFNPs as an ideal delivery carrier. Due to the temperature responsive self assembly behaviour, at 25 °C, the DOX molecules were entered inside the self assembled nanoparticles through the extended polymeric shell while with increasing temperature above the LCST, the shell was contracted, imparting the compact structure of DOX–FA–Poly-MFNPs, entrapping the loaded DOX molecules.27,46 When these compact DOX–FA–Poly-MFNPs were delivered to tumor cells, at the lysosomal pH ∼ 5.5, the proton sponge effect of PEI exhibited protonation in the –NH2 groups of PEI segments of DOX–FA–Poly-MFNPs, which resulted in enhanced electrostatic repulsion followed by swelling between the particles. This phenomenon subsequently triggered the release of drugs followed via the disruption of DOX–FA–Poly-MFNPs and the DOX molecules would come out in a controlled fashion in cancer cells at body temperature.2,38 In contrast, at pH 7.4, the almost neutrally charged DOX particles displayed negligible extent of release restraining the degradation of these DOX–FA–Poly-MFNPs (4) during blood circulation. Thus, at body temperature, the DOX–FA–Poly-MFNPs i.e.4 nanoparticles were highly stable at pH 7.4 but were degraded when the pH was lowered to 5.5, mimicking lysosomal pH of acidic tumor cells. The drug release pattern reflected that the dual responsive polymer exhibited a key role in both the loading as well as release of loaded drug in a desired fashion.
2.3. Biological evaluation of targeted, non-targeted and DOX-loaded nanoparticles
In contrast, the DOX–FA–Poly-MFNPs (4) and DOX–Poly-MFNPs showed significant cytotoxicity to cancer HeLa (HeLa-HFAR) cells after 24 h incubation. It was observed that the DOX–FA–Poly-MFNPs (4) induced almost 81% killing of HeLa cells [as shown in Fig. 3(B)]. DOX–Poly-MFNPs without FA caused more than 50% growth inhibition at 25 μg ml−1, although it was comparatively lower than DOX–FA–Poly-MFNPs. Whereas, at the same concentration (25 μg ml−1), the DOX–FA–Poly-MNPs (4) are almost cytocompatible in normal HaCaT cells. The half maximum inhibition concentration (IC50) value of 4 is 12.5 μg ml−1 in HeLa cells.
Further to check whether the higher death in HeLa cells was due to the FA targeted receptor mediated endocytosis,52 the same concentration dependent study has been performed using synthesized DOX–FA–Poly-MFNPs and DOX–Poly-MFNPs without FA in HeLa-HFAR and HeLa cells expressing a low folic acid receptor (HeLa-LFAR) for 2 h treatment (shown in Fig. 4).
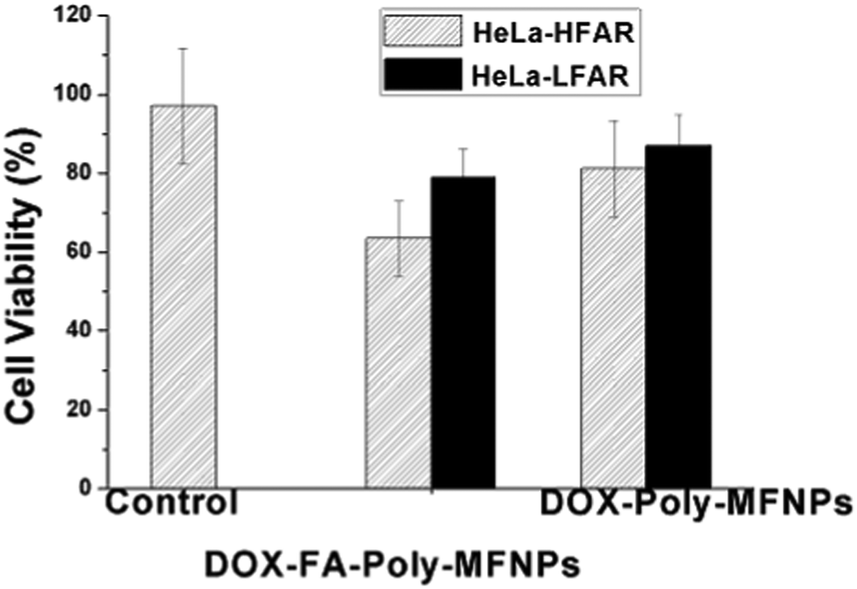 | ||
| Fig. 4 Study of a folic acid receptor (FAR) targeting efficiency of DOX–FA–Poly-MFNPs and DOX–Poly-MFNPs into HeLa-HFAR (highly FAR overexpressed) and HeLa-LFAR (low FAR overexpressed) cells. | ||
It was shown that after 2 h incubation, the DOX–FA–Poly-MFNPs (25 μg ml−1) showed effective toxicity (around 63% viable cells) in HeLa-HFAR cells, while the DOX–Poly-MFNPs were significantly less toxic to HeLa-HFAR (81% viable cells) signifying that this higher toxicity of DOX–FA–Poly-MFNPs was mainly controlled due to FA modification because both were incubated with the same origin of HeLa cells. On the other hand, treated with either DOX–FA–Poly-MFNPs or DOX–Poly-MFNPs, in both the cases, HeLa-LFAR showed almost a considerable amount of viable cells, indicating a less targeting effect to HeLa-LFAR cells. Therefore, it could be authenticated that the targeting efficiency of DOX–FA–Poly-MFNPs was due to FAR mediated internalization to HeLa cells over expressing high percentage of Folate receptors (HeLa-HFAR) and induced higher cell death.
 | ||
| Fig. 5 (a–e) Fluorescence images of the uptake of HeLa-HFAR cells incubated with RITC–Poly-MFNPs and RITC–FA–Poly-MFNPs after (a) 0 min, (b) 30 min, (c) 1 h, (d) 2 h and (e) 4 h incubation. | ||
 | ||
| Fig. 6 (A) T2-weighted spin-echo MR phantom images of HeLa cells and HaCaT cells incubated with 0–30 μg ml−1 of various nanoparticle suspensions (FA–Poly-MNPs, Poly-MFNPs and FA–Poly-MFNPs) for 4 h. FA–Poly-MNPs are the abbreviated form of FA targeted polymer modified magnetite (Fe3O4) nanoparticles to compare the enhanced MR activity between mixed ferrite magnetic nanoparticles (MFNPs) and magnetite nanoparticles (MNPs). (B) T2 relaxation analysis of HeLa and HaCaT cell suspensions of the FA–Poly-MFNPs, Poly-MFNPs and FA–Poly-MNPs. (C) r2 Relaxivity analysis of FA–Poly-MFNPs with increasing Fe concentrations. | ||
The relative T2 relaxation times of target FA–Poly-MFNPs (2) as well as control Poly-MFNPs (1) in HeLa-HFAR and HaCaT cells were also evaluated with increasing concentrations (0–30 μg ml−1). Fig. 6(B) shows the T2-relaxation time as a function of particle concentration (1 and 2) in cell-culture media. HeLa cells cultured with target (2) nanoparticles possessed a shorter T2-relaxation time compared to HaCaT cells, which was probably arising due to enhanced magnetism, germinated probably from the higher internalization of 2 by HeLa cells in comparison to the same in HaCaT cells. It was also observed that HeLa cells cultured targeted 2 had a shorter T2 relaxation time than the targeted-Poly-MNPs due to enhanced magnetism of similar sized Mn-doped mixed ferrite nanoparticles. It was further observed that the relaxation rate 1/T2 varied linearly with the iron concentration (eqn (2) given in the Experimental section). The transverse relaxivity, r2, of the targeted 2 in HeLa cells (corresponding to the slope of the line) was found to be 184.22 mM−1 s−1 and it was found more than three times higher compared to FA–Poly-MNPs (60.12 mM−1 s−1) in HeLa cells as shown in Fig. S11(B) (ESI†). All these results unequivocally established the potential of our as-prepared FA–Poly-MFNPs as cancer-targeted, MRI probes.
From Fig. 7(A–F), it is evident that only FA–Poly-MFNPs (without DOX) has no apoptotic effect on HeLa cells in comparison whereas in free DOX, both the S phase population (24.44%) along with an apoptotic Sub G0/G1 phase (6.0%) population count has been increased. In the case of DOX–FA–Poly-MFNPs, increasing concentrations of DOX–FA–Poly-MFNPs showed the maximum cell intensification at the S phase, exhibiting cell cycle arrest in the same phase. In addition, the apoptotic Sub G0/G1 phase population also increases in a dose dependent manner in comparison to 2. Therefore, DOX–FA–Poly-MFNPs has inhibited the growth of HeLa cells by arresting cell cycle progression at the S phase and followed to apoptosis or cell death. According to the previous reports, the DOX prevents cancer cell growth by inducing DNA breakage and arresting the cell cycle at the G2/M phase53 in HeLa cells. The deviation observed in our work, from previous reports may not be erroneous because free DOX also induced S phase arrest in our study. Again there are also reports that DOX molecules induced S phase arrest in other cancers cells like MCF-7 breast cancer cell line,54 so the possibility of S phase arrest by DOX treatment cannot be nullified.55
 | ||
| Fig. 7 Flow-cytometric analysis of cell cycle phase distribution. HeLa cells treated with (A) control FA-Poly-MFNPs, (B) bulk DOX 10 μg ml−1, (C) target DOX–FA–Poly-MFNPs (3.75 μg ml−1), (D) 6.25 μg ml−1, (E) 12.5 μg ml−1 and (F) 25.0 μg ml−1 for 24 h. Diva software after the cells were labelled by PI preceding RNAase treatment and the percentage of apoptosis was calculated. | ||
With the aim of examining the nuclear morphology and identifying cell death in the path of apoptosis, HeLa cell nuclei were further stained with 4′-6-diamidino-2-phenylindole (DAPI), and the data are presented in Fig. 8(A–E). As predicted from the cytotoxicity experiments, the fluorescence images of HeLa cells, incubated with target nanoparticles without DOX (2) has shown that nuclear structures were well preserved without any visible abnormalities [Fig. 8(A)]. However, while treated with increasing concentration of DOX–Poly-FA–MFNPs, there was significant nuclei fragmentation which included condensed nucleus, membrane blebbing and the formation of apoptotic bodies [Fig. 8(B–E)]. It was also clearly visible that the quantity of apoptotic nuclei enhanced with an increasing concentration of DOX–Poly-FA–MFNPs (apoptotic nuclei shown by arrow in the figure). There were previously acclaimed reports that doxorubicin interacts with DNA topoisomerase II (topo II) and can cause the accumulation of enzyme-DNA adducts that ultimately lead to double-strand breaks followed by programmed cell death or apoptosis.56,57 In our case, the same pattern of nuclei fragmentation was observed after treatment of DOX–FA–Poly-MFNPs with HeLa cells.
 | ||
| Fig. 8 Apoptosis study of DOX–FA–Poly-MFNPs by morphological changes in cell nuclei, determined by means of fluorescence microscopy after DAPI staining. HeLa cells were treated with (A) FA–Poly-MFNPs without DOX for 24 h and (B) 3.75, (C) 6.25, (D) 12.5 and (E) 25 μg ml−1 of DOX–FA–Poly-MFNPs for 24 h. Arrows indicate apoptotic nuclei. | ||
3. Conclusion
In summary, stimuli responsive polymer integrated targeted nanomedicine was designed for combined cancer-targeted therapy and multimodal imaging by a facile preparation method. These nanoprobes were found to be potentially capable of dual pH and temperature responsive drug delivery, specifically targeted to cancer cells and also shows intracellular MR based diagnostic/tracking ability. These nanoprobes display a significant amount of drug entrapment inside the polymeric shell and initiate the release of the drugs preferably at pH 5.5 and temperature 37 °C. The in vitro biological studies revealed that these nanoprobes are able to provide a single nanoconstruct, which is capable of simultaneously targeting, tracking and destroying folate-receptor over expressing cancer cells compared to the free drug. They specifically accumulate inside the cancer cells through receptor mediated endocytosis, and the payloads can be released via the stimuli triggered nature of the fabricated nanoprobes. From the biological studies, it is envisioned that the prepared nanohybrids comprised of cancer specific targeting, imaging, and therapeutic application in a single entity could be used as a theranostic platform for cancer treatment and may be of particular interest in the near future.4. Experimental section
4.1. Materials
Pluronic F127 (F127, Mw: 12600 kDa) were specially ordered from BASF, China. Succinic anhydride (SA), triethylamine (TEA), 4-dimethylaminopyridine (DMAP), branched poly-(ethylenimine) (PEI, 25 kDa), ferric chloride (FeCl3), ferrous sulphate (FeSO4·7H2O), trisodium citrate, MnCl2·4H2O, EDBE, folic acid (FA), N-hydroxysuccinimide (NHS), 1-[3-(dimethylamino)propyl]-3-ethyl carbodiimide hydrochloride (EDC), trinitrobenzene sulfonic acid (TNBS), rhodamine isothiocyanate (RITC), doxorubicin, 4′-6-diamidino-2-phenylindole (DAPI), propidium iodide (PI), RNase and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were purchased from Sigma-Aldrich Chemicals, USA. Commercially available dimethyl sulfoxide (DMSO) and N,N′-dimethyl formamide (DMF) were purified by vacuum distillation and dried 1,4-dioxane was distilled over sodium. Chloroform (CHCl3) was procured from Merck, Germany. Fetal bovine serum and minimum essential medium (MEM) were procured from Hyclone, USA and Himedia, India, respectively.
4.2. Methods
:1:1 ratio were dissolved in 40 ml deoxygenated Milli-Q water containing trisodium citrate (1 mg ml−1) in a three necked flask equipped with an argon flow and a mechanical stirrer. To this, ammonium hydroxide (5 ml) was added dropwise and the mixture was stirred for 4 h at 70 °C. After completion of the reaction, black CA-MFNPs were recovered and washed thoroughly with Milli-Q water followed by drying.
The previously synthesized PEI-PF127 was covalently conjugated to the CA-MFNPs via the well established EDC/NHS method. In brief, 0.1 g of CA-MFNPs was dispersed ultrasonically in 20 ml Milli-Q water for 30 min and was activated with 0.1 g of NHS and 0.1 g of EDC under dark for 4 h. To this solution, 20 ml of PEI-PF127 (5 mg ml−1) was added dropwise under ultrasonication at pH around 5–6 followed by stirring for 24 h. Subsequently, these aminated Poly-MFNPs (1) were finally recovered through magnetic separation, washed with Milli-Q water, and followed by drying.
:1 v/v) and pH was maintained at 8–10 using dilute NaOH. To the above FA solution, activation was carried out using EDC (100 mg, 0.48 mM) and NHS (56 mg, 0.48 mM) for 4 h under dark conditions. Subsequently, 100 mg aqueous dispersion of Poly-MFNPs (1) was added to it and the resulting mixture was stirred overnight in the dark at RT. At last, the FA modified Poly-MFNPs (FA–Poly-MFNPs) were magnetically concentrated, washed several times with Milli Q water as well as DMSO and at last, recovered.


With the aim to confirm the targeting efficacy of the DOX–FA-Poly-MFNPs,52 approximately 1 × 105 cells per well were seeded in 96-well plates the day before the experiments to bring the cells to confluence. Then, the DOX–FA–Poly-MFNPs or DOX–Poly-MFNPs with the same concentration (25 μg ml−1) were separately added to both HeLa-HFAR and HeLa-LFAR cells, the medium in wells containing the complexes was totally taken out and replenished with the same volume of fresh medium after 2 h incubation. The cells were then incubated for 48 h at 37 °C. After that, an MTT assay was used to quantify the viability of cells. The assays were carried out according to the manufacturer's instructions. Mean and standard deviation for the triplicate wells were reported.
| SI = Ae−TE/T2 + B, | (1) |
| 1/T2 = 1/T2(0) + r2[Fe], | (2) |
5. Characterization
The surface chemistry and chemical structures of products were analyzed from FTIR and 1H NMR (Bruker, 400 MHz) spectra. The FTIR of the biofunctionalized nanoparticles were recorded in KBr in the range of 400–4000 cm−1 using a model Nexus-870, Thermo Nicolet Corporation, Wisconsin, USA. The phase analysis of the synthesized mixed ferrite nanopowder was performed on an X'pert Pro Phillips X-ray diffractometer. The sample for XRD was prepared by the deposition of well dispersed nanoparticles on a glass slide and, after drying, the analysis was performed by using Ni-filtered Cu-Kα radiation (I = 1.54 Å). The size and morphology of the nanoparticles were observed by high-resolution transmission electron microscopy (HRTEM) (JEOL 3010, Japan) operated at 200 kV. The nanoparticles were thoroughly dispersed in water by ultra-sonication (1 mg/50 ml Milli Q water) and a drop of the solution was placed on a carbon coated copper grid. The average particle size from TEM micrographs was analyzed using image J software. The surface composition of nanoparticles was obtained from X-ray photoelectron spectroscopic (XPS) analysis using an Al Ka excitation source in an ESCA-2000 Multilab apparatus (VG microtech). The sample used in XPS was prepared in the similar procedure as used in TEM. A TNBS assay was used to determine the percentage of primary amines in the NP solution. The –NH2 group concentration was calculated by a graph originated by taking glycine (0–2 mmol) as standard.61 To determine the extent of folate conjugation, UV-Vis spectra were recorded by spectrophotometric analysis of absorbance at 286 nm and 360 nm and also cross-checked by determination of residual amine concentration via the TNBS assay. The dynamic light scattering (DLS) measurements at different temperatures and pHs were done using a Brookhaven 90 Plus particle size analyzer. The surface charge of the nanoparticles was investigated through zeta potential measurements (Zetasizer 4, Malvern Instruments, UK). Thermal analysis was done using a thermal analyzer (Pyris Diamond TG/DTA) with a heating rate of 8 °C min−1 within the temperature range of 50 °C to 1000 °C. Magnetic measurements of nanoparticles were performed using a SQUID-VSM instrument (Ever cool SQUID VSM DC Magnetometer).Acknowledgements
The authors gratefully acknowledge Dr K. R. Patil, Centre of Material Characterization, National Chemical Laboratory (NCL), Pune, for assistance with the XPS studies. The authors are thankful to Eco MRI, Kolkata, for in vitro MR imaging studies. The authors are grateful to the both CSIR New Delhi and DST Nano Mission for providing research fellowship and financial support for this work.Notes and references
- L. Y. Zhang, R. Guo, M. Yang, X. Q. Jiang and B. R. Liu, Adv. Mater., 2007, 19, 2988 CrossRef CAS.
- K. S. Soppimath, D. C. W. Tan and Y. Y. Yang, Adv. Mater., 2005, 17, 318 CrossRef CAS.
- V. Bagalkot, L. F. Zhang, E. Levy-Nissenbaum, S. Jon, P. W. Kantoff, R. Langer and O. C. Farokhzad, Nano Lett., 2007, 7, 3065 CrossRef CAS PubMed.
- H. S. Choi, K. M. Huh, T. Ooya and N. Yui, J. Am. Chem. Soc., 2003, 125, 6350 CrossRef CAS PubMed.
- O. J. Cayre, N. Chagneux and S. Biggs, Soft Matter, 2011, 7, 2211 RSC.
- M. C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293 CrossRef CAS PubMed.
- X. Wu, Z. Wang, D. Zhu, S. Zong, L. Yang, Y. Zhong and Y. Cui, ACS Appl. Mater. Interfaces, 2013, 5, 10895 CAS.
- X. Liu, D. Yu, C. Jin, X. Song, J. Cheng, X. Zhao, X. Qi and G. Zhang, New J. Chem., 2014, 38, 4830 RSC.
- K. Yan, P. Li, H. Zhu, Y. Zhou, J. Ding, J. Shen, Z. Li, Z. Xu and P. K. Chu, RSC Adv., 2013, 3, 10598 RSC.
- H. Meng, M. Xue, T. Xia, Y. L. Zhao, F. Tamanoi, J. F. Stoddart, J. I. Zink and A. E. Nel, J. Am. Chem. Soc., 2010, 132, 12690 CrossRef CAS PubMed.
- H. Ai, C. Flask, B. Weinberg, X. T. Shuai, M. D. Pagel, D. Farrell, J. Duerk and J. Gao, Adv. Mater., 2005, 17, 1949 CrossRef CAS.
- J. H. Park, G. v. Maltzahn, L. Zhang, M. P. Schwartz, E. Ruoslahti, S. N. Bhatia and M. J. Sailor, Adv. Mater., 2008, 20, 1630 CrossRef CAS PubMed.
- P. Guardia, R. D. Corato, L. Lartigue, C. Wilhelm, A. Espinosa, M. G. Hernandez, F. Gazeau, L. Manna and T. Pellegrino, ACS Nano, 2012, 6, 3080 CrossRef CAS PubMed.
- X. Yang, S. Pilla, J. J. Grailer, D. A. Steeber, S. Gong, Y. Chen and G. Chen, J. Mater. Chem., 2009, 19, 5812 RSC.
- J. M. Yang, C. H. Lee, H. J. Ko, J. S. Suh, H. G. Yoon, K. Lee, Y. M. Huh and S. Haam, Angew. Chem., Int. Ed., 2007, 46, 8836 CrossRef CAS PubMed.
- M. K. Yu, Y. Y. Jeong, J. Park, S. Park, J. W. Kim, J. J. Min, K. Kim and S. Jon, Angew. Chem., Int. Ed., 2008, 47, 5362 CrossRef CAS PubMed.
- W. H. Chiang, V. T. Ho, W. C. Huang, Y. F. Huang, C. S. Chern and H. C. Chiu, Langmuir, 2012, 28, 15056 CrossRef CAS PubMed.
- D. Y. Chen, X. W. Xia, H. W. Gu, Q. F. Xu, J. F. Ge, Y. G. Li, N. J. Li and J. M. Lu, J. Mater. Chem., 2011, 21, 12682 RSC.
- L. E. Gerweck and K. Seetharaman, Cancer Res., 1996, 56, 1 Search PubMed.
- X. Hu, X. Hao, Y. Wu, J. Zhang, X. Zhang, P. C. Wang, G. Zou and X. J. Liang, J. Mater. Chem. B, 2013, 1, 1109 RSC.
- C. S. Brazel, J. Pharm. Sci., 2009, 26, 644 CAS.
- D. Yoo, J. H. Lee, T. H. Shin and J. Cheon, Acc. Chem. Res., 2011, 44, 863 CrossRef CAS PubMed.
- A. Popat, J. Liu, G. Q. M. Lu and S. Z. Qiao, J. Mater. Chem., 2012, 22, 11173 RSC.
- X. Mei, D. Y. Chen, N. J. Li, Q. F. Xu, J. F. Ge, H. Li, B. X. Yang, Y. J. Xu and J. M. Lu, Soft Matter, 2012, 8, 5309 RSC.
- H. C. Dong, V. Mantha and K. Matyjaszewski, Chem. Mater., 2009, 21, 3965 CrossRef CAS.
- T. Isojima, M. Lattuada, J. B. Vander Sande and T. A. Hatton, ACS Nano, 2008, 2, 1799 CrossRef CAS PubMed.
- S. Chen, Y. Li, C. Guo, J. Wang, J. H. Ma, X. F. Liang, L. R. Yang and H. Z. Liu, Langmuir, 2007, 23, 12669 CrossRef CAS PubMed.
- J. J. Lai, J. M. Hoffman, M. Ebara, A. S. Hoffman, C. Estournes, A. Wattiaux and P. S. Stayton, Langmuir, 2007, 23, 7385–7391 CrossRef CAS PubMed.
- S. Bhattacharya, F. Eckert, V. Boyko and A. Pich, Small, 2007, 3, 650 CrossRef CAS PubMed.
- F. Zhang and C. C. Wang, Langmuir, 2009, 25, 8255 CrossRef CAS PubMed.
- B. Sahoo, S. Devi, R. K. Banerjee, T. K. Maiti, P. Pramanik and D. Dhara, ACS Appl. Mater. Interfaces, 2013, 5, 3884 CAS.
- G. Wanka, H. Hoffmann and W. Ulbricht, Macromolecules, 1994, 27, 4145 CrossRef CAS.
- S. Patra, E. Roy, P. Karfa, S. Kumar, R. Madhuri and P. K. Sharma, ACS Appl. Mater. Interfaces, 2015, 7, 9235 CAS.
- A. V. Kabanov, E. V. Batrakova and V. Y. Alakhov, J. Controlled Release, 2002, 82, 189 CrossRef CAS PubMed.
- S. H. Choi, S. H. Lee and T. G. Park, Biomacromolecules, 2006, 7, 1864 CrossRef CAS PubMed.
- S. H. Lee, S. H. Choi, S. H. Kim and T. G. Park, J. Controlled Release, 2008, 125, 25 CrossRef CAS PubMed.
- W. Zhang, K. Gilstrap, L. Wu, K. C. R. Bahadur, M. A. Moss, Q. Wang, X. Lu and X. He, ACS Nano, 2010, 4, 6747 CrossRef CAS PubMed.
- L. Yang, C. Guo, L. Jia, X. Liang, C. Liu and H. Liu, J. Colloid Interface Sci., 2010, 350, 22 CrossRef CAS PubMed.
- M. Thomas and A. M. Klibanov, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 14640 CrossRef CAS PubMed.
- J. W. Park, K. H. Bae, C. Kim and T. G. Park, Biomacromolecules, 2011, 12, 457 CrossRef CAS PubMed.
- U. I. Tromsdorf, N. C. Bigall, M. G. Kaul, O. T. Bruns, M. S. Nikolic, B. Mollwitz, R. A. Sperling, R. Reimer, H. Hohenberg, W. J. Parak, S. Fo1rster, U. Beisiegel, G. Adam and H. Weller, Nano Lett., 2007, 7, 2422 CrossRef CAS PubMed.
- S. D. Li and L. Huang, Mol. Pharmaceutics, 2008, 5, 496 CrossRef CAS PubMed.
- B. Chertok, A. E. David and V. C. Yang, Biomaterials, 2010, 31, 6317 CrossRef CAS PubMed.
- P. Singh, U. Gupta, A. Asthana and N. K. Jain, Bioconjugate Chem., 2008, 19, 2239 CrossRef CAS PubMed.
- D. Bhattacharya, M. Das, D. Mishra, I. Banerjee, S. K. Sahu, T. K. Maiti and P. Pramanik, Nanoscale, 2011, 3, 1653 RSC.
- Y. L. Su, J. Wang and H. Z. Liu, J. Phys. Chem. B, 2002, 106, 11823 CrossRef CAS.
- K. Nawara, J. Romiszewski, K. Kijewska, J. Szczytko, A. Twardowski, M. Mazur and P Krysinski, J. Phys. Chem. C, 2012, 116, 5598 CAS.
- J. Zhang, S. Rana, R. S. Srivastava and R. D. K. Misra, Acta Biomater., 2008, 4, 40 CrossRef CAS PubMed.
- X. Yang, J. J. Grailer, I. J. Rowland, A. Javadi, S. A. Hurley, D. A. Steeber and S. Gong, Biomaterials, 2010, 31, 9065 CrossRef CAS PubMed.
- X. Yang, J. J. Grailer, I. J. Rowland, A. Javadi, S. A. Hurley, V. Z. Matson, D. A. Steeber and S. Gong, ACS Nano, 2010, 4, 6805 CrossRef CAS PubMed.
- J. H. Lee, Y. M. Huh, Y. W. Jun, J. W. Seo, J. T. Jang, H. T. Song, S. E. Kim, J. Cho, H. G. Yoon, J. S. Suh and J. Cheon, Nat. Med., 2007, 13, 95 CrossRef CAS PubMed.
- S. Wen, H. Liu, H. Cai, M. Shen and X. Shi, Adv. Healthcare Mater., 2013, 2, 1267 CrossRef CAS PubMed.
- Y. H. Ling, A. K. el-Naggar, W. Priebe and R. Perez-Soler, Mol. Pharmacol., 1996, 49, 832 CAS.
- O. Bar-On, M. Shapira and D. D. Hershko, Anticancer Drugs, 2007, 18, 1113 CrossRef CAS PubMed.
- T. J. Westmoreland, S. M. Wickramasekara, A. Y. Guo, A. L. Selim, T. S. Winsor, A. L. Greenleaf, K. L. Blackwell, J. R. Marks and C. B. Bennett, PLoS One, 2009, 4, e5830 Search PubMed.
- A. Bodley, L. F. Liu and M. Israel, Cancer Res., 1989, 49, 5969 CAS.
- I. G. Cowell, A. L. Okorokov, S. A. Cutts, K. Padget, M. Bell, J. Milner and C. A. Austin, Exp. Cell Res., 2000, 255, 86 CrossRef CAS PubMed.
- Y. H. Bae, K. M. Huh, Y. Kim and K. H. Park, J. Controlled Release, 2000, 64, 3 CrossRef CAS PubMed.
- K. M. Park, J. W. Bae, Y. K. Joung, J. W. Shin and K. D. Park, Colloids Surf., B, 2008, 63, 1 CrossRef CAS PubMed.
- D. Bhattacharya, A. Baksi, I. Banerjee, R. Ananthakrishnan, T. K. Maiti and P. Pramanik, Talanta, 2011, 86, 337 CrossRef CAS PubMed.
- F. E. Levy, M. C. Andry and M. C. Levy, Int. J. Pharm., 1993, 96, 85 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed tables of DLS after modification with PEI-PF127 and FA, and amine quantification results, change in zeta potential with pH, dual pH and temperature responsive transition of HDs of FA–Poly-MFNPs and Poly-MFNPs at fixed temperature as well as pH, FT-IR, UV-Vis Spectra, TGA, XPS, VSM analysis are given. Cell viability assays of Poly-MFNPs and FA–Poly-MFNPs in cancer HeLa and normal HaCaT cell lines and intracellular uptake of RITC labeled FA–Poly-MFNPs in the presence and in the absence of free FA. 1HNMR of the block copolymer PEI-PF127 is given as Fig. S12 which was used for the surface modification with CA-MFNPs. See DOI: 10.1039/c5nj02504d |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |